

Abstract
In animal models of experimental cerebral malaria (ECM), neuropathology is associated with an overwhelming inflammatory response and sequestration of leucocytes and parasite-infected red blood cells in the brain. Here we explored the effect of vitamin D (VD, cholecalciferol) treatment on host immunity and outcome of ECM in C57BL/6 mice during Plasmodium berghei ANKA (PbA) infection. We observed that oral administration of VD both before and after PbA infection completely protected mice from ECM. VD administration significantly dampened the inducible systemic inflammatory responses with reduced circulating cytokines IFN-γ and TNF and decreased expression of these cytokines by the spleen cells. Meanwhile, VD also resulted in decreased expression of the chemokines CXCL9 and CXCL10 and cytoadhesion molecules (ICAM-1, VCAM-1 and CD36) in the brain, leading to reduced accumulation of pathogenic T cells in the brain and ultimately substantial improvement of the blood-brain barriers of PbA-infected mice. In addition, VD inhibited the differentiation, activation and maturation of splenic dendritic cells. Meanwhile, regulatory T cells and IL-10 expression levels were upregulated upon VD treatment. These data collectively demonstrated the suppressive function of VD on host inflammatory responses, which provides significant survival benefits in the murine ECM model.
Keywords: vitamin D, Plasmodium berghei, cerebral malaria, Th1 response
Introduction
Malaria is widespread throughout the tropical and subtropical regions, causing more than 300 million acute illnesses and resulting in more than 600,000 deaths annually. Cerebral malaria (CM) is the most severe complication of Plasmodium falciparum infection and a major cause of death in children under the age of 5. The mechanisms leading to CM in humans is not well understood and appears to be multifactorial. Cytoadherence of parasitized red blood cells (pRBCs) to the brain endothelium is thought to cause mechanical obstruction of the brain microvessels leading to CM pathology (1). In addition, excessive inflammatory responses characterized by high levels of proinflammatory cytokines are also thought to contribute to CM (1). Inflammatory cytokines up-regulate expression of the adhesion molecules such as ICAM-I and VCAM-I on brain endothelial cells, further enhancing cytoadherence and sequestration of pRBCs in the brain. A better understanding of the mechanisms of CM and identification of effective adjunct therapies of CM are of high priority.
Rodent malaria infections such as the Plasmodium berghei ANKA (PbA) infection in C57BL/6 mice have been used widely as animal CM models because they share several features with human CM (2–4). In the mouse CM models, T helper type 1 (Th1) responses play a critical role in CM pathogenesis. Th1 responses are characterized by the increased production of IFN-γ and decreased production of the Th2 cytokines such as IL-4. Appropriate induction of Th1 cytokines is needed for successful control of parasitemia and resolution of malaria infection (5, 6), whereas excessive levels of these cytokines are implicated in the pathogenesis of CM (7, 8). Thus, regulation of the magnitude and timing of the Th1 response is essential for producing optimized immune responses that inhibit the malaria parasites without causing immunopathology. Regulatory T cells (Tregs) are important player participating in the control of overwhelming responses to infections (9–12). In the mouse CM model of infection, Treg expansion inhibits the development of pathogenic Th1 cells and CM (13, 14).
Vitamin D (VD) is a fat-soluble vitamin that is either synthesized in the skin after exposure to solar ultraviolet B radiation or provided in the diet. In addition to its traditionally known roles in regulation of bone metabolism and calcium-phosphorus homeostasis, VD has been increasingly recognized to have prominent regulatory functions on both innate and adaptive immune systems (15). The active form of VD [1,25(OH)2D3, 1,25D3] primarily affects dendritic cell (DC) maturation and macrophage differentiation (16, 17), and inhibits the production of the cytokines IL-12 and IL-23. In addition, 1,25D3 inhibits the production of Th1 cytokines (IL-2 and IFN-γ) and Th17 cytokines (IL-17 and IL-21), but stimulates Th2 cytokine production (e.g., IL-4) (18), thereby indirectly shifting the polarization of T cells from a Th1 and Th17 phenotype towards a Th2 phenotype. Moreover, 1,25D3 favors development of Tregs via modulation of DCs (19). Since many autoimmune diseases such as inflammatory bowel disease, multiple sclerosis, and arthritis are the result of overwhelming Th1 responses, 1,25D3 treatments suppressed Th1 responses and ameliorated Th1 mediated experimental autoimmunity (20). Paradoxically, even though VD inhibits Th1 and Th17 responses, a number of infectious diseases are not made more severe by treatments with active VD (21).
The immunoregulatory functions of VD especially its inhibitory effect on Th1 responses have prompted us to examine the role of VD in experimental CM (ECM). In P. falciparum malaria, plasma VD level did not vary during the course of infection and VD status was not associated with incident malaria (22, 23). In a rodent malaria model, oral VD treatment of mice for two weeks prior to P. berghei infection has been reported to decrease parasite growth and expand the life span of infected mice (24). However, a recent study showed that three weekly intraperitoneal injections of 0.5 μg/kg VD had no effect on susceptibility of wild-type mice to PbA infection (25). In this report, we explored the effect of VD on ECM and showed that oral supplementation with VD protected mice from ECM. Oral VD administration before and after PbA infection completely prevented the occurrence of ECM. We show that the protective effect of VD was through the inhibition of a strong host pro-inflammatory (IFN-γ and TNF) response mediated directly and indirectly through reduction of DC activation, IL-10 production and expansion of the Tregs.
Materials and Methods
Animals and Experimental Infection
Female C57BL/6 mice (6 to 8 weeks old) were obtained from Beijing Animal Institute and maintained at the Animal Care Facilities of the China Medical University. Infections were initiated by intraperitoneal (i.p.) injection of 1×106 PbA pRBCs into each mouse. Parasitemia was monitored by counting the number of pRBCs per 1000 RBCs by light microscope examination of Giemsa-stained thin smears from tail blood. Mice were monitored daily for survival and neurological signs of CM such as ataxia, paralysis and coma. For the mortality and parasitemia experiment, 10 mice were used in each group and the experiments were repeated for four times. All experiments were performed in compliance with local animal ethics committee requirements.
VD Treatment of Mice
VD (Cholecalciferol) was obtained from Sigma (St. Louis, MO, USA) and delivered in mice using two routes: injection and feeding. For injection, VD was dissolved in 100% ethanol and diluted to a working solution of 0.1 μg/ml in 0.02% Tween 80. Mice in the treatment group was injected with a 0.5 μg/kg every other day, starting three days before the PbA infection (25), while mice in the control group received equal volumes of 0.02% Tween 80. For oral administration, VD was dissolved in soybean oil before use. For the dose ranging experiment, mice were orally administered a daily dosage of 0, 10, 50 and 250 μg/kg of VD for four days before PbA infection. For subsequent experiments, mice were randomly divided into six groups: naïve mice (uninfected), VD supplemented before PbA infection (VD+PbA), two groups with VD supplemented after PbA infection (PbA+VD), and two uninfected but VD treated control groups. Mice in the VD+PbA group were orally administered a daily dosage of 50 μg/kg of VD for four consecutive days before PbA infection, whereas the PbA+VD group mice were orally administered 50 μg/kg of VD daily for four successive days at 2 days (PbA+VD2d) or 5 days (PbA+VD5d) after PbA infection. The two control groups received the same volume of soybean oil at the identical time points as the VD+PbA and PbA+VD groups. On day 5, blood was collected and VD content was measured using the 25(OH)-Vitamin D direct ELISA Kit (Immundiagnostik AG, Bensheim, Germany) according to the manufacturer’s instructions. Samples were measured in duplicate and averaged.
Histopathology and Immunohistochemistry
When PbA-infected mice began showing neurological symptoms on day 6, five mice from each of the experimental groups were selected for histological examination. Brains were removed, fixed in 4% paraformaldehyde for 24 h, and embedded in paraffin. Serial 4-μm-thick horizontal sections were made, stained with hematoxylin and eosin (H & E), and examined for microvascular obstruction and leakage. To detect ICAM-1, VCAM-1, and CD36 along the endothelial lining, immunohistochemical staining was performed with specific rabbit polyclonal antibodies against ICAM-1 (Abcam, USA), VCAM-1 and CD36 (Santa Cruz Biotechnology, USA) as previously described (26), which was followed by biotin-conjugated goat anti-rabbit IgG antibody. Finally, streptavidin-conjugated peroxidase was added and color development was done using 3-amino-9 -ethylcarbazole as the substrate. Finally, the sections were counterstained with hematoxylin, washed and mounted. ICAM-1-, VCAM-1-, and CD36-positive vessels were visualized by microscopy at ×400. The number of positive vessels in 20 fields was counted for each mouse.
Blood Brain Barrier (BBB) Integrity
To evaluate integrity of the BBB, 200 μl of 2% (wt/vol) solution of Evans blue (Sigma) in phosphate-buffered saline (PBS, pH 7.0) was injected intravenously (i.v.) into each mouse. One hour later, mice were euthanized and perfused with 20 ml of PBS. Brains were isolated and incubated in 2 ml of formamide for 48 h at 37°C. The amount of Evans blue in 100 μl of the brain tissue extracts was determined by measuring absorbance at 630 nm (27).
RNA Extraction and Real-Time PCR
Total RNA was extracted from isolated brains by Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Contaminating DNA was removed by subjecting 2 μg total RNA to DNase I digestion. The resulting total RNA was used for reverse transcription (RT) with the oligo(dT) primer. One-fifth volume of the RT reaction mixture was used for real-time PCR with the primer pairs for CXCL9, CXCL10, IFN-γ, TNF, and IL-10 (Table S1). PCR was performed with the SYBR Green PCR Master Mix for 40 cycles in an ABI PRISM 7700 apparatus (Applied Biosystems, Foster City, CA). Threshold values were obtained using PE Biosystem software and mRNA was quantified. β-actin was used as an internal control and the ratio of each target gene to β-actin was determined. An untreated control sample value was taken as 100% and treated values were calculated based on the control. The specificity of the PCR was confirmed by melting-curve analysis.
Quantification of Cytokine Production by Splenocytes
Splenocyte culture was performed as previously described (28). Briefly, spleens from control and infected mice were removed aseptically and pressed through a sterile fine-wire mesh with 10 ml RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), 25 mM Hepes, 0.12% gentamicin and 2 mM glutamine. Cell suspensions were collected by centrifuging at 350 ×g for 10 min. Erythrocytes were lysed with cold 0.17 M NH4Cl and cells were washed twice with fresh medium. Spleen cell viability was determined by trypan blue exclusion and was >90%. Spleen cells were adjusted to a final concentration of 107 cells/ml in RPMI-1640 supplemented with 10% heat-inactivated FCS. Aliquots of 5×106 cells/well were incubated in 24-well flat-bottom tissue culture plates (Falcon) in triplicate for 48 h at 37°C in a humidified 5% CO2 incubator. Cytokines (IFN-γ, TNF and IL-10) in the culture supernatant and plasma samples were measured by enzyme-linked immunosorbent assays (ELISA) (R&D Systems, Minneapolis, MN, USA).
Flow Cytometry
For each experimental group, five mice were sacrificed at the indicated time points for flow cytometric measurement of Th1 cells, Tregs, DCs, and CD11c+DCs expressing the costimulatory marker MHC II, CD86, TLR-4 and TLR9 in splenocytes. Unless otherwise indicated, antibodies were obtained from BD Biosciences (San Jose, CA, USA). To measure Th1 type cells (CD4+ T-bet+ IFN-γ+), 107 fresh splenocytes were stimulated in 12-well plates with 50 ng/ml PMA and 1 mM ionomycin (Sigma) for 5 h at 37°C and 5% CO2 in the presence of 1 μM monensin to inhibit cytokine secretion. Cells were harvested and surface stained with FITC-conjugated anti-CD4 mAb (clone H1.2F3). After fixation and permeabilization with the intracellular fixation kit (eBioscience, San Diego, CA, USA), cells were stained with anti-T-bet-PE (clone eBio4B10), and anti-IFN-γ-APC (XMG1.2). Tregs were surface-stained with FITC-anti-CD4 and PE-anti-CD25 antibodies in 100 μL of PBS supplemented with 3% FCS, and were then stained for intracellular Foxp3 with APC-anti-Foxp3 (clone FJK16s) as described earlier (29). For DCs, cells were double-stained with FITC-conjugated CD11c mAb (clone HL3) and PE-conjugated anti-CD11b (clone M1/70), CD86 (clone GL1), MHCII mAb (clone M5/114.15.2), PerCP-conjugated anti-CD45R/B220 (clone RA3-6B2), or anti-TLR4 (clone MTS510). To assess the expression of TLR9 in CD11c+ DCs, spleen cells were stained first with FITC-conjugated CD11c mAb. After fixation and permeabilization, cells were incubated with biotinylated anti-TLR9 mAb (clone 5G5, Hycult biotech, Plymouth Meeting, PA, US) followed by PE-conjugated streptavidin (Biolegend, San Diego, CA, US).
To determine the migration of CD4+ and CD8+ T cells to the brain, brain mononuclear cells were isolated from the brains on day 5 p.i. following a published procedure (30). A single-cell suspension was obtained by grinding the tissues and resuspending the resulting cells in 5 ml of RPMI1640, which contained 100 U/ml IV collagenase (Invitrogen, USA), and incubated at 42°C for 45 min. Cells were pelleted at 300×g for 10 min, resuspended in 30% Percoll in PBS (Sigma), layered on 70% Percoll, and centrifuged at 515×g for 30 min at room temperature. Cells at the interface were isolated, washed twice, resuspended in PBS, and labeled with the following antibodies: FITC-anti-CD4, PerCP-anti-CD8 (clone 53-6.7), and APC-anti-CD3 (clone 145-2C11). Cells were incubated at 4°C for 30 min and washed twice with PBS. This brain cell isolation technique collects both intravascular and extravascular cells, thus allowing the evaluation of both already trafficked and recruited cells. Flow cytometric analysis was performed using a FACS Calibur, and data were analyzed with the FlowJo software.
Statistical Analysis
Data are presented as the means ± standard errors of the means (SEM). VD levels in human participant were compared among three groups: patients infected with P. falciparum, patients infected with P. vivax, and healthy participants with no malaria infection. Kruskal-Wallis analysis of variance test was used to look for statistically significant median values across the three groups, while an analysis of variance was conducted on the log-transformed VD values. Difference in survival of the mice among experimental groups was assessed using the Kaplan-Meier test. For comparisons between two groups, statistical significance was analyzed by either a t test or Mann-Whitney U test, depending on normality of the data. For comparisons between three or more groups, statistical significance was determined using a one-way ANOVA. The data were analyzed using GraphPad Prism software (version 6.01), and a value of P<0.05 was considered significant.
Results
VD Administration Protects Mice against ECM
The effect of VD on PbA infection depended on the delivery route and dose of the VD. Earlier work showed that there was no effect of injection of vitamin D on P. berghei infection in mice (24, 25). Injection using the same protocol reported (25) offered no protection against ECM (Fig. S1A, B). Preliminary experiments using a dose response of oral VD showed that there was no effect of 10 μg/kg, and 250 μg/kg VD as compared to the controls. In contrast, the mice treated orally with 50 μg/kg VD did not develop ECM (Fig. S1C). Therefore, we selected the 50 μg/kg dosage for subsequent experiments.
We applied three oral VD treatment schemes: daily oral VD supplement of 50 μg/kg for four days prior to PbA infection (VD+PbA), daily oral VD supplement of 50 μg/kg for four days beginning 2 days (PbA+VD2d) and 5 days (PbA+VD5d) after PbA infection. VD supplementation prior to and 2 days after infection was effective at raising the serum 25(OH)D3 levels of mice (Fig. S1A). On day 5 p.i., the 25(OH)D3 levels in the VD-treated groups were significantly higher than that in the untreated PbA group. The four doses of VD in the VD-treated groups were enough to significantly raise serum 25(OH)D3 levels (Fig. S1D). There was no effect of the infection on serum 25(OH)D3 levels since vitamin D status was the same in the healthy control mice and the PbA-infected mice (data not shown).
The three VD treatment schemes offered significant protection of mice against ECM. In the control group, PbA infection resulted in the development of neurological symptoms on days 5–6 and mice began to die on days 6–7, with all of the mice dead by day 11 p.i. (Fig. 1A). Death following PbA infection occurred in mice that had relatively low parasitemia (<12%) (Fig. 1B). In PbA-infected mice, the vehicle control groups (soybean oil) had symptoms (pathology, survivorship, or parasitemia) identical to the untreated group (data not shown). VD treatments either before or 2 days after PbA infection protected the mice from early mortality and all of the VD treated mice survived beyond day 16 post-infection (Fig. 1A). VD administration beginning on day 5 p.i. also offered protection of the mice from early mortality with no mortality in this group before day 11. Regardless of the VD treatment all of the PbA-infected mice progressed to death from anemia with high parasitemia after ~3 weeks (Fig. 1A, B). There was an advantage to VD supplementation on day 2 p.i. since the PbA+VD2d group survived significantly longer (Kaplan-Meier test, P<0.05) than the other two VD treatment groups (Fig. 1A). In addition, the parasitemias in both the PbA+VD2d and PbA+VD5d groups were lower than that of the VD+PbA group (Fig. 2B). For subsequent experiments, we focused on the PbA+VD2d group (referred hitherto as PbA+VD) for more detailed pathological and immunological analyses.
FIGURE 1.
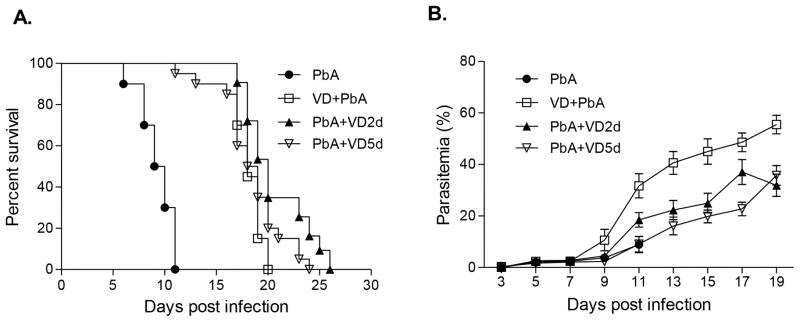
Treatment with VD improves survival in mice with P. berghei ANKA (PbA)-induced ECM. The survival curves of infected mice (A) and parasitemia following VD treatment in three groups of mice were compared: PbA control (PbA) (n=40); daily VD administration for four successive days prior to PbA infection (VD+PbA) (n=28); daily VD administration for four days 2 days after PbA infection (PbA+VD2d) (n=32), and daily VD administration for four days 5 days after PbA infection (PbA+VD5d) (n=40). Shown here are the cumulative results of four separate experiments. Parasitemia is shown as mean ± SEM.
FIGURE 2.

VD treatment decreased endothelium activation and improved BBB integrity. On day 5 p.i., five mice from each of the three groups were processed for histology with H & E staining and immunohistochemical analysis with anti-ICAM-1, -VCAM-1, and -CD36 antibodies. Top four panels: Images show representative brain sections with the microvessels (arrows), while the corresponding bar graphs indicate quantification of leukocytes-, ICAM-1-, VCAM-1-, and CD36-positive microvessels, respectively. Microvessels per microscopic field were quantified in 20 fields per mouse, and values are mean ± SEM from five mice in each group. Bottom panel: Representative brain images show the extent of vascular leakage using the Evans blue extravasation method, while the corresponding bar graph shows quantitative assessment of BBB leakage, expressed as the optical density (OD) of brain extracts at 630 nm (mean ± SEM, n=5). ** indicates significant difference at P<0.05 (t-test) between uninfected control group and PbA-infected group, while # and ## indicate significant difference between the PbA and PbA+VD groups at P=0.05 and P=0.01, respectively.
VD Treatment Reduces Brain Pathology
The pathological changes of ECM that mark the early lethality of mice following PbA infection includes adhesion of leukocytes in the brain microvasculature, microhemorrhages, disruption of BBB integrity, and extravasation of pRBCs into the brain parenchyma (31, 32). Activation and upregulation of endothelial adhesion molecules are responsible for these pathologies (33, 34). Thus, we assessed the brain pathology and expression levels of adhesion molecules in cerebral vessels. On day 5 p.i., the number of vessels containing leukocytes per microscopic field in the PbA+VD group was significantly lower than that in the untreated group (Fig. 2). Immunostaining for VCAM-1, ICAM-1 and CD36 on the endothelium of the microvessels was less intense in PbA+VD than the PbA group (Fig. 2). Meanwhile, a substantial loss of BBB integrity was evident in the PbA group, which showed high levels of dye extravasation. In comparison, VD treatment offered significant protection of the integrity of BBB in PbA-infected mice (P <0.05) (Fig. 2). VD treatment beginning 24 h after PbA infection protected the brain from pathologic signs of ECM.
VD Treatment Reduces T Cell Trafficking to the Brain
Increased trafficking and accumulation of pathogenic CD8+ T cells in the brain mediated through the IFN-γ inducible chemokines CXCL9 and CXCL10 are associated with ECM (35–37). To determine whether VD treatment affected the migration and accumulation of T cells in the brains of PbA-infected mice, brain mononuclear cells were isolated and quantified for CD8+ and CD4+ cells. Compared to uninfected naive mice, PbA infection promoted the accumulation of both CD8+ and CD4+ T cells (Fig. 3A). In contrast, the numbers of both T cell populations in the brains of VD-treated PbA-infected mice did not differ significantly from those in uninfected naive mice (Fig. 3A). To determine whether reduced T cell trafficking to the brain was associated with decreased expression of chemokines, we compared the expression of CXCL9 and CXCL10 mRNA in the brains of VD-treated and untreated mice. On day 5 p.i., VD treatment significantly reduced the expression of both chemokines in the brains (Fig. 3B,C). These results indicate that VD treatment is sufficient to modulate the cerebral environment to down-regulate expression of key chemokines, leading to decreased accumulation of pathogenic T cells in the brain.
FIGURE 3.
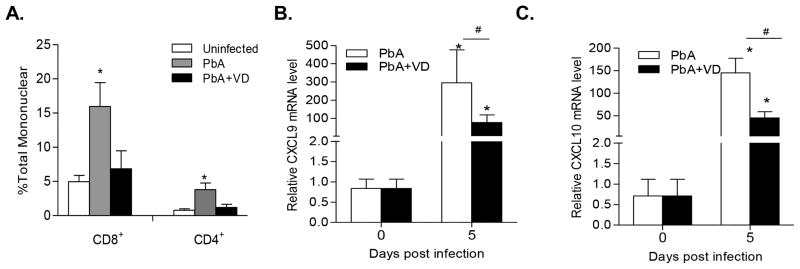
VD treatment reduced pathogenic T cell sequestration in the brain. (A) VD treatment of PbA-infected mice reduced the number of CD4+ and CD8+ T cells in the brains (n=5). Values are means plus SEMs (error bars). (B,C) VD treatment significantly reduced the expression of the chemokines CXCL9 and CXCL10 in the brains of PbA-infected mice. The mRNA levels for the two chemokines were normalized to the β-actin mRNA level, and fold changes were calculated against control uninfected mice. * indicates significant difference between mRNA levels in infected mice on day 5 and their corresponding baseline levels on day 0 (P<0.05). # indicates significant difference (P<0.05) between the PbA and PbA+VD groups on day 5 p.i.
VD Treatment Dampens the Th1 Immune Response
An overwhelming pro-inflammatory (Th1) response is essential for ECM (7, 38, 39). To test whether VD treatment inhibits Th1 cells during ECM, we quantified the IFN-γ-producing CD4+T-bet+ cells in the spleens of infected mice and two Th1 cytokines IFN-γ and TNF on day 3 and 5 p.i. In association with the appearance of ECM pathology, IFN-γ and TNF levels were significantly increased in the serum of PbA-infected mice on day 5 p.i. (P<0.01) (Fig. 4A, B). Significantly increased production of these cytokines was also observed by splenocytes isolated from PbA-infected mice during their in vitro culture (P<0.01) (Fig. 4C,D). VD treatment led to a significant reduction of the levels of these two cytokines in both serum and cultured splenocytes of PbA-infected mice (Fig. 4A–D). In addition, the mRNA levels of IFN-γ and TNF in the brains of infected mice on day 5 p.i. also experienced significant declines in VD-treated mice as compared with untreated mice (Fig. 4E,F).
FIGURE 4.

VD treatment reduced expression of pro-inflammatory cytokines (IFN-γ and TNF). (A, B) Levels of circulating cytokines in serum. (C,D) Production of IFN-γ and TNF by splenocytes. (E,F) mRNA levels of IFN-γ and TNF in the brain of mice. Each experiment was repeated three times (n=5 per group). Values represent mean ± SEM. * and ** indicate significant differences between the values in PbA-infected mice and the baseline levels on day 0 at P<0.05 and P<0.01, respectively. # and ## indicate significant difference between the PbA and PbA+VD groups at P<0.05 and P<0.01.
PbA infection was associated with increased expansion of CD4+T-bet+IFN-γ+ T cells. On both days 3 and 5 p.i., the total population as well as the proportion of this Th1 cell subtype in the spleens of infected mice were substantially increased (Fig. 5). VD treatment, however, significantly reduced this Th1 cell subtype in the spleens of infected mice on day 5 p.i. (P<0.01) (Fig. 5).
FIGURE 5.
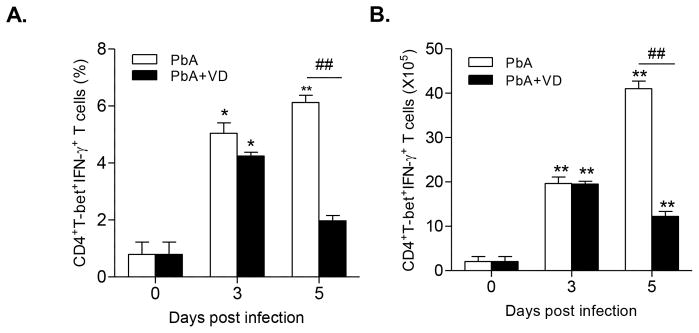
VD reduces the expansion of CD4+T-bet+IFN-γ+ T cells. Both proportion (A) and the absolute number (B) of the CD4+T-bet+IFN-γ+ T cells in the spleens were quantified. Each experiment was repeated three times. For each experiment, five mice were used per group. Values represent the mean ± SEM (n=5 mice per group). * and ** indicate significant differences between the values in PbA-infected mice and the baseline levels on day 0 at P<0.05 and P<0.01, respectively. # and ## indicate significant difference between the PbA and PbA+VD groups at P<0.05 and P<0.01.
VD Promotes Expansion of Tregs and the Production of IL-10 in Spleen
Both Tregs and IL10 contribute to the improvement of PbA-specific Th1 response involved in the induction of ECM. Overall, compared with the untreated group, VD treatment promoted the expansion of CD4+CD25+Foxp3+ Tregs in PbA-infected mice, albeit significance (P<0.01) was only observed in total Treg populations on day 3 p.i. (Fig. 6A,B). However, the secretion of the anti-inflammatory cytokine IL-10 by cultured splenocytes was significantly higher in VD-treated groups than untreated PbA groups (P<0.05) (Fig. 6C). Meanwhile, the plasma level of IL-10 was also significantly increased in VD-treated mice on day 5 p.i. (P<0.01) (Fig. 6D).
FIGURE 6.

VD stimulated the expansion of Tregs and increased IL-10 production. The absolute number (A) and the proportion (B) of CD4+CD25+Foxp3+ Tregs were quantified by flow cytometry. The concentrations of IL-10 secreted by cultured splenocytes (C) and in the mouse serum (D) were determined by ELISA. Results are representatives of three independent experiments. Values are presented as the mean ± SEM (n = 5 mice per group). * and ** indicate significant differences between the values in PbA-infected mice and the baseline levels on day 0 at P<0.05 and P<0.01, respectively. # and ## indicate significant difference between the PbA and PbA+VD groups at P<0.05 and P<0.01, respectively.
VD Inhibits DC Differentiation, Maturation, and Function
The nature of the immune response is critically dependent on the interplay between the innate and adaptive immune systems. Central to this interaction are the DCs (40). We found that the proportions of both mDCs (Fig. 7A,C) and pDCs (Fig. 7B,D) were significantly higher than those in the VD-treated groups on day 5 p.i. (P<0.05, t-test), indicating that VD inhibited differentiation of DCs. In addition, VD treatment also led to a decrease of expression of the co-stimulatory molecules MHCII on DCs on day 5 p.i. (Fig. 8A,D). Furthermore, the expression of TLR4 on DCs was drastically reduced in VD-treated groups on day 5 p.i. (Fig. 8B,E), while TLR9 expression in the DCs was also reduced to some extent (Fig. 8C,F). Meanwhile, the total numbers of DC populations within the spleen of mDCs, pDCs, and DCs expressing MHC II, CD86, TLR4, and TLR9 were mostly down-regulated by VD treatment (Fig. S2). To further illustrate that VD treatment affected the function of DCs, we measured cytokine production by DCs and found that VD treatment resulted in decreased production of IL12p40 on both day 3 and 5 p.i. and increased production of IL10 on day 5 (data not shown). Taken together, these results indicate that VD treatment inhibited the differentiation, maturation and activation of DCs.
FIGURE 7.

VD treatment inhibited the expansion of DC subsets. The frequency of CD11c+CD11b+ DCs (mDCs), and CD11c+B220+ DCs (pDCs) were measured by flow cytometry. (A,B) Representative plots showing the proportions of the two DC populations. (C,D) Bar graphs show the frequencies of the two DC populations within the spleens. Data are presented as the mean ± SEM (n=5 mice per group). Results are representatives of three independent experiments. * indicates significant difference between the values in PbA-infected mice and the baseline levels on day 0 (P<0.05). # indicates significant difference between the PbA and PbA+VD groups (P<0.05).
FIGURE 8.

VD treatment inhibited DC differentiation, maturation and activation. The frequency of CD11c+ DCs expressing MHC II (A,D), TLR-4 (B,E) and intracellular TLR-9 (C,F) within the spleen were determined by flow cytometry. (A–C) Representative dot plots show the proportion of each cell subsets within the spleen at day 5 p.i. (D–E) Graphs show the frequencies of the respective DC populations. Data are presented as the mean ± SEM (n=5 per group). Results are representatives of three independent experiments. * and ** indicate significant differences between the values in PbA-infected mice and the baseline levels on day 0 at P<0.05 and P<0.01, respectively. ## indicates significant difference between the PbA and PbA+VD groups (P<0.01).
4. Discussion
Susceptible mice infected with PbA developed a severe CM syndrome characterized by hyperinflammation, intravascular accumulation of immune T cells, and BBB leakage (38). While it has been extensively shown that in vivo treatments with 1,25(OH)2D3 inhibit Th1 mediated immune responses (20, 21) and suppress experimental autoimmunity, this is the first study to demonstrate the effectiveness of short-term VD treatments to suppress in vivo Th1 mediated inflammation. It is well established that vitamin D and 1,25(OH)2D3 directly and indirectly via actions on DCs and macrophages inhibit Th1 cell production of IFN-γ. We demonstrated that oral administration of VD improved serum 25(OH)D3 levels and down-regulated the levels of circulating inflammatory cytokines (IFN-γ and TNF). The inhibition of Th1 responses in addition with the reduced expression of chemokines CXCL9 and CXCL10, and cell adhesion molecules ICAM-1, VCAM-1 and CD36, resulted in the decreased accumulation of CD8+ T cells in the brain, and improved the integrity of the BBB. Interestingly, the timing of the VD supplementation looks to be critical and dosing animals just after infection seems to be more effective than before infection. There was no effect of infection on serum 25(OH)D3 levels in mice. Analysis of plasma VD levels in humans infected with P. falciparum or P. vivax malaria from a subtropical area (Kachin State, Myanmar) found highly variable 25(OH)D3 levels including frank vitamin D deficiency (<12 ng/ml) in 10% and vitamin D deficiency <20 ng/ml in 28% of the population (data not shown). It would be interesting to determine whether lower VD status would predict development of CM. Although individuals in tropical malaria regions may not suffer from low sunlight exposure, VD deficiency is still prevalent in tropical climates despite high amounts of sunlight (41–43). Vitamin D production in the skin following UV exposure is significantly less in dark skin and dietary sources for vitamin D is uncommon in most parts of the world. This study suggests that VD might be an effective treatment for reducing lethality from CM in humans, albeit the potential difference in VD physiology between humans and mice demands further investigation.
An overwhelming pro-inflammatory Th1 response characterized by excessive production of the cytokines IFN-γ and TNF is involved in CM pathogenesis (4). The cerebral pathology during ECM is an IFN-γ-dependent process as evidenced by the complete resistance of the IFN-γ deficient mice to ECM (39). IFN-γ is responsible for the induction of chemokines CXCL9 and CXCL10 during ECM (44), which subsequently recruit CD4+ and CD8+ T cells to the brain via CXCR3 on the cell surface (35, 36, 45). In addition, the proinflammatory cytokine TNF is proposed to up-regulate the production of adhesive molecules in the brain vasculature. Here we showed that VD treatment significantly reduced the levels of circulating IFN-γ and TNF, secretion of these cytokines by the immune cells in the spleen, as well as expression of these cytokines in the brains of infected mice. Further, VD inhibited the differentiation of CD4+ IFN-γ+T-bet+ Th1 cells. Vitamin D deficiency is associated with an increased risk of disease in Th1 mediated diseases like inflammatory bowel disease (20). VD via the vitamin D receptor (VDR) and 1,25(OH)2D3 directly inhibits IFN-γ transcription in human and mouse T cells (46, 47). In addition, TNF production is inhibited by 1,25(OH)2D3 in macrophage and T cells (20, 48). Direct inhibition of TNF and IFN-γ following VD supplementation protects against cerebral malaria.
VD exerts its immunomodulatory effects through interaction with VDR, a member of the superfamily of nuclear hormone receptors, which is expressed in numerous human immune cells such as T cells, B cells, iNKT cells, macrophages, and DCs (49). Thus, VD directly acts on T cells to inhibit T cell proliferation and IFN-γ production (50). DCs, the highly specialized APCs, are critical for stimulating the differentiation of effector T cells from naive T-cell precursors. There is ample evidence showing that VD also hampers DC maturation from monocytes (51–54). Our data show that VD treatment decreased the number of mDCs and pDCs during PbA infection. This decrease may be due to changes in DC migration and homing. In addition, VD suppressed the expression of antigen presenting molecules such as MHCII and CD86 on DCs, changes in surface marker with corresponding change in DC function such as antigen presentation (55). These results are consistent with the role of VD for inhibiting differentiation, maturation and activation of DCs. Furthermore, we found decreased expression of TLR4 and TLR9 in DCs upon VD treatment during PbA infection. Since DCs are essential to induce Th1 cell development as well as Th1 cytokine production upon malaria infection (8), our data demonstrate that VD also results in decreased expression of IL-12 and increased production of IL-10 in these APCs, consistent with a decrease of the Th1 response (56). Collectively, these data are in line with earlier descriptions of the suppressive activities of VD with respect to the stimulation of Th1 mediated immunity.
Tregs expand during Plasmodium infection (57, 58) and have been shown to inhibit the development of Th1 immune responses (59). Given the critical role of Tregs and the anti-inflammatory cytokine IL-10 in inhibiting CM, expansion of Tregs and elevation of IL-10 level would improve the outcome of the ECM. VD has been shown to be required for the optimal development and function of Tregs. VD increased the Treg population (Foxp3) (60), resulting in the blockade of the Th1 response (61). Importantly, treatment of naive CD4+T cells with VD potently induced the development of Tregs (62, 63). Meanwhile, VD alone or in combination with dexamethasone induced IL-10-producing Tregs in an APC-free in vitro system (64, 65). Also, a VD analog triggered the emergence of a CD4+CD25highCD127low Treg phenotype and selectively induced IL-10 expression within the CD4+ T cell subset (66). These results highlighted the impacts of VD on the differentiation of Tregs and expression of IL-10. Our results provided further evidence to show that VD treatment expanded the Treg population, which was accompanied by increased levels of IL-10, conditions that favor host protection during CM.
The role of VD regulation of infection has been more difficult to determine. For many infections including malaria strong Th1 responses are needed to clear the parasite. Our work suggests that vitamin D is protective against ECM but not PbA infection since the VD treated mice survive the ECM but then several weeks later show increased parasitemia and eventual death caused by anemia. Interestingly, murine models of infectious diseases, where IFN-γ are required for host protection, were not affected by 1,25(OH)2D3 treatments (20, 21). Our data support a model where vitamin D and production of 1,25(OH)2D3 act to limit the extent of the Th1 and inflammatory response, which protects against CM.
In summary, we demonstrated that oral VD treatment completely prevented the occurrence of ECM during the PbA infection in susceptible mice. The protective effect is consistent with the role of VD in the inhibition of the Th1 immune responses. Immune cells express the VDR and are targeted by VD. Therefore, dampening of the host Th1 response by VD is mediated directly by its action on the Th1 cells as well as indirectly through the inhibition of the APCs and innate immunity. In addition, our results demonstrate that VD treatments expanded Tregs and inhibited differentiation, maturation and functioning of DCs, which together resulted in increased expression of IL10. Because of the direct effects of VD on IFN-γ and TNF and the indirect effects of VD on DCs and production of IL-10 that serve to suppress the Th1 driven ECM following VD treatment. Importantly, administration of VD either before or after PbA infection was effective in preventing ECM, suggesting that both prophylactic treatment and post-infection therapy could benefit human CM patients.
Supplementary Material
Acknowledgments
We would like to thank Jun Liu for technical support.
This study was supported by National Institutes of Health (R01AI099611 and U19AI089672), USA and Grant-in-Aid (LJQ2011084) from the Liaoning Provincial Development Program for outstanding young scholars, China.
References
- 1.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- 3.de Souza JB, Riley EM. Cerebral malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 2002;4:291–300. doi: 10.1016/s1286-4579(02)01541-1. [DOI] [PubMed] [Google Scholar]
- 4.Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5:722–735. doi: 10.1038/nri1686. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell AJ, Hansen AM, Hee L, Ball HJ, Potter SM, Walker JC, Hunt NH. Early cytokine production is associated with protection from murine cerebral malaria. Infect Immun. 2005;73:5645–5653. doi: 10.1128/IAI.73.9.5645-5653.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley EM, Wahl S, Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol. 2006;28:35–49. doi: 10.1111/j.1365-3024.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 7.Grau GE, Bieler G, Pointaire P, De Kossodo S, Tacchini-Cotier F, Vassalli P, Piguet PF, Lambert PH. Significance of cytokine production and adhesion molecules in malarial immunopathology. Immunol Lett. 1990;25:189–194. doi: 10.1016/0165-2478(90)90113-5. [DOI] [PubMed] [Google Scholar]
- 8.Grau GE, Frei K, Piguet PF, Fontana A, Heremans H, Billiau A, Vassalli P, Lambert PH. Interleukin 6 production in experimental cerebral malaria: modulation by anticytokine antibodies and possible role in hypergammaglobulinemia. J Exp Med. 1990;172:1505–1508. doi: 10.1084/jem.172.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mills KH. Regulatory T cells: friend or foe in immunity to infection? Nature Rev. 2004;4:841–855. doi: 10.1038/nri1485. [DOI] [PubMed] [Google Scholar]
- 10.Belkaid Y, Sun CM, Bouladoux N. Parasites and immunoregulatory T cells. Curr Opin Immunol. 2006;18:406–412. doi: 10.1016/j.coi.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Suvas S, Rouse BT. Treg control of antimicrobial T cell responses. Curr Opin Immunol. 2006;18:344–348. doi: 10.1016/j.coi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Demengeot J, Zelenay S, Moraes-Fontes MF, Caramalho I, Coutinho A. Regulatory T cells in microbial infection. Springer Seminars in Immunopathol. 2006;28:41–50. doi: 10.1007/s00281-006-0024-5. [DOI] [PubMed] [Google Scholar]
- 13.Nie CQ, Bernard NJ, Schofield L, Hansen DS. CD4+ CD25+ regulatory T cells suppress CD4+ T-cell function and inhibit the development of Plasmodium berghei-specific TH1 responses involved in cerebral malaria pathogenesis. Infect Immun. 2007;75:2275–2282. doi: 10.1128/IAI.01783-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haque A, Best SE, Amante FH, Mustafah S, Desbarrieres L, de Labastida F, Sparwasser T, Hill GR, Engwerda CR. CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo. PLoS Pathog. 2010;6:e1001221. doi: 10.1371/journal.ppat.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hewison M. Vitamin D and the immune system: new perspectives on an old theme. Rheum Dis Clin North Am. 2012;38:125–139. doi: 10.1016/j.rdc.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Soruri A, Gieseler RK, Peters JH. 1,25-Dihydroxyvitamin D3 exerts opposing effects to IL-4 on MHC class-II antigen expression, accessory activity, and phagocytosis of human monocytes. Scand J Immunol. 1993;38:535–540. doi: 10.1111/j.1365-3083.1993.tb03237.x. [DOI] [PubMed] [Google Scholar]
- 17.Berer A, Stockl J, Majdic O, Wagner T, Kollars M, Lechner K, Geissler K, Oehler L. 1,25-Dihydroxyvitamin D(3) inhibits dendritic cell differentiation and maturation in vitro. Exp Hematol. 2000;28:575–583. doi: 10.1016/s0301-472x(00)00143-0. [DOI] [PubMed] [Google Scholar]
- 18.de Sa MS, Costa JF, Krettli AU, Zalis MG, Maia GL, Sette IM, Camara A, de C, Filho JM, Giulietti-Harley AM, Ribeiro Dos Santos R, Soares MB. Antimalarial activity of betulinic acid and derivatives in vitro against Plasmodium falciparum and in vivo in P. berghei-infected mice. Parasitol Res. 2009;105:275–279. doi: 10.1007/s00436-009-1394-0. [DOI] [PubMed] [Google Scholar]
- 19.Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M, Walker LS, Lammas DA, Raza K, Sansom DM. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol. 2009;183:5458–5467. doi: 10.4049/jimmunol.0803217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Y, Mahon BD, Froicu M, Cantorna MT. Calcium and 1 alpha,25-dihydroxyvitamin D3 target the TNF-alpha pathway to suppress experimental inflammatory bowel disease. Eur J Immunol. 2005;35:217–224. doi: 10.1002/eji.200425491. [DOI] [PubMed] [Google Scholar]
- 21.Bruce D, Ooi JH, Yu S, Cantorna MT. Vitamin D and host resistance to infection? Putting the cart in front of the horse. Exp Biol Med. 2010;235:921–927. doi: 10.1258/ebm.2010.010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newens K, Filteau S, Tomkins A. Plasma 25-hydroxyvitamin D does not vary over the course of a malarial infection. Trans Roy Soc Trop Med Hyg. 2006;100:41–44. doi: 10.1016/j.trstmh.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 23.Sudfeld CR, Giovannucci EL, Isanaka S, Aboud S, Mugusi FM, Wang M, Chalamilla G, Fawzi WW. Vitamin D status and incidence of pulmonary tuberculosis, opportunistic infections, and wasting among HIV-infected Tanzanian adults initiating antiretroviral therapy. J Infect Dis. 2013;207:378–385. doi: 10.1093/infdis/jis693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sergacheva I, Sokhanenkova TL, Soprunov FF, Lur’e AA. Effect of vitamin D and E on the development of Plasmodium berghei infection in mice. Med Parasitol (Mosk) 1986:15–18. [PubMed] [Google Scholar]
- 25.Waisberg M, Vickers BK, Yager SB, Lin CK, Pierce SK. Testing in mice the hypothesis that melanin is protective in malaria infections. PloS One. 2012;7:e29493. doi: 10.1371/journal.pone.0029493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, 3rd, Pober JS, Lowenstein CJ. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, Intaglietta M, van der Heyde HC. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 28.Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J Immunol. 2002;168:1348–1355. doi: 10.4049/jimmunol.168.3.1348. [DOI] [PubMed] [Google Scholar]
- 29.Zheng W, Wang QH, Feng H, Liu J, Meng HR, Cao YM. CD4+CD25+Foxp3+ regulatory T cells prevent the development of Th1 immune response by inhibition of dendritic cell function during the early stage of Plasmodium yoelii infection in susceptible BALB/c mice. Fol Parasitol (Praha) 2009;56:242–250. doi: 10.14411/fp.2009.028. [DOI] [PubMed] [Google Scholar]
- 30.Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, Hooks JJ, Detrick B. Beta interferon suppresses the development of experimental cerebral malaria. Infec Immun. 2011;79:1750–1758. doi: 10.1128/IAI.00810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renia L, Wu Howland S, Claser C, Charlotte Gruner A, Suwanarusk R, Hui Teo T, Russell B, Ng LF. Cerebral malaria: mysteries at the blood-brain barrier. Virulence. 2012;3:193–201. doi: 10.4161/viru.19013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medana IM, Turner GD. Human cerebral malaria and the blood-brain barrier. Int J Parasitol. 2006;36:555–568. doi: 10.1016/j.ijpara.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Favre N, Da Laperousaz C, Ryffel B, Weiss NA, Imhof BA, Rudin W, Lucas R, Piguet PF. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. 1999;1:961–968. doi: 10.1016/s1286-4579(99)80513-9. [DOI] [PubMed] [Google Scholar]
- 34.Bauer PR, Van Der Heyde HC, Sun G, Specian RD, Granger DN. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation. 2002;9:463–470. doi: 10.1038/sj.mn.7800159. [DOI] [PubMed] [Google Scholar]
- 35.Belnoue E, Potter SM, Rosa DS, Mauduit M, Gruner AC, Kayibanda M, Mitchell AJ, Hunt NH, Renia L. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 2008;30:544–553. doi: 10.1111/j.1365-3024.2008.01053.x. [DOI] [PubMed] [Google Scholar]
- 36.Campanella GS, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, Colvin RA, Luster AD. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A. 2008;105:4814–4819. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van den Steen PE, Deroost K, Van Aelst I, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, Van Damme J, Opdenakker G. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur J Immunol. 2008;38:1082–1095. doi: 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- 38.Engwerda C, Belnoue E, Gruner AC, Renia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103–143. [PubMed] [Google Scholar]
- 39.Amani V, Vigario AM, Belnoue E, Marussig M, Fonseca L, Mazier D, Renia L. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol. 2000;30:1646–1655. doi: 10.1002/1521-4141(200006)30:6<1646::AID-IMMU1646>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 40.Wilson NS, Villadangos JA. Regulation of antigen presentation and cross-presentation in the dendritic cell network: facts, hypothesis, and immunological implications. Adv Immunol. 2005;86:241–305. doi: 10.1016/S0065-2776(04)86007-3. [DOI] [PubMed] [Google Scholar]
- 41.Gebreegziabher T, Stoecker BJ. Vitamin D insufficiency in a sunshine-sufficient area: southern Ethiopia. Food Nutr Bull. 2013;34:429–433. doi: 10.1177/156482651303400408. [DOI] [PubMed] [Google Scholar]
- 42.Nurbazlin M, Chee WS, Rokiah P, Tan AT, Chew YY, Nusaibah AR, Chan SP. Effects of sun exposure on 25(OH) vitamin D concentration in urban and rural women in Malaysia. Asia Pac J Clin Nutr. 2014;22:391–399. doi: 10.6133/apjcn.2013.22.3.15. [DOI] [PubMed] [Google Scholar]
- 43.Djennane M, Lebbah S, Roux C, Djoudi H, Cavalier E, Souberbielle JC. Vitamin D status of schoolchildren in Northern Algeria, seasonal variations and determinants of vitamin D deficiency. Osteoporos Int. 2014;25:1493–1502. doi: 10.1007/s00198-014-2623-7. [DOI] [PubMed] [Google Scholar]
- 44.Carter SL, Muller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-gamma but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55:1728–1739. doi: 10.1002/glia.20587. [DOI] [PubMed] [Google Scholar]
- 45.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Renia L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 46.Rigby WF, Denome S, Fanger MW. Regulation of lymphokine production and human T lymphocyte activation by 1,25-dihydroxyvitamin D3. Specific inhibition at the level of messenger RNA. J Clin Invest. 1987;79:1659–1664. doi: 10.1172/JCI113004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O’Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001;167:4974–4980. doi: 10.4049/jimmunol.167.9.4974. [DOI] [PubMed] [Google Scholar]
- 48.Hakim I, Bar-Shavit Z. Modulation of TNF-alpha expression in bone marrow macrophages: involvement of vitamin D response element. J Cell Biochem. 2003;88:986–998. doi: 10.1002/jcb.10453. [DOI] [PubMed] [Google Scholar]
- 49.Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10:482–496. doi: 10.1016/j.coph.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Reichel H, Koeffler HP, Tobler A, Norman AW. 1 alpha,25-Dihydroxyvitamin D3 inhibits gamma-interferon synthesis by normal human peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1987;84:3385–3389. doi: 10.1073/pnas.84.10.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Voisine C, Mastelic B, Sponaas AM, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol. 2010;40:711–719. doi: 10.1016/j.ijpara.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol Immunother CII. 2005;54:1153–1161. doi: 10.1007/s00262-005-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeda M, Yamashita T, Sasaki N, Nakajima K, Kita T, Shinohara M, Ishida T, Hirata K. Oral administration of an active form of vitamin D3 (calcitriol) decreases atherosclerosis in mice by inducing regulatory T cells and immature dendritic cells with tolerogenic functions. Arterioscler Thromb Vasc Biol. 2010;30:2495–2503. doi: 10.1161/ATVBAHA.110.215459. [DOI] [PubMed] [Google Scholar]
- 54.Enioutina EY, Bareyan D, Daynes RA. TLR-induced local metabolism of vitamin D3 plays an important role in the diversification of adaptive immune responses. J Immunol. 2009;182:4296–4305. doi: 10.4049/jimmunol.0804344. [DOI] [PubMed] [Google Scholar]
- 55.Gleisner MA, Rosemblatt M, Fierro JA, Bono MR. Delivery of alloantigens via apoptotic cells generates dendritic cells with an immature tolerogenic phenotype. Transplant Proc. 2011;43:2325–2333. doi: 10.1016/j.transproceed.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 56.Imazeki I, Matsuzaki J, Tsuji K, Nishimura T. Immunomodulating effect of vitamin D3 derivatives on type-1 cellular immunity. Biomed Res. 2006;27:1–9. doi: 10.2220/biomedres.27.1. [DOI] [PubMed] [Google Scholar]
- 57.Berretta F, St-Pierre J, Piccirillo CA, Stevenson MM. IL-2 contributes to maintaining a balance between CD4+Foxp3+ regulatory T cells and effector CD4+ T cells required for immune control of blood-stage malaria infection. J Immunol. 2011;186:4862–4871. doi: 10.4049/jimmunol.1003777. [DOI] [PubMed] [Google Scholar]
- 58.Goncalves RM, Salmazi KC, Santos BA, Bastos MS, Rocha SC, Boscardin SB, Silber AM, Kallas EG, Ferreira MU, Scopel KK. CD4+ CD25+ Foxp3+ regulatory T cells, dendritic cells, and circulating cytokines in uncomplicated malaria: do different parasite species elicit similar host responses? Infect Immun. 2010;78:4763–4772. doi: 10.1128/IAI.00578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorman S, Kuritzky LA, Judge MA, Dixon KM, McGlade JP, Mason RS, Finlay-Jones JJ, Hart PH. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol. 2007;179:6273–6283. doi: 10.4049/jimmunol.179.9.6273. [DOI] [PubMed] [Google Scholar]
- 60.Urry Z, Xystrakis E, Richards DF, McDonald J, Sattar Z, Cousins DJ, Corrigan CJ, Hickman E, Brown Z, Hawrylowicz CM. Ligation of TLR9 induced on human IL-10-secreting Tregs by 1alpha,25-dihydroxyvitamin D3 abrogates regulatory function. J Clin Invest. 2009;119:387–398. doi: 10.1172/JCI32354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94:26–34. doi: 10.1210/jc.2008-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vigario AM, Gorgette O, Dujardin HC, Cruz T, Cazenave PA, Six A, Bandeira A, Pied S. Regulatory CD4+ CD25+ Foxp3+ T cells expand during experimental Plasmodium infection but do not prevent cerebral malaria. Int J Parasitol. 2007;37:963–973. doi: 10.1016/j.ijpara.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 63.Chambers ES, Hawrylowicz CM. The impact of vitamin D on regulatory T cells. Curr Allergy Asthma Rep. 2011;11:29–36. doi: 10.1007/s11882-010-0161-8. [DOI] [PubMed] [Google Scholar]
- 64.Roelen DL, van den Boogaardt DE, van Miert PP, Koekkoek K, Offringa R, Claas FH. Differentially modulated dendritic cells induce regulatory T cells with different characteristics. Transplant Immunol. 2008;19:220–228. doi: 10.1016/j.trim.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 65.Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. 2009;39:3147–3159. doi: 10.1002/eji.200839103. [DOI] [PubMed] [Google Scholar]
- 66.Baeke F, Korf H, Overbergh L, Verstuyf A, Thorrez L, Van Lommel L, Waer M, Schuit F, Gysemans C, Mathieu C. The vitamin D analog, TX527, promotes a human CD4+CD25highCD127low regulatory T cell profile and induces a migratory signature specific for homing to sites of inflammation. J Immunol. 2011;186:132–142. doi: 10.4049/jimmunol.1000695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.