

Summary
Lung cancer in never-smokers is an important disease often characterized by mutations in EGFR, yet risk reduction measures and effective chemopreventive strategies have not been established. We identify mTOR as a new and potentially valuable target for EGFR mutant lung cancer, as mTOR was activated in human lung cancers with EGFR mutations, which increased with acquisition of T790M mutation. In a mouse model of EGFR mutant lung cancer, activation of mTOR was an early event. As a single agent, the mTOR inhibitor rapamycin, prevented tumor development, prolonged overall survival, and improved outcomes after treatment with an irreversible EGFR TKI. These studies support clinical testing of mTOR inhibitors to prevent the development and progression of EGFR mutant lung cancers.
Introduction
Lung cancer is the leading cause of cancer-related death in both males and females in the United States, which is mostly related to smoking. Nonetheless, 25% of all lung cancer cases worldwide (15% of lung cancers in males and 53% in females) are not attributable to smoking (Sun et al., 2007), which makes lung cancer in never-smokers an important and common problem. Molecular differences in lung cancers between smokers and never-smokers have been identified. For example, epidermal growth factor receptor (EGFR) mutations, echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusions, ROS1 rearrangements (CD74-ROS1 fusion), and kinesin family member 5B (KIF5B)-ret proto-oncogene (RET) fusions are more likely to be identified in never-smokers (Johnson et al., 2012).
Somatic mutations of EGFR in non-small cell lung cancer (NSCLC), such as point mutations in exon 21 (e.g. L858R) and exon 19 deletions, are gain-of-function and enhance autophosphorylation of EGFR, resulting in EGFR-addicted lung cancers that are sensitive to EGFR specific tyrosine kinase inhibitors (TKIs) (Sordella et al., 2004). Although profound responses are observed with EGFR TKIs in patients with the activating somatic EGFR mutations, these therapies are not curative. Continued exposure to EGFR TKIs selects for resistant populations and/or induces EGFR TKI-resistant mechanisms in tumor cells (Pao and Chmielecki, 2010). Emergence of tumors with a secondary EGFR mutation in exon 20, T790M, is the most common mechanism of resistance to reversible EGFR TKIs, which is a vexing clinical problem (Kobayashi et al., 2005; Pao et al., 2005). Strategies to overcome EGFR TKI-resistance by T790M include 1) prevention of emergence of the resistant populations; 2) rechallenge with TKIs after a drug holiday that may allow dilution or disappearance of T790 mutation in the absence of the TKI-selection pressure; and 3) treatment of the resistant population with second or third-generation irreversible EGFR TKIs (Hirsch et al., 2013). The concept of maintenance therapy for patients with EGFR mutations treated with first-line EGFR TKI has not been established.
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that controls cell proliferation and survival (Laplante and Sabatini, 2012). mTOR activity also contributes to resistance to EGFR TKI in lung cancer cells (Fei et al., 2013). Previously, we reported that rapamycin, an mTOR inhibitor, prevented the development of tobacco carcinogen–induced lung tumors in a mouse model (Granville et al., 2007). These data prompted us to hypothesize that mTOR inhibition will prevent the emergence of EGFR TKI-resistant populations with T790M mutation. Here, we show that human NSCLC specimens with EGFR mutations that subsequently acquire T790M mutation have increased mTOR activation. In a series of preclinical studies with a doxycycline-inducible, mutant EGFRL858R+T790M (mEGFRL+T) lung cancer mouse model, we demonstrate that rapamycin prevents the growth of T790M tumors, which was associated with improved overall survival (OS), as well as increased progression-free survival (PFS) and OS after treatment with an irreversible EGFR TKI.
Results
mTOR is activated in human NSCLC with EGFR mutations
We analyzed a series of human NSCLC specimens with mutant EGFRL858R (mEGFRL) or mEGFRL+T for phosphorylation of the ribosomal protein, S6, using immunohistochemistry. All eleven NSCLC specimens showed mTOR activation (Figure 1A and Figures S1A and S1B). mEGFRL+T specimens had increased phosphorylation of S6 than those with mEGFRL (p=0.0006, Figure 1B), indicating greater mTOR activation in lung cancers expressing mEGFRL+T, and suggesting that mTOR inhibitors might prevent emergence of mutant EGFR lung cancers with T790M mutation.
Figure 1. mTOR is activated in human NSCLC with EGFR mutations.
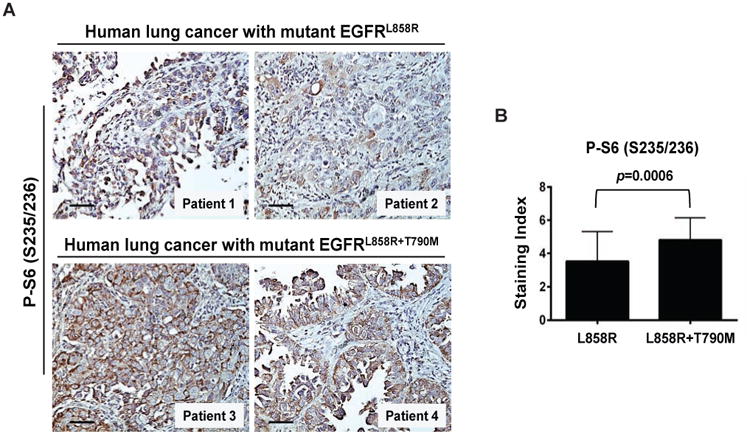
(A) IPhotomicrographs show representative staining for phosphorylated S6 (Ser235/236) as a marker of mTOR activation in 2 specimens with mEGFRL (upper) or mEGFRL+T (lower). IHC was performed as described in Supplemental Experimental Procedures. The scale bars represent 50 μm. See also Figures S1A and S1B.
(B) Increased activation of mTOR in lung tumor tissues with mEGFRL+T. The phosphorylated S6 staining index was established as described in Supplemental Experimental Procedures. Columns, mean of total 40 fields in lung tumor tissues from 4 patients with mEGFRL (10 fields per patient) or from 4 patients with mEGFRL+T. bars, standard deviation. Two-tailed p value was obtained from Unpaired t test.
Role of mTOR in established mutant EGFR-driven lung tumors and during lung tumor regression caused by doxycycline withdrawal
To determine if mTOR could be a therapeutic target in established mutant EGFR-driven lung tumors, mEGFRL+T mice were treated with vehicle or rapamycin. Disease progression was observed in both groups (Figure 2A), despite the fact that mTOR was inhibited by rapamycin (Figure 2B). This demonstrates that mTOR inhibition alone is insufficient to cause regression of established mutant EGFR-driven lung tumors.
Figure 2. Role of mTOR in established mutant EGFR-driven lung tumors and during lung tumor regression caused by doxycycline withdrawal.
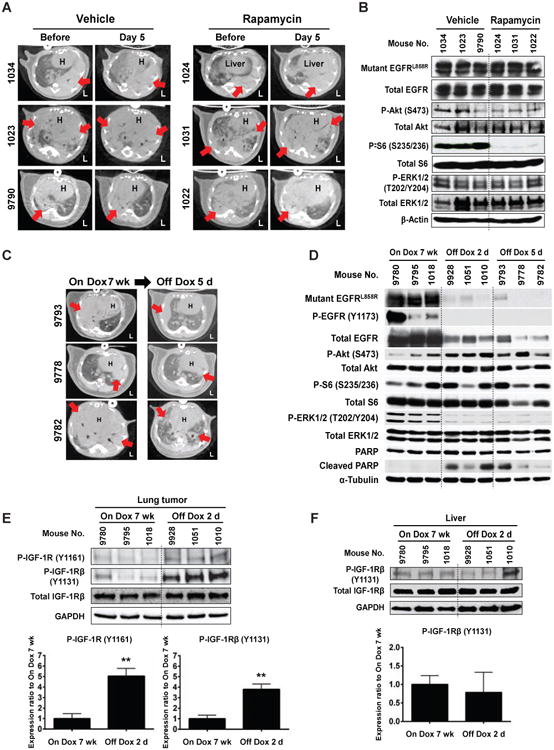
(A) Treatment with rapamycin alone is ineffective in the established mutant EGFR-driven lung tumors. 6 C/L858R+T790M mice were given Dox for 7 weeks, scanned by micro-CT, and were treated with vehicle or rapamycin for 5 days and then rescanned. The dosing and schedule of rapamycin were the same as the chemoprevention study. H, heart. L, left side. Red arrows indicate dense areas of lung tumors.
(B) mTOR inhibition in lung tumors extracted after 3 hours after last rapamycin administration in (A). IB was performed using the indicated antibodies.
(C) Dox withdrawal induces regression of mEGFRL+T-driven lung tumors. CT images show tumor regression before and after Dox withdrawal for 5 days (5d) in 3 mice administrated on Dox for 7 weeks. Red arrows indicate dense areas of lung tumors. See also Figure S1C.
(D) The activation of Akt/mTOR pathway is maintained during mEGFRL+T-driven lung tumor regression. After Dox administration for 7 weeks, lung tumors from C/L858R+T790M mice were extracted on the indicated days after Dox withdrawal. IB was performed using the indicated antibodies. See also Figure S1C.
(E and F) IGF-1R is activated by Dox withdrawal for 2 days (2d) in lung tissues (E), but not liver tissues (F). IB was performed using the indicated antibodies. Densitometry analysis was performed using ImageJ software, and levels of each marker were normalized to total expression and/or GAPDH for each sample. Columns, mean from all three mice examined in On Dox 7 wk or Off Dox 2d group. bars, standard deviation. Statistical analysis of the densitometry was performed with two-tailed Unpaired t test. **p<0.01.
The continued expression of EGFRL+R in this mouse model is dependent on the administration of doxycycline (Dox). To assess the role of mTOR in mutant EGFR-induced tumor maintenance, we measured activation of EGFR and the downstream signaling pathways upon Dox withdrawal. Lung tumor regression was observed after 5 days of Dox withdrawal (Figures 2C and S1C). Dox withdrawal decreased expression and activation of EGFR, as well as extracellular signal-regulated kinases (ERK1/2), which was associated with increased cleaved PARP, a marker of apoptosis. In contrast to ERK, activation of Akt/mTOR was not decreased by Dox withdrawal (Figure 2D), indicating that the Akt/mTOR pathway is independent of EGFR signaling. To assess the mechanism by which activation of Akt and mTOR are maintained during regression, activation of the insulin-like growth factor 1 receptor (IGF-1R) was assessed. Increased activation of IGF-1R was observed 2 days after Dox withdrawal (P<0.01, Figure 2E), implicating activated IGF-1R in the maintenance of Akt/mTOR activation. To confirm that activation of IGF-1R was a local event related to lung tumor regression, the status of IGF-1R in liver samples from the same mice in Figure 2E was assessed. IGF-1R was not activated in liver tissues (Figure 2F). Taken together, these data suggest that activation of the Akt/mTOR pathway is maintained by IGF-1R activation to compensate for EGFR/ERK1/2 deactivation during regression of mutant EGFR-driven lung tumors in a futile attempt to maintain survival of tumor cells.
Rapamycin prevents tumorigenesis in mEGFRL+T mice
Previously, we reported that rapamycin prevented the development of tobacco carcinogen–induced lung tumors in a mouse model (Granville et al., 2007). If mTOR inhibition was to prevent tumorigenesis in mEGFRL+T mice, we hypothesized that it should be an early event. Therefore, we assessed mTOR activation in lung lysates prior to tumor development. Dox induced expression of mutant EGFR and activated the EGFR/Akt/mTOR pathway (Figure 3A), but not EGFR/ERK1/2 signaling, suggesting that Akt/mTOR activation is an early event in EGFR-addicted lung tumorigenesis. These results strengthened our hypothesis that mTOR inhibition will prevent mutant EGFR-driven lung cancers.
Figure 3. Rapamycin prevents tumorigenesis in mEGFRL+T mice.
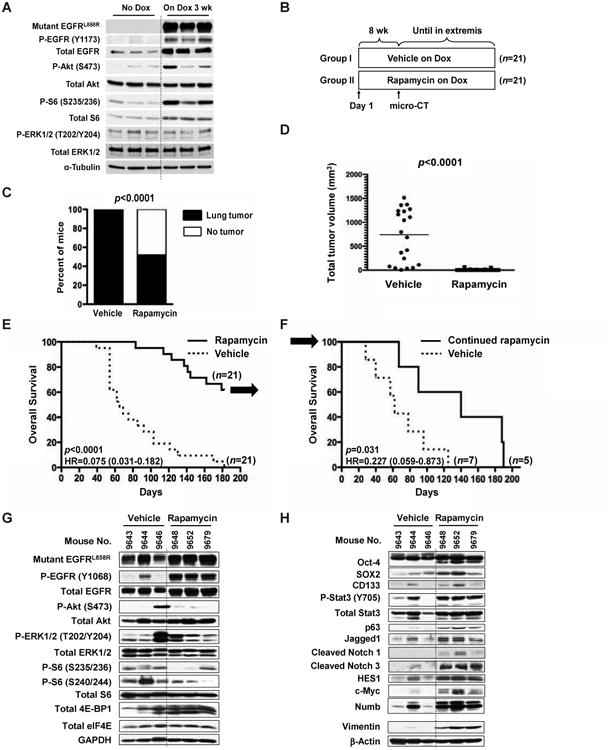
(A) mTOR activation following the induction of mutant EGFR precedes lung tumor development. Six C/L858R+T790M mice received normal diet or Dox for 3 weeks, and then the normal lungs were extracted. IB was performed using the indicated antibodies. mTOR activation was measured by phosphorylated S6.
(B) Schema of chemoprevention study. See also Table S1A.
(C) Rapamycin decreases lung tumor incidence. After 8 weeks of vehicle or rapamycin treatment, lung tumor-bearing mice were identified by micro-CT. Lung tumors were detected in 100% of mice (21/21) in vehicle-treated and in 52.4% of rapamycin-treated mice (11/21). Fisher's exact test, two-tailed, p< 0.0001. See also Table S1B.
(D) Rapamycin decreases TV. TV was calculated after 8 weeks of vehicle or rapamycin treatment as described in Experimental Procedures. Mann-Whitney test, two-tailed, p< 0.0001. For the dot plots, each point represents a mouse and the lines represent the median values. See also Table S1B.
(E) Rapamycin prolongs OS in the chemoprevention study. Median survival in vehicle group was 65 days, but had not been reached in rapamycin group. Arrow represents 12 remaining mice in rapamycin group at 182 days for a rapamycin withdrawal study. HR, hazard ratio. See also Table S1B.
(F) Continuation of rapamycin is required for OS prolongation in the rapamycin withdrawal study. Arrow represents 12 remaining mice in rapamycin group from Figure 3E. Median survival in vehicle and continued rapamycin group was 62 days and 140 days, respectively. HR, hazard ratio. See also Figure S2C and Table S1C.
(G and H) Tumors from mice that progressed on rapamycin maintain mTOR inhibition, but show the increased expression of lung stem cell markers, Notch signaling, and vimentin. Three mice in vehicle group were from the chemoprevention study in (B). See also Table S1B. On the other hand, three mice in rapamycin group were from the rapamycin withdrawal study in (F). See also Table S1C. Lung tumors were extracted, and IB was performed using the indicated antibodies.
To test this, a study was performed with rapamycin using a dosing regimen that yields trough drug levels that are well tolerated in humans (Granville et al., 2007). After confirming that rapamycin did not decrease transgene expression (Supplemental Results and Figures S1D and S1E) in the mEGFRL+T mouse model, mEGFRL+T mice were randomized to vehicle or rapamycin (Figure 3B and Table S1A). At 8 weeks, rapamycin decreased lung tumor incidence by 47.6% (p<0.0001, Figure 3C) and total tumor volume (TV) by 98% (p<0.0001, Figure 3D). Treatment with rapamycin was continued until signs of distress appeared, which mandated euthanasia (Figure S2A). Median OS was not reached in the rapamycin group, but compared to the vehicle group, the median OS was prolonged by more than three fold (p<0.0001, Figure 3E), which was inversely correlated to TV (Figure S2B and Table S1B), suggesting that prolongation of survival was due to inhibition of tumor growth. When the last mouse in the vehicle group died, the remaining mice in rapamycin group were evenly distributed based on tumor size and randomized to continued rapamycin or vehicle (Figures S2C to S2E and Table S1C). Mice switched to vehicle had shorter OS than continued rapamycin (p=0.031, Figure 3F), indicating that continuous exposure to rapamycin is required for clinical benefit and prolongation of survival. Collectively, these findings show that rapamycin is highly effective to prevent lung tumorigenesis in mEGFRL+T mice.
Markers of rapamycin resistance
Because the mice on rapamycin treatment eventually die of lung cancers, resistance mechanisms to rapamycin were examined. Rapamycin-resistant tumors maintained mTOR inhibition, as assessed by two phosphorylation sites of S6, as well as increased expression of hypophosphorylated forms of 4E-BP1 (Figure 3G). Feedback activation of Akt was not observed. Because mTOR signaling induces differentiation of human embryonic stem cells (Easley et al., 2010), we assessed markers of undifferentiated tumor cells. Rapamycin-resistant tumors had increased expression of lung stem cell markers such as Oct-4 and SOX2, as well as increased activation of a Notch pathway comprised of Stat3/p63/Jagged1/Notch/HES1/c-Myc/Numb (Ma et al., 2010). Increased expression of vimentin, a mesenchymal marker, was also observed (Figure 3H). Other mechanisms reported to be related to EGFR TKI-resistance such as increased expression of SKP2, Bim, and her3 were not observed (data not shown). These results suggest that rapamycin resistance is associated with the selection or induction of pluripotent tumor cells.
Rapamycin prolongs PFS and OS after treatment with an irreversible EGFR TKI
The issue of discontinuous therapy with an EGFR TKI to delay emergence of T790M mutation is controversial. For example, patients with acquired resistance to reversible EGFR TKIs showed re-response to the TKI therapy after a drug holiday (Hirsch et al., 2013), whereas Chaft et al reported discontinuation of erlotinib or gefitinib (first-generation reversible EGFR TKIs) led to accelerated disease progression referred to “disease flare” (Chaft et al., 2011). Because rapamycin prevented development of mutant EGFR lung tumors with T790M mutation, we hypothesized that rapamycin might be beneficial as an interventional therapy during a TKI-free period following EGFR TKI treatment. Such an approach might prevent disease flare and prolong PFS.
To test this hypothesis, we performed a switch maintenance study with rapamycin following treatment of mEGFRL+T mice with a third-generation irreversible EGFR TKI, WZ4002 (Zhou et al., 2009) (Figure 4A). WZ4002 rapidly and efficiently inhibited EGFR activation, as well as activation of Akt, ERK, and mTOR (Figure 4B). The design of the maintenance study is as follows. 24 C/L858R+T790M mice were treated with Dox for 5 weeks, at which time lung tumors were documented by micro-computed tomography (CT). Mice were then treated with WZ4002 for 5 days and divided into treatment groups based on equal distribution of tumor volumes. After 5 days, WZ4002 was stopped and mice were randomized to vehicle or rapamycin (Figures S2F to S2H and Table S2). WZ4002 effectively treated established tumors, with a 100% response rate and average reduction in tumor volume of approximately 80% (Figure S2H). Mice randomized to vehicle showed rapid progression, but mice treated with rapamycin had delayed disease progression (Figure 4C). Rapamycin prevented disease flare (p=0.0006, Table S2), delayed the percent increase of TV over time (p=0.030, Figure S2I), and prolonged PFS (p=0.014, Figure 4D). Treatment with rapamycin also prolonged OS (p=0.0007, Figure 4E) and was associated with sustained mTOR inhibition during this maintenance study (Figure S2J). To assess whether rapamycin exerted direct effects on epithelial cells or worked independently, in vitro experiments were performed in H1975 and H820 cells that have T790M EGFR mutation. In each cell line, an LD80 was established for WZ4002. After treatment and subsequent washout with WZ4002, cells were treated with vehicle or rapamycin. Rapamycin markedly diminished cell proliferation (Supplemental Results and Figures S3 and S4A), which correlated with inhibition of EGFR as well as mTOR signaling (Figure S4B), suggesting that rapamycin exerts direct effects on EGFR mutant lung cancer cells. Taken together, our results show that rapamycin prevents disease flare during an EGFR TKI-free period following TKI treatment, and prolongs time to disease progression as well as OS.
Figure 4. Rapamycin prolongs PFS and OS after treatment with an irreversible EGFR TKI.
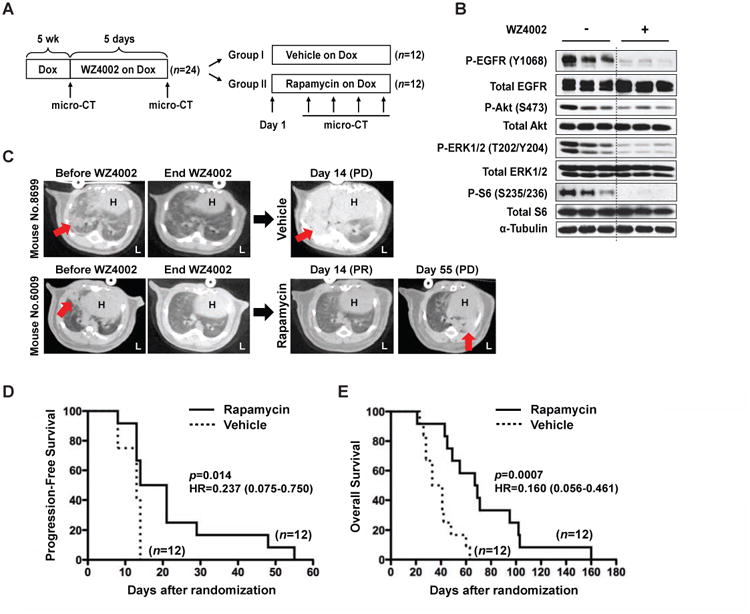
(A) Schema of switch maintenance therapy. See also Table S2.
(B) WZ4002 inhibits EGFR and the downstream Akt/mTOR pathway in mEGFRL+T-driven lung tumors. As a biomarker study, 6 lung tumor-bearing C/L858R+T790M mice on Dox for 7 weeks were given 2 doses of vehicle or WZ4002 (25 mg/kg once a day) by gavage. Lung tumors were extracted 3 hours after last administration. IB was performed using the indicated antibodies. (Note- the 2 doses administration of WZ4002 did not induce tumor regression (data not shown)).
(C) Two representative mice from the switch maintenance study. Mouse No. 8699 in vehicle group showed PD by day 14; on the other hand, rapamycin prevented disease progression to day 55 in mouse No. 6009. H, heart. L, left side. Red arrows indicate dense areas of lung tumors. See also Table S2.
(D) Rapamycin prolongs PFS. PD of mice in vehicle or rapamycin groups was assessed by criteria to classify tumor responses as described in Supplemental Experimental Procedures. Median PFS in vehicle and rapamycin group was 13.0 days and 17.5 days, respectively. HR, hazard ratio. See also Table S2.
(E) Rapamycin prolongs OS. Median OS in vehicle and rapamycin group was 37 days and 68 days, respectively. HR, hazard ratio. See also Table S2.
Discussion
mTOR is progressively activated in human lung cancers with T790M mutation, and is an early event in a murine model of lung tumorigenesis. We demonstrate that rapamycin decreases the incidence of mutant EGFR-driven lung tumors and prolongs survival, and prevents disease progression of lung cancers after EGFR TKI treatment. This is the first study describing the involvement of mTOR activation in EGFR-addicted lung tumorigenesis, and the first to show that rapamycin prolongs PFS and OS. Although rapamycin can extend the lifespan of mice (Harrison et al., 2009), we contend that rapamycin prolongs OS in our studies by inhibiting lung cancer mortality rather than by extension of the overall lifespan, because mTOR inhibition was still observed in tumors from mice that progressed on rapamycin treatment (Figures 3G and S2J), and OS was inversely correlated to TV (Figure S2B).
Although T790M mutation accounts for approximately 50% of acquired EGFR TKI-resistance, when tumor cells acquire the resistant mutation remains unclear. Recent reports have identified T790M mutation in 25.2% and 31.5% of EGFR TKI-naïve and pre-TKI samples, respectively, implying that the incidence of T790M may be higher than expected (Su et al., 2012) and that the presence of somatic T790M mutation before treatment might cause primary resistance to reversible EGFR TKIs (Ohashi et al., 2013). This suggests that an mTOR inhibitor might prevent outgrowth of tumors with T790M mutation. Moreover, rapamycin prevented disease flare during a TKI-free period following EGFR TKI treatment and prolonged PFS in our preclinical switch maintenance study (Figure 4D and Table S2), suggesting that inhibition of mTOR might prevent disease flare caused by “drug holidays” or intermittent therapy in advanced NSCLC patients with EGFR mutations.
Although rapamycin decreased tumor incidence and TV, and markedly prolonged OS in this study, mice on rapamycin eventually die of lung cancer. Unexpectedly, we did not observed mechanisms of resistance such as feedback activation of Akt (Laplante and Sabatini, 2012) (Figures 3G and S2J). Inhibition of mTOR was sustained, even at the point of death, demonstrating that rapamycin maintained adequate intracellular levels to inhibit downstream signaling from mTOR. Because prolonged stimulation of mTOR can lead to stem cell depletion through the activation of senescence programs, the use of mTOR inhibitors might protect adult stem cells from initiating premature cell senescence programs (Iglesias-Bartolome and Gutkind, 2012). This is consistent with our observation that long-term treatment with rapamycin increased expression of stem cell markers such as Oct-4 and SOX2, increased activation of a Notch pathway, and increased expression of mesenchymal markers such as vimentin (Figure 3H). This implicates pluripotent tumor cells in rapamycin resistance.
In summary, our study highlights the importance of mTOR activation as an early event in EGFR-addicted lung tumorigenesis and the efficacy of rapamycin as an agent to prevent the development or progression of EGFR mutant lung cancers. These results can be used to develop strategies to incorporate mTOR inhibitors into clinical trials with EGFR TKI to prevent the emergence of lung cancers with T790M mutation.
Experimental Procedures
Mouse cohort and genotyping
All experiments were conducted under a protocol approved by the NCI animal care and use committee. NIH is an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-certified facility. The generation of bi-transgenic progeny (harboring the CCSP-rtTA and tet-regulated EGFRL858R+T790M transgenes; “C/L858R+T790M”) has been previously described, (Regales et al., 2007). CCSP-rtTA and EGFR genotype was assessed by PCR from tail clips (Floyd et al., 2005; Politi et al., 2006). The expression of mEGFRL+T was assessed by immunoblotting (IB) and immunohistochemical analysis (IHC) with an antibody to EGFR (L858R mutant specific, 43B2). All mice were housed in specific pathogen-free housing with NIH-31 regular diet and autoclaved water ad libidum. For all experiments, mice were fed doxycycline-impregnated food pellets (NIH-31 regular diet with 650 ppm doxycycline, Harlan-Teklad, Madison, WI, USA) to induce expression of mutant EGFR.
Chemoprevention study
42 fourteen-week-old C/L858R+T790M mice were randomized to vehicle or rapamycin group based on both their gender and age. Rapamycin (LC Laboratories, Woburn, MA, USA) was formulated in vehicle (5% Tween 80 and 5% polyethylene glycol in 0.9% NaCl to yield a final concentration of 4% DMSO from Sigma, St. Louis, MO, USA) and injected intraperitoneally at 1.5 mg/kg every other day following a 4.5 mg/kg loading dose as described previously (Granville et al., 2007). Tumor-bearing mice were identified by micro-CT after 8 weeks of rapamycin treatment (primary end point: lung tumor incidence). The vehicle or rapamycin treatment was continued to assess OS as the secondary end point. For all studies, mice that showed respiratory distress and reduced motility were euthanized. Day 1 for OS was the date when the 42 C/L858R+T790M mice were randomized. Day 182 was the end date of this chemoprevention study when the last mouse in vehicle group died.
Rapamycin withdrawal study
After all mice in the vehicle group in the chemoprevention study had died, 12 remaining C/L858R+T790M mice in rapamycin group were evenly distributed based on tumor size and randomized to vehicle or continued rapamycin group. The dosing and schedule of rapamycin were the same as the chemoprevention study. The primary end point was OS. Day 1 for OS was the date when the 12 mice were randomized. Day 190 was the end date of rapamycin withdrawal study when the last mouse in continued rapamycin group died.
Switch maintenance study
Micro-CT scans were performed on 24 eleven-week-old C/L858R+T790M mice on Dox for 5 weeks. All mice had lung tumors and were treated with WZ4002 by gavage at 50 mg/kg twice a day for 5 days, and then were rescanned by micro-CT to assess tumor regression. WZ4002 was formulated in vehicle (10% 1-Methyl-2-pyrrolidinone and 90% Polyethylene glycol-300 from Sigma). All WZ4002 responding mice were randomized to vehicle or rapamycin group based on the status of lung tumors. The dosing and schedule of rapamycin were the same as the chemoprevention study. To assess PFS as the primary end point, mice were monitored by micro-CT once a week until disease progression. When mice showed progression, monitoring was stopped, but the switch maintenance therapy with rapamycin was continued to assess OS as a secondary end point. Once a mouse showed respiratory distress and reduced motility, it was euthanized. Disease flare in this preclinical study was defined as progressive disease (PD) happen earlier than median PFS in vehicle group (13.0 days). Day 1 for both PFS and OS was the date when the 24 WZ4002-responding mice were randomized. Day 160 in OS was the end date of the switch maintenance study when the last mouse in rapamycin group died.
Patients
Lung cancer specimens of patients with NSCLC were obtained from the Dana-Farber Cancer Institute, Boston, MA, USA. Tumor tissues with mEGFRL were from 4 patients in pre-erlotinib treatment, whereas those with mEGFRL+T were from other 7 patients treated with erlotinib, which showed erlotinib-resistance. Information on gender, age, and histology was available for most samples. The approval of the institutional review board of Dana-Farber Cancer Institute was obtained for all studies.
Statistics
All analyses were performed using the GraphPad Prism software version 5.0c (GraphPad Software, Inc. La Jolla, CA, USA). OS and PFS curves were created by the Kaplan-Meier method. Comparisons of two survival curves were performed with the Log-rank test. Correlation analyses were performed with Pearson (ρ: Pearson r). P values of less than 0.05 were considered significant.
Supplementary Material
Highlights.
mTOR activation is detected in human lung cancers with EGFR mutations
mTOR activation is an early event in a mouse model of EGFR-mutant lung cancer
Rapamycin prevents growth of EGFR T790M mutant tumors and prolongs overall survival
Rapamycin improves progression-free and overall survival after EGFR TKI treatment
Acknowledgments
The authors would like to thank Maiga Emmanuel (Office of the Director, NCI) for assistance with genotyping; Morales-Contreras Juan, Dumas Tarra, and Dr. John U. Dennis (Laboratory Animal Medicine, NCI) for veterinary services; Dr. Jeffrey A. Whitsett (University of Cincinnati College of Medicine, Cincinnati, OH, USA) for providing Clara cell secretory protein (CCSP)-reverse tetracycline responsive transactivator (rtTA) transgenic mice; Dr. Nathanael S. Gray (Harvard Medical School, Boston, MA, USA) for providing WZ4002 compound; and the NIH Fellows Editorial Board for editorial assistance. This research was supported by the Intramural Research Program of the NIH, NCI, CCR (P.A. Dennis) and NIH R01CA135257 (P.A. Jänne). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. Publication of this article was funded in part by the Open Access Promotion Fund of the Johns Hopkins University Libraries.
Footnotes
Supplemental Information: The Supplemental Information includes four Supplemental Figures and Legends, two Supplemental Tables, Supplemental Experimental Procedures, Supplemental Results, and Supplemental References and can be found with this article online.
Author Contributions: S.K. designed and performed the experiments, collected and analyzed data, and wrote the manuscript; M.C.H. established the mouse lung-specific tumor models and wrote the manuscript; J.R.M. and W.W. performed the experiments, collected and analyzed data; D.D., S.K., and J.R.M performed micro-CT scan; L.R. and W.P. provided the double (L858R+T790M) mutant EGFR transgenic mice and H1975 cell line, and advised on the establishment of the mouse lung-specific tumor models; K.K.W. and P.A.J. advised on the study design; M.B. and P.A.J. collected and provided human NSCLC specimens; P.A.D designed the experiments, analyzed data, wrote the manuscript, and supervised the project. All authors discussed the results and implications and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011;17:6298–6303. doi: 10.1158/1078-0432.CCR-11-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley CAt, Ben-Yehudah A, Redinger CJ, Oliver SL, Varum ST, Eisinger VM, Carlisle DL, Donovan PJ, Schatten GP. mTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells. Cell Reprogram. 2010;12:263–273. doi: 10.1089/cell.2010.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei SJ, Zhang XC, Dong S, Cheng H, Zhang YF, Huang L, Zhou HY, Xie Z, Chen ZH, Wu YL. Targeting mTOR to overcome epidermal growth factor receptor tyrosine kinase inhibitor resistance in non-small cell lung cancer cells. PloS one. 2013;8:e69104. doi: 10.1371/journal.pone.0069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd HS, Farnsworth CL, Kock ND, Mizesko MC, Little JL, Dance ST, Everitt J, Tichelaar J, Whitsett JA, Miller MS. Conditional expression of the mutant Ki-rasG12C allele results in formation of benign lung adenomas: development of a novel mouse lung tumor model. Carcinogenesis. 2005;26:2196–2206. doi: 10.1093/carcin/bgi190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila RI, Dennis PA. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007;13:2281–2289. doi: 10.1158/1078-0432.CCR-06-2570. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch FR, Janne PA, Eberhardt WE, Cappuzzo F, Thatcher N, Pirker R, Choy H, Kim ES, Paz-Ares L, Gandara DR, et al. Epidermal growth factor receptor inhibition in lung cancer: status 2012. J Thorac Oncol. 2013;8:373–384. doi: 10.1097/JTO.0b013e31827ed0ff. [DOI] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, Gutkind SJ. Exploiting the mTOR paradox for disease prevention. Oncotarget. 2012;3:1061–1063. doi: 10.18632/oncotarget.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Pillai S, Chellappan SP. Genetic and biochemical alterations in non-small cell lung cancer. Biochem Res Int. 2012;2012:940405. doi: 10.1155/2012/940405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Meng Y, Kwiatkowski DJ, Chen X, Peng H, Sun Q, Zha X, Wang F, Wang Y, Jing Y, et al. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. The Journal of clinical investigation. 2010;120:103–114. doi: 10.1172/JCI37964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31:1070–1080. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, Koutcher JA, Solit DB, Rosen N, Zakowski MF, Pao W. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS One. 2007;2:e810. doi: 10.1371/journal.pone.0000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Su KY, Chen HY, Li KC, Kuo ML, Yang JC, Chan WK, Ho BC, Chang GC, Shih JY, Yu SL, Yang PC. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30:433–440. doi: 10.1200/JCO.2011.38.3224. [DOI] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.