

Abstract
Despite the proven efficacy of current pharmacotherapies for tobacco dependence, relapse rates continue to be high, indicating that novel medications are needed. Currently, several smoking cessation agents are available, including varenicline (Chantix®), bupropion (Zyban®), and cytisine (Tabex®). Varenicline and cytisine are partial agonists at the α4β2* nicotinic acetylcholine receptor (nAChR). Bupropion is an antidepressant but is also an antagonist at α3β2* ganglionic nAChRs. The rewarding effects of nicotine are mediated, in part, by nicotine-evoked dopamine (DA) release leading to sensitization, which is associated with repeated nicotine administration and nicotine addiction. Receptor antagonists that selectivity target central nAChR subtypes mediating nicotine-evoked DA release should have efficacy as tobacco use cessation agents with the therapeutic advantage of a limited side-effect profile. While α-conotoxin MII (α-CtxMII)-insensitive nAChRs (e.g., α4β2*) contribute to nicotine-evoked DA release, these nAChRs are widely distributed in the brain, and inhibition of these receptors may lead to nonselective and untoward effects. In contrast, α-CtxMII-sensitive nAChRs mediating nicotine-evoked DA release offer an advantage as targets for smoking cessation, due to their more restricted localization primarily to dopaminergic neurons. Small drug-like molecules that are selective antagonists at α-CtxMII-sensitive nAChR subtypes that contain α6 and β2 subunits have now been identified. Early research identified a variety of quaternary ammonium analogs that were potent and selective antagonists at nAChRs mediating nicotine-evoked DA release. More recent data have shown that novel, non-quaternary bis-1,2,5,6-tetrahydropyridine analogs potently inhibit (IC50<1 nM) nicotine-evoked DA release in vitro by acting as antagonists at α-CtxMII-sensitive nAChR subtypes; these compounds also decrease NIC self-administration in rats.
1. INTRODUCTION
Cigarette smoking is the most preventable cause of death with regard to global health. Despite the known health consequences of chronic smoking, compulsive tobacco use still persists throughout the world. Despite the proven efficacy of some current pharmacotherapies for tobacco dependence, relapse rates continue to be high (George & O’Malley, 2004; Hajek, McRobbie, & Myers, 2013; Hajek, Stead, et al., 2013; Hurt et al., 2003; Wileyto et al., 2004), indicating a need for alternative and more efficacious pharmacotherapies. Cessation therapies are available utilizing either nicotine replacement therapy (i.e., the nicotine patch and gum) or the use of nicotinic acetylcholine receptor (nAChR) partial agonists (e.g., varenicline).
Tobacco dependence is described as a chronic, relapsing disorder in which compulsive drug use persists despite negative consequences (George & O’Malley, 2004; Le Foll & Goldberg, 2009; Rose, 2008). About 80% of those who attempt to quit smoking relapse within the first month, and only 3% remain abstinent at 6 months (Benowitz, 2009), indicating that new medications are needed to specifically address the problem of relapse (George & O’Malley, 2004; Harmey, Griffin, & Kenny, 2012; Hurt et al., 2003; Irvin, Hendricks, & Brandon, 2003). The rewarding effects of nicotine are mediated, at least in part, by nicotine-evoked dopamine (DA) release leading to sensitization, which is associated with repeated nicotine administration and NIC addiction. Based on findings that the nonselective nAChR antagonist mecamylamine (MEC) has efficacy as a tobacco use cessation agent but is limited by peripherally mediated side effects, antagonist molecules with selectivity for central nAChR subtypes mediating nicotine-evoked DA release should have efficacy as tobacco use cessation agents with the therapeutic advantage of a limited side-effect profile.
2. CURRENT SMOKING CESSATION THERAPIES THAT TARGET nAChRs
2.1. Varenicline and cytisine
Varenicline was developed by Pfizer, Inc. and approved by the FDA as a smoking cessation agent in 2006. Varenicline not only acts as a partial agonist at α4β2* nAChRs but also interacts weakly with α3β2* and α6β2* nAChRs and is a full agonist at α7 nAChRs (Mihalak, Carroll, & Luetje, 2006). Varenicline has been shown to substitute for nicotine in drug discrimination studies in rats and blocks nicotine self-administration. In a randomized controlled clinical trial, carried out after 1 year of varenicline therapy, the rate of continuous tobacco use abstinence was 10% for placebo and 23% for varenicline; this compared favorably with the rate for bupropion (15%) (Jorenby et al., 2006). A subsequent meta-analysis of 101 clinical studies investigating varenicline showed it to be more effective than both bupropion and nicotine replacement therapy (~1.5 odds ratio) (Mills, Wu, Spurden, Ebbert, & Wilson, 2009).
A public health advisory notice was issued by the FDA in 2008 indicating that patients on varenicline experience serious neuropsychiatric symptoms, including depressed mood, suicidal ideation, and attempted or completed suicide (FDA public health advisory, 2008). In 2009, the FDA required both varenicline and bupropion to carry black box warnings, due to public reports of side effects, including depression, suicidal thoughts, and suicidal actions (FDA public health advisory, 2009). However, a recent a study of the medical records of 119,546 adults who had used a smoking cessation product between 1st September 2006 and 31st October 2011 (Thomas et al., 2013) found no clear evidence of an increased risk of treated depression or suicidal behavior for patients prescribed with varenicline or bupropion when compared to those taking nicotine replacement therapies. The FDA has also issued a recent safety announcement that the use of varenicline may be associated with a small, increased risk of certain cardiovascular adverse events in patients with cardiovascular disease (FDA drug safety communication, 2011).
Cytisine is a natural product isolated from Cytisus laborinum that is marketed in Europe as a smoking cessation agent under the trade name Tabex® (Etter, 2006; Zatonski, Cedzynska, Tutka, & West, 2006). Varenicline is a synthetic analog of cytisine. Cytisine was approved as a smoking cessation drug in 2006 in eastern Europe but is currently unavailable in the United States. Cytisine has a long-standing record of efficacy in the treatment of nicotine addiction in Europe and, similar to varenicline, is a selective partial agonist of human α4β2* nAChRs (Lukas, 2007). In addition to its utility in smoking cessation treatments, cytisine has shown antidepressant activity in male C57BL/6J mice (Mineur, Somenzi, & Picciotto, 2007). A recent meta-analysis of eight clinical studies indicated that cytisine has comparable effectiveness to other smoking cessation agents marketed in the United States (Hajek, McRobbie, et al., 2013; Hajek, Stead, et al., 2013).
2.2. Bupropion
Bupropion was originally marketed as an antidepressant molecule due to its ability to inhibit DA and norepinephrine (NE) transport into the presynaptic nerve terminal. Importantly, bupropion inhibits DA uptake into striatal synaptosomes and NE uptake into hypothalamic synaptosomes; it also dose-dependently increases presynaptic vesicular DA uptake and redistributes vesicular monoamine transporter-2 (VMAT2) protein (Rau et al., 2005). Bupropion has also been shown to act as an antagonist at human α3β4 ganglionic nAChRs via an allosteric inhibitory mechanism (Fryer & Lukas, 1999) and inhibits α3β2, α4β2, and α7 nAChRs expressed in Xenopus oocytes (Slemmer, Martin, & Damaj, 2000). These studies indicate that bupropion-induced decreases in smoking likely result from both its nAChR antagonism and its DAT and NET inhibition properties (Dwoskin, Pivavarchyk, et al., 2009; Dwoskin, Smith, et al., 2009). The clinical effectiveness of bupropion as a smoking cessation agent has been evaluated (Wu, Wilson, Dimoulas, & Mills, 2006). After 1 year of treatment, the odds of sustaining smoking cessation are 1.5 times higher in the bupropion group compared to the placebo group. The evidence indicates that bupropion is comparable to nicotine replacement therapy but is less effective than varenicline.
2.3. Mecamylamine
MEC is a nonselective noncompetitive channel blocker of nAChRs and was originally introduced as an antihypertensive drug in 1955 (Bacher, Wu, Shytle, & George, 2009). It has more recently been shown to dose-dependently decrease nicotine self-administration in laboratory animal models and to block cue-induced reinstatement of nicotine-seeking behavior (DeNoble & Mele, 2006; Click, Visker, & Maisonneuve, 1996). Clinical studies with MEC, which acts at both central and peripheral nAChRs, provide precedence for the use of nAChR antagonists as tobacco use cessation agents. MEC reverses both positive and negative subjective effects of i.v. nicotine in smokers (Lundahl, Henningfield, & Lukas, 2000). In a randomized, double-blind placebo-controlled study, MEC combined with a nicotine transdermal patch improved cessation outcome for up to 1 year compared to nicotine alone (Rose, 2006, 2008; Rose et al., 1994). Also, MEC is beneficial in reducing smoking satisfaction (Rose, 2008); however, MEC’s clinical utility is limited by anticholinergic side effects (e.g., constipation and hypotension) due to the lack of nAChR selectivity and inhibition of peripheral nAChRs (Rose, 2009). Thus, the development of selective small-molecule antagonists targeting central nAChRs mediating nicotine-evoked DA release constitutes a novel approach to the development of treatments for tobacco dependence and relapse prevention.
3. EMERGING POTENTIAL THERAPEUTICS FOR TREATMENT OF TOBACCO DEPENDENCE
Other partial agonists of nAChRs are currently under development. Dianicline, a drug developed by Sanofi-Aventis, is a selective partial agonist, binding primarily to the α4β2* nAChR subtype, and has a similar pharmacological profile to varenicline (Cohen et al., 2003). In drug discrimination studies, dianicline substituted for nicotine but at doses that decreased the rate of responding (Cohen et al., 2003). In clinical studies, dianicline exhibited a 16% success rate compared to 8% for placebo (Fagerstrom & Balfour, 2006). Clinical development of dianicline has been discontinued after reports of unfavorable results in phase 3 studies (Sanofi Pipeline, 2012).
Sazetidine A is a nicotine analog that also acts as a subtype-selective partial agonist at α4β2* nAChRs. Interestingly, this drug interacts as an agonist at (α4)2(β2)3 subtypes and as an antagonist at (α4)3(β2)2 subtypes (Xiao et al., 2006; Zwart et al., 2008). These studies have also shown that both α4β2* and α62* nAChRs mediate sazetidine A-evoked DA release, which can be blocked by both MEC and dihydro-β-erythroidine (Zwart et al., 2008). The α6-selective antagonist, α-conotoxin MII (α-CtxMII), also inhibits sazetidine A-evoked DA release (Imax = 50%). Sazetidine A substitutes completely for nicotine in drug discrimination studies in the rat and appears to act as a partial agonist at nAChRs in hippocampus and as a full agonist at nAChRs in striatum (Dwoskin, Pivavarchyk, et al., 2009; Dwoskin, Smith, et al., 2009; Xiao, Woolverton, Sahibzada, Yasuda, & Kellar, 2007).
UCI-30002 is a novel antagonist that acts as a positive allosteric modulator of GABAA receptors (Johnstone et al., 2004) and, as such, may also have efficacy as a negative allosteric modulator of α4β2 nAChRs, since this drug inhibits nicotine-evoked currents in Xenopus oocytes expressing neuronal α4β2, α7, and α3β4 nAChRs (Yoshimura et al., 2007). UCI-30002 has been shown to decrease nicotine self-administration utilizing both a fixed ratio and a progressive ratio schedule (Yoshimura et al., 2007), suggesting an effect on nicotine reward, and does not alter food-maintained responding (Yoshimura et al., 2007). Negative allosteric modulators of nAChRs represent a new area of drug discovery in the search for novel smoking cessation agents.
Recendy, AT-1001 N-(2-bromophenyl)-9-methyl-9-azabicyclo[3.3.1] nonan-3-amine has been reported as a selective, high-affinity α3β4* nAChR antagonist that potently and dose-dependently blocks nicotine self-administration in rats without altering responding for food (Toll et al., 2012). AT-1001 is the first ligand reported with a Ki below l0 nM at α3β4* nAChRs and a 90-fold selectivity over α4β2* and α7* nAChRs. Interestingly, AT-1001 is a poor inhibitor of NIC-induced DA release, compared to MEC and α-CtxMII, suggesting that its inhibition of nicotine self-administration in rats is not directly due to a decrease in DA release in nucleus accumbens, but may involve an indirect pathway mediated by α3 β4* nAChR inhibition. Nevertheless, these findings highlight the emergence of the α3β4* nAChR as a new potential target for the development of clinically useful smoking cessation agents.
4. SMALL-MOLECULE SURROGATES OF α-CtxMII AS SMOKING CESSATION AGENTS
We began our nAChR antagonist research program because we believed that subtype-selective nAChR antagonists that block reward-relevant mesocorticolimbic and nigrostriatal DA release evoked by nicotine might offer patients who do not respond to existing smoking cessation therapies alternative treatment options. Thus, we set out to discover and develop molecules that were selective antagonists at those nAChR subtypes that mediate nicotine-evoked DA release, that is, nAChR subtypes associated with reward produced by tobacco smoking (Crooks, Ravard, Teng, & Dwoskin, 1995; Dwoskin & Crooks, 2001). At the time we initiated this research program, this was considered to be a new and somewhat controversial approach to therapeutic intervention in tobacco addiction.
The seminal discovery that the neuropeptide α-CtxMII blocks nicotine-stimulated DA release in rat striatal synaptosomes (Kulak, Nguyen, Olivera, & Mcintosh, 1997) was a key finding of great relevance to the discovery of nAChR antagonists that block nicotine reward. The specific goal was to identify small, drug-like molecules that act as selective antagonists at α-CtxMII-sensitive nAChR subtypes, which contain α6 and β2 subunits (i.e., α6β2*, α6β2β3*, α6α4β2β3*, and α4α6β2*). These α-CtxMII-sensitive nAChRs were selected as targets for drug discovery because they are localized primarily to DA neurons that mediate nicotine-evoked DA release leading to nicotine reward. While α-CtxMII-insensitive nAChRs (e.g., α4β2* ) contribute to nicotine-evoked DA release, these nAChRs have a wide distribution in brain, which may lead to nonselective effects. In contrast, α-CtxMII-sensitive nAChRs mediating nicotine-evoked DA release offer an advantage as a target for smoking cessation due to their more restricted localization primarily to DA neurons. Currently, there are few selective drug-like compounds that differentiate α-CtxMII-sensitive nAChRs from other central and peripheral nAChR subtypes (e.g., α4β2*, α7*, and α3β4* nAChRs).
We considered our approach of developing small-molecule surrogates of α-CtxMII to be innovative for a number of reasons. First, no new chemical entities have been synthesized that are small molecules acting as α-CtxMII surrogates. Second, identification of α6β2* nAChRs as targets for the discovery of novel tobacco dependence medications was a novel approach at the time we commenced our studies (Crooks et al., 1995). Third, the overall general strategy is innovative, since currently, there are no specific nAChR antagonists approved for smoking cessation, and we believed that antagonist treatment could be useful in providing an alternative clinical option for highly motivated individuals undergoing cessation. Current pharmacotherapeutic approaches are based primarily on replacement therapies, in which full or partial nAChR agonists (e.g., nicotine and varenicline) interact predominantly with α4β2* nAChRs (Coe et al., 2005; Paterson et al., 2010; Rose, Salley, Behm, Bates, & Westman, 2010). nAChR agonists are generally well tolerated and produce good patient compliance, substituting for the reinforcing effect of tobacco use (Garwood & Potts, 2007; Shahan, Odum, & Bickel, 2000). However, partial agonist or agonist replacement therapy is less than optimal (Schuh, Schuh, & Henningfield, 1996); a potential disadvantage is that continued activation of nAChRs maintains receptor desensitization and nicotine dependence. Fourth, the ability to selectively inhibit central nAChRs offers new tools to provide insights on mechanisms involving α6β2* nAChRs. Fifth, our focus on the development of pharmacotherapies to thwart relapse is of great significance with regard to developing effective strategies to combat nicotine addiction. While the various factors involved in relapse in humans are complex, including the co-occurrence of alcohol consumption and factors related to impulsivity and stress (Bourque, Mendrek, Dinh-Williams, & Potvin, 2013; Brunzell, 2012; VanderVeen, Cohen, & Watson, 2012), it is also known that smoking-related cues play a role (Le Foll & Goldberg, 2005; Perkins, 1999). To the extent that cue-induced craving is associated with activation of α6β2* nAChRs, it may be possible to develop an antagonist pharmacotherapy that enhances relapse prevention by blocking the effect of these cues.
nAChRs, members of the Cys-loop family of ligand-gated ion channel receptors, consist of pentameric transmembrane proteins with diverse composition (Anand, Conroy, Schoepfer, Whiting, & Lindstrom, 1991; Millar & Gotti, 2009). Although nAChR subtype predominance does not necessarily reflect functional importance, the α4β2 subtype, probed by high-affinity [3H]-nicotine binding, is predominant in the CNS (Flores, Rogers, Pabreza, Wolfe, & Kellar, 1992; Whiting & Lindstrom, 1987; Zoli, Lena, Picciotto, & Changeux, 1998). Immunoprecipitation studies indicate that more than two different subunits form functional nAChRs and individual neurons elaborate multiple subtypes (Conroy, Vernallis, & Berg, 1992; Forsayeth & Kobrin, 1997; Turner & Kellar, 2005), increasing the complexity associated with elucidation of specific nAChR subtype function. Mammalian homomeric nAChRs consist of α7 or α9 subunits (Flores et al., 1992; Gotti, Zoli, & Clementi, 2006; Mcintosh et al., 2005; Wada et al., 1989). The exact subunit composition and stoichiometry of native nAChRs remain to be elucidated. Nevertheless, subunit composition has an important impact on pharmacological sensitivity, including antagonist affinity (Cachelin & Rust, 1995; Harvey & Luetje, 1996; Harvey, Maddox, & Luetje, 1996). Nicotine activation of nAChRs increases extracellular DA in nucleus accumbens and striatum, mediating, at least in part, nicotine reward (Corrigall, Franklin, Coen, & Clarke, 1992; Rahman et al., 2008). nAChRs modulate synaptic activity and neurotransmitter release (Collins, Salminen, Marks, Whiteaker, & Grady, 2009; Dani & Bertrand, 2007; McGehee & Role, 1995; Quarta et al., 2007). Rat nigral DA neurons express mRNA for α3, α4, α5, α6, α7, β2, β3, and β4 (Arroyo-Jimenez et al., 1999; Wada et al., 1989). Studies using β2 knockout mice have revealed that β2 is necessary for nicotine-evoked DA release (Grady et al., 2001, 2007; Salminen et al., 2004; Whiteaker et al., 2000). Subtype assignment of native nAChRs mediating nicotine-evoked DA release is based largely on the inhibition of agonist-induced responses by subtype-selective antagonists, defined by their inhibitory activity in cell systems expressing nAChR subunits of known composition. A major role for α6β2* in nicotine-evoked striatal DA release is based on α-CtxMII inhibition of nicotine-evoked [3H]-DA release, as well as knockout and gain-of-function studies (Cartier et al., 1996; Cui et al., 2003; Drenan et al., 2010; Drenan, Grady et al., 2008; Drenan, Nashmi, et al., 2008; Kaiser, Soliakov, Harvey, Luetje, & Wonnacott, 1998); α6 and β2 subunits are highly expressed in DA cell bodies (Cm et al., 2003; Goldner, Dmeley, & Patrick, 1997; Le Novère, Zoli, & Changeux, 1996).
Results from comprehensive molecular genetics gene deletion studies implicate the α4α6β2β3* subtype in nicotine-evoked DA release and nicotine reward. These studies suggested that nicotine-evoked DA release is mediated by 6 different subtypes, that is, α6β2β3*, α4α6β2β3*, α6β2*, and α4α6β2* (α-CtxMII-sensitive subtypes) and α4β2* and α4α5β2* (a-CtxMII-insensitive subtypes) (Gotti et al., 2005; Salminen et al., 2004). The α4α6β2β3* subtype had the highest sensitivity to nicotine of any native nAChR and constituted approximately 50% of a6-containing nAChRs on DA terminals of wild-type mice (Salminen et al., 2007). From these studies, it is clear that multiple subtypes can mediate nicotine-evoked DA release. The ability to design small-molecule antagonists that can selectively target these subtypes to inhibit nicotine-evoked DA release represents a primary goal for our laboratory. DA neurons have special chaperones for assembling accessory subunits with α6 for transporting nAChRs to the cell surface; α6 concatamers will increase understanding of the functional properties of α6-containing nAChRs (Kuryatov & Lindstrom, 2011). It is important to note that the subtype composition and relative contribution of nicotine-evoked DA release nAChRs are species-dependent, and relative contributions of different subtypes are species-dependent. In mice, α6-containing nAChRs represent 30% of presynaptic nAChRs that mediate nicotine-evoked DA release, while in nonhuman primates, 70% of nAChRs that mediate nicotine-evoked DA release are α6-containing nAChRs (Kulak et al., 1997; McCallum, Collins, Paylor, & Marks, 2006; Salminen et al., 2004). CNS location is also an important factor; α6β2* nAChRs are the predominant subtype that mediates nicotine-evoked DA release in nucleus accumbens, whereas α6β2* nAChRs only contribute in striatum (Exley, Clements, Hartung, Mcintosh, & Cragg, 2008). These studies demonstrate that α6β2* nAChRs are major subtypes that mediate nicotine-evoked DA release and nicotine self-administration leading to dependence (Brunzell, 2012; Brunzell, Boschen, Hendrick, Beardsley, & Mcintosh, 2010; Drenan et al., 2010; Gotti et al., 2010; Jackson, Mcintosh, Brunzell, Sanjakdar, & Damaj, 2009; Moretti et al., 2010; Pons et al., 2008).
Neurotoxin peptides acting as subtype-selective nAChR antagonists are unlikely to be easily developed into treatments for tobacco use cessation, since they are high-molecular-weight peptides and unlikely to not cross the blood–brain barrier. Our current approach aimed to provide potent small-molecule antagonists that inhibit the same nAChR subtypes as α-CtxMII. Pharmacological tools, which differentiate among specific high-affinity heteromeric nAChR subtypes, are few. This situation represents a major hindrance in increasing our understanding of which specific subtypes contribute to nicotine addiction and in the development of effective pharmacotherapies for tobacco dependence. We hypothesize that drug-like nAChR antagonists inhibiting α6β2* nAChR subtypes will decrease nicotine-evoked DA release and nicotine reward, thus providing a new class of cessation agents that prevent relapse. In support of this hypothesis, our drug discovery effort focused initially on the development of nAChR subtype-selective antagonists based on structural modification of the nicotine molecule (Dwoskin & Bardo, 2009; Dwoskin & Crooks, 2001; Dwoskin, Smith, et al., 2009; Dwoskin et al., 2004).
5. MONOQUATERNARY AMMONIUM SALTS DERIVED FROM N-METHYLNICOTINIUM ION AS NICOTINIC RECEPTOR ANTAGONISTS
Based upon early findings that the N-methylnicotinium ion inhibited DA uptake into rat striatal slices (Dwoskin, Leibee, Jewell, Fang, & Crooks, 1992), we initially synthesized and evaluated a series of N-n-alkylnicotinium analogs and related compounds for their ability to inhibit nicotine-evoked DA release from rat striatal slices and to displace [3H] -nicotine binding from rat striatal membranes (α4β2* nAChRs) (Crooks et al., 1995). We were particularly interested in the nAChR mediating nicotine-evoked DA release, since we believed that it constituted an excellent therapeutic target for the development of clinical candidates for treating nicotine addiction. The exact subunit composition and stoichiometry of this nAChR receptor were not known at the time our studies began and even today have still not been fully elucidated. The receptor was initially termed the α3β2 subtype but was later renamed as a group of α6* or more recently α6β2* subtypes.
The most effective antagonists screened in our nicotine-evoked DA release assay were those quaternary ammonium nicotine analogs that incorporated an N-n-alkyl substituent of three carbons or more in length. The introduction of an aromatic or unsaturated residue into the N-alkyl substituent also afforded antagonist molecules. The most potent compound was S-(−)-N-n-octylnicotinium iodide (NONI, Fig. 13.1), which had full antagonist potency. Importantly, NONI had low affinity for the high-affinity [ 3H] -nicotine binding site (Ki = 20 µM) and appeared to be somewhat more selective as an antagonist at α6β2* nAChRs (IC50 = 0.62 µM) (Crooks et al., 2004). Interestingly, some of the N-alkylnicotinium analogs had differential properties at α4β2* , α7* , and α6β2* nAChRs. The related compound S-(−)-N-n-decylnicotinium iodide (NDNI; Fig. 13.1) was not active as an antagonist at α6β2* nAChRs (IC50 > 100 µM) but afforded a Ki value of 90 nM in the [ 3H] -nicotine binding assay. The N-n-dodecyl analog, NDDNI (Fig. 13.1), was a very potent antagonist at α6β2* nAChRs (IC50 = 9 nM) but was nonselective, exhibiting a Ki of 140 nM at α4β2* nAChRs. In the nicotine-evoked 86Rb+ efflux assay, a functional assay for the α4β2* nAChR subtype, NDNI and NONI had IC50s of 30 pM and 85 nM, respectively (Dwoskin, Wilkins, Pauly, & Crooks, 1999).
Figure 13.1.

Chemical structures of the N-n-alkyl-substituted analogs of S-(−)-nicotine, NONI (S-(−)-N-n-octylnicotinium iodide), NDNI (S-(−)-N-n-decylnicotinium bromide, and NDDNI (S-(−)-N-n-dodecylnicotinium iodide. NONI exhibited an IC50 of 0.62 µM at α6-containing nAChRs.
These results were considered key findings, since NONI represented our first lead compound in the search for new tobacco use cessation agents (Dwoskin et al., 2004). These early data also indicated that while NONI was a selective α6β2* nAChR subtype antagonist, NDNI was a potent and selective α4β2* nAChR subtype antagonist and that both compounds might be valuable tools for probing the consequences of activating different subtypes of nicotinic receptor. More importantly, the N-n-alkylnicotinium analogs as a group were found to have good affinity for the blood–brain barrier choline transporter, and [3H]-NONI was shown to act as a substrate for this transporter in brain uptake studies in the rat. NONI exhibited the following saturable blood–brain barrier penetration parameters— Vmax = 603 ± 80 nmol/min/g, Km + 661± 75 µM, and Kd = 7.3±3.1 × 10−4 ml/g—compared to choline, Vmax = 3±0.3 nmol/ min/g, Km = 42±6µM, and Kd = 1.0±0.06 × 10−4 ml/g (Crooks et al., 2004; Lockman et al., 2008), indicating that this molecule, although cationic and polar, is brain-bioavailable. However, the general lack of selectivity of this class of analogs for α6β2* nAChRs prompted us to explore other structural modifications of the nicotine molecule.
5.1. Bis-Quaternary ammonium analogs
During the course of the structure–activity studies on the N-alkylnicotinium analogs, it was observed that significant structural changes could be made to the nicotinium moiety (i.e., the quaternary ammonium “headgroup”) with retention of antagonist potency at nAChRs mediating nicotine-evoked DA release. It was found that simple azaheterocycles, such as pyridine and 3-picoline, could be substituted for the S-(−)-nicotine moiety with retention of nAChR inhibitory potency (Dwoskin et al., 2004). The classical bis-quaternary ammonium salts, hexamethonium chloride (N,N′-hexane-l,6-bis-trimethyammonium chloride) and decamethonium chloride (N,N′-decane-l,10-bis-trimethyammonium chloride), which are regarded as simplified analogs of the more conformationally retrained natural product, d-tubocurarine, have been utilized in the past to differentiate between subtypes of different peripheral nicotinic receptors. Thus, decamethonium is a selective inhibitor of neuromuscular nicotinic receptors, while hexamethonium inhibits ganglionic nicotinic receptors (Koelle, 1975). Recent studies (Papke, Wecker, & Stitzel, 2010) have since shown that decamethonium and hexamethonium interact with both peripheral and central nAChRs expressed in Xenopus oocytes. Thus, although decamethonium was shown to be a selective depolarizing blocker of muscle-type nAChRs in these studies, it was also an effective antagonist of mouse neuronal nAChRs and was particularly potent at α7 homomeric receptors. Similarly, hexamethonium exhibited comparable inhibitory potency at both α3-containing nAChRs (ganglionic) and α4β2 nAChRs. These data indicate that the earlier classification of these channel-blocking agents did not take into account the inability of these molecules to cross the blood–brain barrier, which results in functional protection of CNS receptors in whole animal studies.
Based on the earlier studies with the trimethylammonium analogs, a second quaternary ammonium moiety was introduced into the scaffold of the N-alkylnicotinium analogs, in order to determine the effect of bis-cationic analogs on antagonist potency and selectivity and to additionally probe the chemical space around the bis-”headgroups” by replacing the nicotinium headgroup with other azaheterocyclic moieties. Thus, a series of bis-analogs bearing nicotinium, pyridinium, picolinium, quinolinium, and isoquinolinium headgroups separated by variable-length n-alkane linkers were initially synthesized and evaluated as ligands for α6β2* , α4β2* , and α7* nAChRs (Ayers et al., 2002; Crooks et al., 2004). From the observed structure-activity trends, it was clear that the majority of the analogs generally had poor affinity for the α4β2* nAChR, with the exception of the bis-nicotinium analogs. One of these, S-(−)-N,N′-decane-l,10-diyl-bis-nicotinium diiodide (Fig. 13.2), exhibited a Ki value of 330 nM at α4β2* nAChRs and functionally inhibited nicotine-evoked 86Rb+ efflux (IC50 = 3.76 µM) while demonstrating no affinity for α7* nAChRs. The bis-quinolinium and bis-isoquinolinium analogs all exhibited affinity for α7* nAChRs, the most potent being N,N′-dodecane-l,12-diyl-bisquinolinium dibromide (Fig. 13.2), with a Ki value of 1.6 µM, which is comparable to nicotine.
Figure 13.2.

Chemical structures of the bis-quaternary ammonium compounds bNDI (S-(−)-N,N-decane-1,10-diyl-bis-nicotinium diiodide), bQDDB (N,N-dodecane-1,12-diyl-bis-quinolinium dibromide), and bPiDDB (N,N-dodecane-1,12-diyl-bis-3-picolinium dibromide). bPiDDB exhibited an IC50 of 5 µM at α6-containing nAChRs.
The most interesting series of bis-analogs were the N,N′-n-alkane-diyl-bis-3-picolinium dibromides. These analogs had n-alkane tethers ranging from C6 to C12, and all of the analogs exhibited antagonism of α6β2* nAChRs (Crooks et al., 2004; Dwoskin et al., 2004). The most potent analog was N,N′-dodecane-l,12-diyl-bis-3-picolinium dibromide (bPiDDB; Fig. 13.2), which exhibited a remarkable IC50 of 5nM (Imax = 60%) and had little or no affinity for either α4β2* or α7* nAChRs. We considered bPiDDB to be an important lead candidate for further study, since it had high potency and selectivity at the nAChR mediating nicotine-evoked DA release and, like NONI, was an excellent substrate for the blood–brain barrier choline transporter (i.e., brain-bioavailable). bPiDDB exhibited the following saturable blood–brain barrier penetration parameters—Vmax = 4.3±1.0 nmol/min/g, Km = 5±2µM, and Kd = 1.1 ±0.06 × 10−4 ml/g—which are comparable to choline (Lockman et al., 2008). Additionally, we also found that bPiDDB diminished nicotine self-administration in proof-of-concept in vivo studies (Fig. 13.3).
Figure 13.3.

General chemical structures of conformationally restricted analogs of bPiDDB that incorporate 1,2-, 1,3-, and 1,4-dialkylphenyl linkers between the two quaternary ammonium headgroups. This structural change generally led to a decrease in the inhibition of nicotine-evoked DA release.
In a subsequent study, we synthesized and evaluated an extended series of bis-azaaromatic quaternary ammonium salts (Zheng, Sumithran, Deaciuc, Dwoskin, & Crooks, 2007). This study focused on investigating the possible binding conformation of these molecules. The design of the model compounds incorporated a phenyl ring into the middle of the N-N′ alkane linker moiety, allowing a variety of arrangements of the two smaller methylene linker units around the aromatic “core” unit (i.e., 1,2-, 1,3-, and 1,4-dialkylphenyl linkers) (Fig. 13.4). This approach constrains these molecules into “extended” or “angular” geometries and is a well-known and extensively used strategy in drug design for locking ligands into a desired conformation or geometry, with the goal of increasing activity and selectivity. To increase conformational restriction even further, a triple bond was also introduced into each of the alkane linker units that were attached to the central phenyl ring. The study indicated that the transition from the more conformationally flexible N,N′-alkanyl-diyl-bis-azaheterocyclic analogs, such as bPiDDB, to more conformationally restricted analogs generally resulted in a decrease in inhibition of nicotine-evoked [3H]-DA release and suggested that maintaining flexibility of the alkane linker is important for high inhibitory potency and optimal interaction with nAChRs. The data also suggested that bPiDDB likely binds in an “extended” conformation, rather than an “angular” conformation. It was hypothesized from molecular modeling studies that the greater potency of the more flexible bis-analogs was due to the binding site of the α6-containing nAChR subtype likely being deep inside the channel of the receptor; thus, ligands must be flexible enough to reach this binding site.
Figure 13.4.

Concentration of 14C-bPiDDB in the plasma and brain after 1 and 3 mg/kg administration of 14C-bpiDDB in the Sprague–Dawley rat.
In a related study (Zhang, Lockman, et al., 2008; Zhang, Zheng, et al., 2008), a series of novel bis-pyridinium cyclophanes were evaluated as conformationally restricted bis-quaternary ammonium compounds. These analogs were poor antagonists at α6β2* nAChR but were considered worthy of evaluating for their affinity for the blood–brain barrier choline transporter. All the cyclophanes examined exhibited high affinity for the choline transporter when compared to the natural substrate, choline. Of the cyclophanes tested, two analogs N,N′-(l,10-decanediyl)3,3′-(l,9-decadiyn-l,10-diyl)-bis-pyridinium diiodide and N,N′-(l,9-nonanediyl)3,3′-(l,9-decadiyn-l,10-diyl)-bis-pyridinium dibromide exhibited Ki values of 0.8 and 1.4 µM, respectively, and are among the most potent blood-brain barrier choline transporter ligands ever reported. We extended this study to include a series of bis-azaaromatic quaternary ammonium compounds containing both flexible and conformationally restricted polymethylenic linker moieties around a central phenyl core (Zheng et al., 2010). The results from this study suggested that incorporating a linear, conformationally restricted linker into the molecule improves affinity for the choline transporter.
5.1.1 Pharmacokinetic studies with bPiDDB
With bPiDDB as the lead molecule, the pharmacokinetics of [14C-methyl]-bPiDDB was investigated in the rat (Albayati, Dwoskin, & Crooks, 2008). We were interested in determining whether bPiDDB was brain-bioavailable, since we had determined earlier that it was a good substrate for the blood–brain barrier choline transporter (Lockman et al., 2008) and should therefore accumulate in the brain after peripheral administration. Plasma concentrations of [14C-methyl]-bPiDDB after peripheral administration (1, 3, and 5 mg/kg) were determined at 10 time points over 3 h to afford absolute plasma bioavailabilities of 80%, 68%, and 104%, respectively, and Cmax values of 0.13, 0.33, and 0.43 µg/ml, respectively (Fig. 13.5). Tmax values were 5.0, 6.7, and 8.8 min, and t1/2 values were 76.0, 54.6, and 41.7 min, respectively. No significant metabolic products of bPiDDB could be detected, and moderate protein binding (63–65%) was observed. As anticipated, bPiDDB penetrated the blood–brain barrier, even though it is an extremely polar, dicationic molecule. The blood–brain ratio at 5 min was 0.18, increasing to 0.51 at 60 min, indicating that clearance of bPiDDB from the brain was slower than clearance from plasma. Overall, the results indicated that bPiDDB is distributed rapidly from the site of administration into plasma, affords good plasma concentrations, and reaches brain tissues through facilitated transport via the blood–brain barrier choline transporter to afford therapeutically relevant concentrations in rat brain.
Figure 13.5.

Acute bPiDDB (1 and 3 mg/kg) decreases NIC self-administration: bPiDDB (0.3–3 mg/kg, s.c, 15 min pretreatment) decreased the number of nicotine infusions (0.03 mg/kg/infusion) earned (active lever).
5.1.2 bPiDDB pharmacology
We have shown that repeated nicotine administration in rats robustly increases in vitro bPiDDB inhibitory potency at α6-containing nAChRs by three orders of magnitude compared to similar in vitro assays in naive rats (Smith et al., 2010) (i.e., IC50 = 5 pM vs. 6 nM). This finding is relevant to smoking cessation therapy, since tobacco smokers self-administer nicotine repeatedly and animal models incorporating repeated nicotine treatment allow for better mechanistic evaluation of therapeutic candidates following neuroadaptive changes. This study demonstrated that repeated nicotine treatment may differentially regulate the stoichiometry, conformation, and/or composition of α6β2* nAChRs.
We have also shown that bPiDDB inhibits nicotine-evoked norepinephrine release from superfused rat hippocampal slices with an IC50 of 430 nM (Imax = 90%) and likely acts as an allosteric inhibitor at this nAChR subtype (putative α3β4) (Smith et al., 2009). Kinetic analysis afforded a Schild regression slope that was different from unity, which is consistent with allosteric inhibition. Thus, bPiDDB has 200-fold more inhibitory potency at α6β2* nAChR subtypes compared to its inhibitory potency at α3β4* nAChRs. This is important because the α3β4* subtype, like the α6β2* subtype, contributes to nicotine reward (Picciotto & Kenny, 2013; Stoker & Markou, 2013).
In a critical behavioral study, we determined that bPiDDB dose-dependently decreased nicotine self-administration, but not sucrose-maintained responding in the rat (Neugebauer, Zhang, Crooks, Dwoskin, & Bardo, 2006); and in locomotor experiments, bPiDDB attenuated the hyperactivity produced by acute and repeated nicotine dosing in the rat. Thus, bPiDDB continued to be our lead molecule in the search for subtype-selective nAChR antagonists. It should be noted that when administered peripherally, several bis-quaternary ammonium antagonists, including bPiDDB, specifically decreased nicotine self-administration and reinstatement of nicotine seeking in rats, providing proof of concept of this representative class of drug as a new pharmacotherapy for nicotine addiction. Additional behavioral studies in the rat have been carried out on other structurally related N,N′-alkane-diyl-bis-3-picoliniums (Dwoskin et al., 2008; Wooters et al., 2011).
In summary, bPiDDB is a selective, noncompetitive inhibitor of α6β2* nAChRs; bPiDDB inhibits [3H]-nicotine and [3H]-MLA binding (α4β2* and α7* nAChRs, respectively) only at high concentrations (>10 µM; Dwoskin et al., 2008) and decreases nicotine self-administration in rats. Furthermore, bPiDDB (1 µM) produced only 15% and 12% inhibition of acetylcholine (1 mM) -induced current at ganglionic-type α3β4 nAChRs and muscle-type α1β1εδ nAChRs, respectively, expressed in Xenopus oocytes (unpublished data). Subsequent optimization studies (see the succeeding text) were carried out to address the likelihood that this general class of compound would have poor oral bioavailability.
5.2. Tris-Quaternary ammonium salts
A related SAR study to assess the effect of introducing a third picolinium headgroup into the bPiDDB scaffold on inhibition of nicotine-evoked DA release was also carried out. The rationale for this structural modification was to determine if the change from one (mono), to two (bis), to three (tris) cationic headgroups would continue to improve affinity and inhibitory potency for the target nAChR protein. An increase in inhibitory potency in the progression from mono, to bis, to tris would indicate that at least three polar interactions between the cationic groupings in these molecules and the corresponding putative anionic sites on the receptor protein can occur. This approach was based on molecular modeling data that indicated that the binding sites that the bis-analogs interacted with were located in the vestibule area of the nAChR ion channel.
Two series of tris-scaffolds were constructed, that is, the l,3,5-tri-(pent-l-ynyl-5-azaaromatic quaternary ammonium) benzene series (scaffold A, unsaturated) (Fig. 13.6A) and the l,3,5-tri-(n-pentanyl-5-azaaromatic quaternary ammonium) benzene series (scaffold B, saturated) (Fig. 13.6B) (Zheng, Zhang, et al., 2007). The tris-azaaromatic quaternary ammonium salts were synthesized utilizing Sonogashira coupling chemistry (Fig. 13.7). A variety of azaaromatic heterocycles were utilized as headgroups. These molecules were then evaluated as antagonists at α6β2 nAChRs and for their affinity for α4β2* and α7* nAChRs. Three compounds emerged from this study as potent and selective inhibitors of α6β2* nAChRs with IC50S in the range 0.2–4 nM. Two of these were unsaturated picolinium analogs, that is, l,3,5-tri-{5-[l-(3-picolinium)]-pent-l-ynyl}benzene tribromide (TRIS-1) (IC50 = 0.2 nM) and 1,3,5-tri- { 5- [1 - (2-picolinium)] -pent-1 -ynyl}benzene tribromide (TRIS-2) (IC50 = 4.0 nM); the third compound was a saturated 3-picolinium analog: l,3,5-tri-{5-[l-(3-picolinium )]-pentyl}benzene tribromide (TRIS-3) (IC50 = 2.0 nM). None of these analogs had any significant affinity for α4β2* and α7* nAChRs. Thus, these very potent and selective tris-analogs represented additional new leads in our search for potent and selective nAChR antagonists as treatments for nicotine addiction. However, subsequent studies showed that although one of the lead tris-analogs was effective in dose-dependently decreasing nicotine self-administration in the rat, it produced toxicity at the higher concentration range of the dose–response. Additionally, the tris-analogs generally exhibited lower affinity for the blood–brain choline transporter and thus were expected to have poor oral and brain bioavailability.
Figure 13.6.

Synthetic scheme for the synthesis of tris-3-picolinium analogs.
Figure 13.7.

Structures of the two tris-quaternary ammonium scaffolds A (1,3,5-tri-{5-[1-(ammonium)-pent-1-ynyl}benzene) and B (1,3,5-tri-{5-[1-(ammonium)-pentyl}benzene); TRIS-1 scaffold A, NR3 = 3-picolinium; TRIS-2 scaffold A, NR3 = 2-picolinium; TRIS-3 scaffold B, NR3 = 3-picolinium.
5.3. Tetrakis-Quaternary ammonium salts
Concomitant with the development of the tris-series of compounds, the incorporation of an additional cationic headgroup into the tris-scaffold was carried out to complete the transition from mono-, to bis-, to tris-, to tetrakis-quaternary ammonium salts. This modification was investigated to determine if further increases in the number of cationic centers in these molecules would enhance antagonist potency and selectivity at α6β2 nAChRs. We generated two series of tetrakis-azaaromatic quaternary ammonium analogs, that is, l,2,4,5-tetrakis-(pent-l-ynyl-5-azaaromatic quaternary ammonium)benzene salts (scaffold A, unsaturated) (Fig. 13.8A) and l,2,4,5-tetrakis-(n-pentanyl-5-azaaromatic quaternary ammonium) benzene salts (scaffold B, saturated) (Fig. 13.8B) (Zhang, Lockman, et al., 2008; Zhang, Zheng, et al., 2008), utilizing a similar Sonogashira coupling strategy to that used in the synthesis of the saturated and unsaturated tris-scaffolds (Fig. 13.9). These analogs were evaluated as antagonists at α6β2* nAChRs and as ligands for α4β2* and α7* nAChRs. Three tetrakis-analogs where identified as potent and selective inhibitors of α6β2* nAChR subtype; these were all members of the saturated tetrakis-series, that is, l,2,4,5-tetrakis-{5-[l-(3-benzylpyridinium)]-n-pentanyl}benzene tetrabrormde (TETRAKIS-1) (IC50 = 28 nM), l,2,4,5-tetrakis-{5-[l-(quinolinium)]-n-pentanyl}benzene tetrabromide (TETRAKIS-2) (IC50 = 56 nM), and l,2,4,5-tetrakis-{5-[l-(3-(3-hydroxypropyl)-pyridinium)]-n-pentanyl}benzene tetrabromide (TETRAKIS-3) (IC50 = 3.4 nM). It is noteworthy that 3-picolinium analogs in both tetrakis-series afforded only weak inhibitory activity at α6β2* nAChR subtypes, while a variety of signif-icandy larger and unique quaternary ammonium headgroups were present in the more active analogs.
Figure 13.8.

Synthetic scheme for the synthesis of tetrakis-3-picolinium and tetrakis-isoquinolinium analogs.
Figure 13.9.

Structures of the two tetrakis-quaternary ammonium scaffolds A (1,2,4,5-tetrakis-{5-[1-(ammonium)-pent-1-ynyl}benzene) and B (1,2,4,5-tetrakis-{5-[1-(ammonium)-pentyl}benzene); TETRAKIS-1 scaffold A, NR3 = 3-benzylpyridinium; TETRAKIS-2 scaffold A, NR3 = quinolinium; TETRAKIS-3 scaffold B, NR3 = 3-(3-hydroxypropyl)-pyridinium.
Unfortunately, the tetrakis-analogs were not substrates for the blood-brain barrier choline transporter and consequently turned out be ineffective in dose-dependently decreasing nicotine self-administration in the rat. Nevertheless, these results and those described earlier for the bis- and tris-analogs suggested that the strategy to identify highly potent nAChR ligands through progressive introduction of cationic moieties around a common phenyl core structure had been successful in affording potent inhibitors of α6β2* nAChRs.
Reviews of our work on the discovery and development of bis-, tris-, and tetrakis-quaternary ammonium salts as potential smoking cessation agents provide a comprehensive account of how new leads from this drug discovery effort were developed in our preclinical pharmacological and behavioral evaluation programs (Dwoskin, Pivavarchyk, et al., 2009; Dwoskin, Smith, et al., 2009).
5.4. QSAR modeling of quaternary ammonium nicotinic receptor antagonists
Since α6β2* nAChRs have not been fully characterized and to gain a better understanding of the relationship between biological activity and antagonist structure, we carried out QSAR modeling studies on the libraries of quaternary ammonium salts as antagonists at α6β2* nAChRs (Zheng et al., 2006). Back-propagation artificial neural networks (ANNs) were trained on a data set of 45 molecules with quantitative IC50 values to model structure–activity relationships. The ANN QSAR models produced a reasonable level of correlation between experimental and calculated log(l/IC50) (r2 =0.76). We have used this predictive model effectively to reduce synthetic and in vitro screening activities by eliminating virtual compounds of predicted low activity from the pool of candidate molecules for synthesis. The application of this ANN QSAR model has led to the successful discovery of six new compounds in this study and continues to be utilized as a useful predictive tool for our drug discovery efforts. We were also able to develop a QSAR model to predict maximal inhibition (Imax) values for quaternary ammonium antagonists interacting at α6β2* nAChRs (Zheng, McConell, Zhan, Dwoskin, & Crooks, 2009; Zheng, Zheng, et al., 2009). This represented the first reported QSAR study to predict Imax values. The study was conducted using multiple linear regression (MLR) analysis and neural network analysis with maximal inhibition values of the antagonists as target values. Both models afforded good correlations (MLR—r2 =0.89, rmsd = 9.01, q2 = 0.83, and loormsd = 11.1 and NN—r2 = 0.89, rmsd = 8.98, q2 = 0.83, and loormsd= 11.2), which provided a basis for rationalizing the selection of virtual compounds for synthesis in the discovery of effective and selective second-generation antagonists of α6β2* nAChRs.
5.5. Bis-, tris-, and tetrakis-tertiary amino analogs
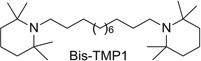
Building on the success of the bis-, tris-, and tetrakis-series of compounds as potent antagonists of α6β2* nAChRs, we initiated a structure–activity study to determine if the azaaromatic headgroups attached to these scaffolds could be replaced with nonaromatic azacyclic tertiary amino headgroups. Our reasoning was based on the properties of the BTMPS molecule [bis-(2,2,6,6,-tetramethyl-4-piperidinyl) sebacate] (Fig. 13.10), a noncompetitive, use-dependent antagonist at nAChRs. This molecule incorporates two azacyclic 2,2,6,6-tetramethyl-4-piperidinol (TMP) headgroups separated by a linear sebacate linker and could be considered a structural analog of bPiDDB. One advantage of replacing azaaromatic headgroups with nonaromatic azacyclic headgroups is that this would allow more efficient partitioning of these molecules through cell membranes via passive diffusion, leading to improved oral and brain bioavailability, while still allowing the headgroups to be protonated at physiological pH (since the headgroup moieties would likely have pKas in the range 7–9). Thus, these molecules, like bPiDDB, would be dicationic at physiological pH and may interact with the same receptor binding sites that bPiDDB interacts with. We considered azacyclic TMP and MEC structures as suitable replacement headgroups for the 3-picolinium headgroups in bPiDDB, since both TMP and MEC have been previously shown to be noncompetitive antagonists at nAChRs. By linking two or three MEC or TMP molecules together via a linear lipophilic bis-methylene linker or a conformationally restricted (unsaturated) tris-scaffold, a series of bis- and tris-tertiary amine analogs were synthesized and evaluated as potent antagonists at α6-containing nAChRs (Zhang et al., 2010). Three analogs were identified that exhibited high antagonist potency (Fig. 13.10): the bis-TMP analog, N,N′-dodecane-l,12-diyl-bis-[l-(2,2,6,6-tetramethylpiperidine)] dihydrochloride (IC50 = 2.2 nM; Imax = 87%); the bis-MEC analog, S,S-(+)-N,N′-dodecane-1,12-diyl-bis-1-mecamylamine dichloride (IC50 = 46nM; Imax = 90%); and the tris-MEC analog, S,S,S-(+)-l,3,5- tri-{5-[l-(mecamylamine)]-pent-1-ynyl}benzene trihydrochloride (IC50 = 107nM; Imax = 62%). Thus, linking TMP and MEC headgroups to a lipophilic C12 n-alkane linker or a conformationally restrained (unsaturated) tris-scaffold with tertiary amino rather than quaternary ammonium headgroups afforded several potent antagonists of α6β2* nAChRs. Such analogs may provide a new strategy for the design of more drug-like molecules that have improved membrane permeation characteristics and high inhibitory potency against α6β2* nAChRs.
Figure 13.10.

Structure of BTMPS and structurally related analogs; IC50 and Imax values of TMP and mecamylamine analogs in the nicotine-evoked dopamine release assay are also provided.
The promising results with the bis- and tris-analogs in which azaaromatic quaternary ammonium headgroups had been replaced with nonaromatic azacyclic headgroups prompted us to expand this library of more drug-like molecules. From the synthesis viewpoint, a means to reductively convert our large library of azaaromatic quaternary ammonium bis-, tris-, and tetrakis-analogs into corresponding analogs containing tertiary amino headgroups was sought. This would allow us to generate a large library of more drug-like molecules very efficiently in a single reductive chemical step. We were mindful of the likelihood that exhaustive reduction of the azaaromatic moiety would, in cases where a substituted aromatic heterocyclic headgroup was present (e.g., a 3-picolinium moiety), introduce a chiral center into the resulting azacyclic product, which would cause serious issues with regard to isomer separation in the final product (i.e., multiple diastereomers and their enantiomers). Also, we were aware that the reductive agent to be utilized should be one that would not compromise linkers and/or scaffolds that incorporated double or triple bonds. A suitable reduction methodology utilizing NaBH4 was developed that rapidly and efficiently converted pyridinium and isoquinolinium headgroups into corresponding 1,2,5,6-tetrahydropyridine and 1,2,3,4-tetrahydroisoquinoline headgroups (Zhang et al., 2011). This methodology was selective for azaaromatic moieties and also useful for the reduction of 3-substituted pyridinium headgroups, since the residual double bond in the reduced headgroup prevented the generation of a chiral carbon at C3 of the pyridine ring in the reduced product.
A small library of these more drug-like bis-, tris-, and tetrakis-tertiary amine analogs was generated and evaluated as antagonists of α6β2 nAChRs. Of the analogs tested, several were identified with IC50 values in the low nM or sub-nM range. It was observed that the IC50 values of these reduced bis-, tris-, and tetrakis-analogs were generally within an order of magnitude of the IC50 values of the corresponding parent quaternary ammonium molecule. Also, all the tertiary amino analogs exhibited incomplete inhibition of nicotine-evoked DA release, with Imax values ranging from 58% to 76%. The results suggest that the quaternary ammonium lead compounds and their reduced tertiary amine analogs may be interacting at a common site in the channel of the α6β2* nAChR and that the tertiary amine analogs likely interact at these sites in their protonated forms via similar ionic interactions to those involving the quaternary ammonium salts. From a developmental perspective, these tertiary amino analogs that possess better drug-like properties than the parent quaternary ammonium salts are expected to permeate biological membranes more easily, leading to improved oral and brain bioavailability.
A published review (Dwoskin, Pivavarchyk, et al., 2009) provides additional information on the preclinical development of both bPiDDB and reduced bPiDDB (r-bPiDDB or bis-THP3) as nicotinic receptor-based therapeutics and candidates for smoking cessation. Both bPiDDB and bis-THP3 (Fig. 13.11) are effective in decreasing nicotine self-administration in the rat. The ability of bis-THP3 to decrease nicotine self-administration was retained without any loss of effect following seven repeated daily treatments.
Figure 13.11.

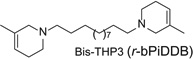
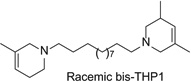
Structures, IC50 and Imax values of the three lead analogs, (±)-1-[12-(3-methyl-1,2,5,6-tetrahydropyridin-1-yl)dodecyl]-3,5-dimethyl-1,2,5,6-tetrahydropyridine (bis-THPI), 1,12-bis(3-methyl-1,2,5,6-tetrahydropyridinyl)dodecane (bis-THP3), and 1,10-bis(3-methyl-1,2,5,6-tetrahydropyridinyl)decane (bis-THP4).
More recent studies have now identified several lead molecules that have a common structural scaffold (i.e., two 1,2,5,6-tetrahydropyridino (THP) moieties connected via a C10 or C12 n-alkane linker) that allows for optimization toward drug-likeness and clinical utility (Table 13.1). This scaffold suggests a likely pharmacophore for the antagonist recognition site on α-CtxMII-sensitive nAChRs mediating nicotine-evoked DA release, bis-THP1 (Fig. 13.11) and bis-THP3 (previously known as r-bPiDDB; Dwoskin, Pivavarchyk, et al., 2009; Smith et al., 2010) potently inhibit nicotine-evoked DA release (IC50 = 0.009–0.058 nM; Imax = 60–74%; Table 13.1 and Fig. 13.11) and inhibit [ H]-nicotine and [3H]-MLAbinding only at µM concentrations; note that bis-THPl is a nonsymmetrical racemic analog and its optical isomers have not yet been evaluated. bis-THP4 (Fig. 13.11), a structurally related analog of bis-THP3 with a C10 linker, exhibits decreased potency (IC50 = 37.4 nM, Imax = 65%) relative to the C12 leads. All the analogs in Table 13.1 are hydrophobic, highly flexible molecules. In terms of drug-likeness, predictive ADMET screening of lead analogs bis-THP1 and bis-THP3 indicates a >75% chance that oral bioavailability will be >30%, with a >20% likelihood that oral bioavailability will be >70%, which is within the acceptable range. However, the LogD7.4 is estimated to be in the range 4.6–5.1 for the leads, which is above the ideal range of 1–3 but is still acceptable. Analogs with a LogD7.4 of 3–5 are predicted to have good cell membrane permeability, but absorption will be lower, due to lower water solubility, and metabolism will increase owing to increased binding to metabolic enzymes.
Table 13.1.
Comparative IC50 values, cLogD7.4, rotatable bonds, and predicted water solubility (Sw) of a series of nonquaternary ammonium, bis-pyridine analogs
| Reduced bis-pyridine Analogs |
Inhibition of NIC-evoked [3DA] release (IC50±SEM, nM) |
cLogD7.4 | Rotatable bonds |
Predicted Sw (mg/ml) |
|---|---|---|---|---|
 |
0.009 ±0.004 | 4.6 | 13 | 0.02 |
 |
0.058 ±0.020 | 5.1 | 13 | 0.02 |
 |
83.5 ±46.8 | 9.9 | 13 | <0.01 |
 |
8.59 ±3.27 | 5.9 | 13 | <0.01 |
 |
3.51 ±1.24 | 7.2 | 13 | 0.05 |
Preliminary results in Dwoskin, Pivavarchyk, et al. (2009), Smith et al. (2010), and Zhang et al. (2010. 2011).
The lead bis-THP analogs potently inhibit (IC50 = 9–58 pM) nicotine-evoked [3H]-DA release at α-CtxMII-sensitive nAChRs (Fig. 13.12, top) (Dwoskin, Pivavarchyk, et al., 2009, unpublished data). Inhibition produced by a maximally effective concentration of bis-THP3 (bPiDDB) was not additive with a maximally effective concentration of α-CtxMII (Fig. 13.12, middle) (Smith et al., 2010), indicating that this lead molecule acts at the same nAChR subtypes as α-CtxMII. Both bis-THPl and bis-THP3 decreased responding for i.v. nicotine self-administration (Fig. 13.12, bottom) in the rat at doses that had no effect on responding for food and at doses that did not produce any overt signs of toxicity (i.e., lethargy and weight loss) (Dwoskin, Pivavarchyk, et al., 2009, unpublished data). While these behavioral data are encouraging, it is not known if either bis-THP3 or bis-THP1 will block cue-induced reinstatement of nicotine seeking or if these analogs will retain behavioral specificity when given orally. Preliminary results for bis-THP3 reveal a brain–plasma ratio = 2 (60 min post-s.c. dose; 58 µmol/kg).
Figure 13.12.

Top: Concentration-dependent inhibition of NIC-evoked [3H]-DA release by bis-THP3 (bPiDDB) and bis-THP1 in rat striatum in vitro (Dwoskin, Pivavarchyk, et al., 2009; Smith et al., 2010). (One-way ANOVAs: bis-THP3, F8, 16 = 33.9, p<0.05; bis-THP1, F7, 27=10.7, p<0.01; n = 9 and 8, respectively.) Center: Inhibition of NIC-evoked [3H]-DA release by maximally effective concentrations of bis-THP3 is not additive with a maximally effective α-CtxMII concentration in vitro (one-way ANOVAs:THP3, F4,15 = 82.1, p<0.001 n = 4. *p<0.05 compared to control; #p<0.05 compared to α-CtxMII + bis-THP3). Bottom: bis-THP3 and bis-THP1 (s.c.) decrease NIC self-administration (i.v.) at doses that do not alter food-maintained responding; n = 4–8 per group. *p<0.05, **p<0.01 compared to control.
6. CONCLUSION
Despite the proven efficacy of some current pharmacotherapies for tobacco dependence, relapse rates continue to be high, indicating that novel medications are needed. The rewarding effects of nicotine are mediated, at least in part, by nicotine-evoked DA release leading to sensitization, which is associated with repeated nicotine administration and nicotine addiction. Based on findings that the nonselective nAChR antagonist MEC has efficacy as a tobacco use cessation agent but is limited by peripherally mediated side effects, we hypothesized that antagonists with selectivity for central nAChR subtypes mediating nicotine-evoked DA release will have efficacy as tobacco use cessation agents with the therapeutic advantage of a limited side-effect profile. While α-CtxMII-insensitive nAChRs (e.g., α4β2* ) contribute to nicotine-evoked DA release, these nAChRs have a wide distribution in brain, which may lead to nonselective effects. In contrast, α-CtxMII-sensitive nAChRs mediating nicotine-evoked DA release offer an advantage as a target for smoking cessation due to their more restricted localization primarily to DA neurons. Our goal is to identify small drug-like molecules that act as selective antagonists at α-CtxMII-sensitive nAChR subtypes that contain α6 and β2 subunits (i.e., α6β2*, α6β2β3*, α6α4β2β3*, and α4α6β2*). Our earlier research identified quaternary ammonium analogs that act as potent and selective nAChR antagonists. The current focus of our research is to enhance the drug-likeness of these nAChR antagonists, while retaining their high potency and selectivity for α6 β2* nAChR subtypes. Our research has shown that novel small molecules can be designed that inhibit (IC50 < 1 nM) nicotine-evoked DA release in vitro by acting as antagonists at α-CtxMII-sensitive nAChR subtypes and by decreasing nicotine self-administration in rats. Thus, our working hypothesis is that novel drug-like bis-THP analogs, which potently and selectively inhibit α-CtxMII-sensitive nAChR subtypes mediating nicotine-evoked DA release, will specifically inhibit nicotine self-administration and/or cue-induced reinstatement of nicotine seeking. Our future research aim is to enhance the drug-likeness of our lead compounds by improving water solubility through introduction of hydrogen-bond acceptor moieties in the linker and by decreasing conformational flexibility toward the goal of identifying orally bioavailable, druglike preclinical leads for the development as smoking cessation and/or relapse prevention.
ACKNOWLEDGMENTS
This research was supported by NIH grant U19DA017548. The University of Kentucky holds patents on the compounds described in the current work.
ABBREVIATIONS
- ANN
artificial neural network
- bNDI
S-(−)-N,N′-decane-1,10-diyl-bis-nicotinium diiodide
- bPiDDB
N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide
- bQDDB
N,N′-dodecane-1,12-diyl-bis-quinolinium dibromide
- BTMPS
bis-(2,2,6,6,-tetramethyl-4-piperidinyl) sebacate
- DA
dopamine
- MEC
mecamylamine
- MLA
methyllycaconitine
- MLR
multiple linear regression
- NAc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- NDDNI
S-(−)-N-n-dodecylnicotinium iodide
- NDNI
S-(−)-N-n-decylnicotinium iodide
- NE
norepinephrine
- NONI
S-(−)-N-n-octylnicotinium iodide
- THP
1,2,5,6-tetrahydropyridine
- TMP
2,2,6,6-tetramethyl-4-piperidinol
- VMAT2
vesicular monoamine transporter-2
- α-CtxMII
α-conotoxin MII
Footnotes
CONFLICT OF INTEREST
A potential royalty stream to PAC and LPD may occur consistent with University of Kentucky policy.
REFERENCES
- Albayati ZAP, Dwoskin LP, Crooks PA. Pharmacokinetics of the novel nicotinic receptor antagonist N, N′-dodecane-l,12-diyl-bis-3-picolinium dibromide in the rat. Drug Metabolism and Disposition. 2008;18:3870–3873. doi: 10.1124/dmd.108.020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Conroy WG, Schoepfer R, Whiting P, Lindstrom J. Neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes have a pentameric quaternary structure. Journal of Biological Chemistry. 1991;266:11192–11198. [PubMed] [Google Scholar]
- Arroyo-Jimenez MM, Bourgeois JP, Marubio LM, Le Sourd AM, Ottersen OP, Rinvik E, et al. Ultrastructural localization of the α4-subunit of the neuronal acetylcholine nicotinic receptor in the rat substantia nigra. Journal of Neuroscience. 1999;19:6475–6487. doi: 10.1523/JNEUROSCI.19-15-06475.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers JT, Dwoskin LP, Deaciuc AG, Grinevich VP, Zhu J, Crooks PA. bis-Azaaromatic quaternary ammonium analogues: Ligands for α4β2 and α7* subtypes of neuronal nicotinic receptors. Bioorganic and Medicinal Chemistry Letters. 2002;12:3067–3071. doi: 10.1016/s0960-894x(02)00687-x. [DOI] [PubMed] [Google Scholar]
- Bacher I, Wu B, Shytle DR, George TP. Mecamylamine—A nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opinion on Pharmacotherapy. 2009;10:2709–2721. doi: 10.1517/14656560903329102. [DOI] [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: Addiction, smoking-induced disease and therapeutics. Annual Review of Pharmacology and Toxicology. 2009;49:57–71. doi: 10.1146/annurev.pharmtox.48.113006.094742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque J, Mendrek A, Dinh-Williams SL, Potvin S. Neural circuitry of impulsivity in a cigarette craving paradigm. Front Psychiatry. 2013;4:1–9. doi: 10.3389/fpsyt.2013.00067. Article 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell DH. Preclinical evidence that activation of mesolimbic alpha 6 subunit containing nicotinic acetylcholine receptors supports nicotine addiction phenotype. Nicotine & Tobacco Research. 2012;14:1258–1269. doi: 10.1093/ntr/nts089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell DH, Boschen KE, Hendrick ES, Beardsley PM, Mcintosh JM. Alpha-conotoxin Mil-sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology. 2010;35:665–673. doi: 10.1038/npp.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachelin AB, Rust G. β-Subunits co-determine the sensitivity of rat neuronal nicotinic receptors to antagonists. Pflugers Archive: European Journal of Physiology. 1995;429:449. doi: 10.1007/BF00374164. [DOI] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, Mcintosh JM. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. Journal of Biological Chemistry. 1996;211:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J. Varenicline: An alpha4beta2 nicotinic receptor partial agonist for smoking cessation. Journal of Medicinal Chemistry. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Cohen C, Bergis OE, Galli F, Lochead AW, Jegham S, Biton B, et al. SSR591813, a novel selective and partial alpha4beta2 nicotinic receptor agonist with potential as an aid to smoking cessation. Journal of Pharmacology and Experimental Therapeutics. 2003;306:407–420. doi: 10.1124/jpet.103.049262. [DOI] [PubMed] [Google Scholar]
- Collins AC, Salminen O, Marks MJ, Whiteaker P, Grady SR. Nicotine psychophar- macology. Handbook of experimental pharmacology. Vol. 192. New York, NY: Springer; 2009. The road to discovery of neuronal nicotinic cholinergic receptor subtypes; pp. 85–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy WG, Vernallis AB, Berg DK. The α5 gene product assembles with multiple acetylcholine receptor subunits to form distinctive receptor subtypes in brain. Neuron. 1992;9:679–691. doi: 10.1016/0896-6273(92)90031-8. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Franklin KBJ, Coen KM, Clarke PBS. The mesolimbic dopaminergic system is implicated in the reinforcing properties of nicotine. Psychophar- macology. 1992;107:285–289. doi: 10.1007/BF02245149. [DOI] [PubMed] [Google Scholar]
- Crooks PA, Ayers JT, Rui X, Sumithran SP, Grinevich VP, Wilkins LW, et al. Development of subtype-selective nicotinic receptor ligands as receptor antagonists. Bioorganic and Medicinal Chemistry Letters. 2004;14:1869–1874. doi: 10.1016/j.bmcl.2003.10.074. [DOI] [PubMed] [Google Scholar]
- Crooks PA, Ravard A, Teng LH, Dwoskin LP. Inhibition of nicotine-evoked [3H] dopamine release by pyridine N-substituted nicotine analogues: A new class of nicotinic antagonist. Drug Development Research. 1995;36:71–82. [Google Scholar]
- Cui C, Booker TK, AUen RS, Grady SR, Whiteaker P, Marks MJ, et al. The β3 nicotinic receptor subunit: A component of α-conotoxin Mil-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. Journal of Neuroscience. 2003;23:11045–11053. doi: 10.1523/JNEUROSCI.23-35-11045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annual Review of Pharmacology and Toxicology. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- DeNoble VJ, Mele PC. Intravenous nicotine self-administration in rats: Effects of mecamylamine, hexamethonium and naloxone. Psycho-pharmacology. 2006;184:266–272. doi: 10.1007/s00213-005-0054-z. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, Mcintosh JM, et al. Cholinergic modulation of locomotion and striatal dopamine release is mediated by α6α4* nicotinic acetylcholine receptors. Journal of Neuroscience. 2010;30:9877–9889. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized high affinity α6* nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled α6 and β3 sub-units. Molecular Pharmacology. 2008;13:27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Bardo MT. Targeting nicotinic receptor antagonists as novel pharmacotherapies for tobacco dependence and relapse. Neuropsychopharmacology. 2009;34:244–246. doi: 10.1038/npp.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwoskin LP, Crooks PA. Competitive neuronal nicotinic receptor antagonists: A new direction for drug discovery. Journal of Pharmacology and Experimental Therapeutics. 2001;298:395–402. [PubMed] [Google Scholar]
- Dwoskin LP, Leibee LL, Jewell AL, Pang Z-X, Crooks PA. Inhibition of [3H]-dopamine uptake into rat striatal slices by quaternary N-methylated nicotine metabolites. Life Sciences. 1992;50:PL233–PL237. doi: 10.1016/0024-3205(92)90533-u. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Pivavarchyk M, Joyce BM, Neugebauer NM, Zheng G, Zhang Z, et al. Targeting reward-relevant nicotinic receptors in the discovery of novel pharmacotherapeutic agents to treat tobacco dependence. In: Bevins RA, Caggiula AR, editors. 55th annual Nebraska symposium on motivation: The motivational impact of nicotine and its role in tobacco use. Vol. 55. New York, NY: Springer; 2009. pp. 31–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwoskin LP, Smith AM, Wooters TE, Zhang Z, Crooks PA, Bardo MT. Nicotinic receptor-based therapeutics and candidates for smoking cessation. Biochemical Pharmacology. 2009;78:732–743. doi: 10.1016/j.bcp.2009.06.002. NIHMS123315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, Crooks PA. Subtype-selective nicotinic receptor antagonists: Potential as tobacco use cessation agents. Bioorganic and Medicinal Chemistry Letters. 2004;14:1863–1867. doi: 10.1016/j.bmcl.2003.10.073. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Wilkins LH, Pauly J, Crooks PA. Development of a novel class of subtype-selective nicotinic receptor antagonist: Pyridine-N-substituted nicotine analogs. In: Rudy B, Seeburg P, editors. Molecular and junctional diversity of ion channels and receptors. Annals of the New York academy of sciences. Vol. 868. New York, NY: Wiley Dwoskin; 1999. (Special Issue) [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Wooters TE, Sumithran SP, Siripurapu KB, Joyce BM, Lockman PR, et al. N, N′-Alkane-diyl-bis-3-picoliniums as nicotinic receptor antagonists: Inhibition of nicotine-induced dopamine release and hyperactivity. Journal of Pharmacology and Experimental Therapeutics. 2008;326:563–576. doi: 10.1124/jpet.108.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter J-F. Cytisine for smoking cessation: A literature review and a meta-analysis. Archives of Internal Medicine. 2006;166:1553–1559. doi: 10.1001/archinte.166.15.1553. [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, Mcintosh MJ, Cragg SJ. α6 Containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychophartnacology. 2008;3:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- Fagerstrom K, Balfour DJ. Neuropharmacology and potential efficacy of new treatments for tobacco dependence. Expert Opinion on Investigational Drugs. 2006;15:107–116. doi: 10.1517/13543784.15.2.107. [DOI] [PubMed] [Google Scholar]
- FDA Drug Safety Communication. Chantix (varenidine) may increase the risk of certain cardiovascular adverse events inpatients with cardiovascular disease. 2011 http://www.fda.gov/Drugs/DrugSafety/ucm259161.htm.
- FDA Public Health Advisory. Public health advisory on Chantix. 2008 http://www.fda.gov/CDER/Drug/early_comm/varenicline.htm.
- FDA Public Health Advisory. FDA requires new boxed warnings for the smoking cessation drugs Chantix and Zyban. 2009 http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHealthcareProfessionals/PublicHealthAdvisories/ucm169988.htm.
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, KeUar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha4 and beta2 subunits and is up-regulated by chronic nicotine treatment. Molecular Pharmacology. 1992;41:31–37. [PubMed] [Google Scholar]
- Forsayeth JR, Kobrin E. Formation of oligomers containing the β3 and β4 sub-units of the rat nicotinic receptor. Journal of Neuroscience. 1997;17:1531–1538. doi: 10.1523/JNEUROSCI.17-05-01531.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Lukas RJ. Noncompetitive functional inhibition at diverse, human nicotinic acetylcholine receptor subtypes by bupropion, phencyclidine, and ibogaine. Journal of Pharmacology and Experimental Therapeutics. 1999;288:88–92. [PubMed] [Google Scholar]
- Garwood CL, Potts LA. Emerging pharmacotherapies for smoking cessation. American Journal of Health-System Pharmacy. 2007;15:1693–1698. doi: 10.2146/ajhp060427. [DOI] [PubMed] [Google Scholar]
- George TP, O’Malley SS. Current pharmacological treatments for nicotine dependence. Trends in Pharmacological Sciences. 2004;25:42–48. doi: 10.1016/j.tips.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Click SD, Visker KE, Maisonneuve JM. An oral self-administration model of nicotine preference in rats: Effects of mecamylamine. Psychopharmacology. 1996;128:426–431. doi: 10.1007/s002130050153. [DOI] [PubMed] [Google Scholar]
- Goldner FM, Dineley KT, Patrick JW. Immunohistochemical localization of the nicotinic acetylcholine receptor subunit α6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. Neuroreport. 1997;8:2739–2742. doi: 10.1097/00001756-199708180-00019. [DOI] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbiolo S, Zanetti L, Moretti M, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: Primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. Journal of Neuroscience. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Clementi F, Rganti F, Mcintosh JM, Collins AC, et al. Expression of nigrostriatal α6-containing nicotinic acetylcholine receptors is selectively reduced, but not eliminated, by β3 subunit gene deletion. Molecular Pharmacology. 2005;61:2007–2015. doi: 10.1124/mol.105.011940. [DOI] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends in Pharmacological Sciences. 2006;21:482–A91. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Grady SR, Meinerz NM, Cao J, Reynolds AM, Picciotto MR, Changeux J-P, et al. Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: A function mediated by a different nAChR than dopamine release from striatum. Journal of Neurochemistry. 2001;16:258–268. doi: 10.1046/j.1471-4159.2001.00019.x. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, Mcintosh JM, Collins AC. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochemical Pharmacology. 2007;14:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek P, McRobbie H, Myers K. Efficacy of cytisine in helping smokers quit: Systematic review and meta-analysis. Thorax. 2013 doi: 10.1136/thoraxjnl-2012-203035. http://dx.doi.org/10.1136/thoraxjnl-2012-203035. [DOI] [PubMed]
- Hajek P, Stead LF, West R, Jarvis M, Hartmann-Boyce J, Lancaster T. Relapse prevention interventions for smoking cessation. Cochrane Database of Systematic Reviews. 2013;(8) doi: 10.1002/14651858.CD003999.pub4. http://dx.doi.org/10.1002/1465188458, Art. No. CD003999. [DOI] [PubMed]
- Harmey D, Griffin PR, Kenny PJ. Development of novel pharmacotherapeutics for tobacco dependence: Progress and future directions. Nicotine & Tobacco Research. 2012;14:1300–1318. doi: 10.1093/ntr/nts201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Luetje CW. Determinants of competitive antagonist sensitivity on neuronal nicotinic receptor beta subunits. Journal of Neuroscience. 1996;16:3798–3806. doi: 10.1523/JNEUROSCI.16-12-03798.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Maddox FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. Journal of Neurochemistry. 1996;61:1953–1959. doi: 10.1046/j.1471-4159.1996.67051953.x. [DOI] [PubMed] [Google Scholar]
- Hurt RD, Krook JE, Croghan IT, Loprinzi CL, Sloan JA, Novotny PJ, et al. Nicotine patch therapy based on smoking rate followed by bupropion for prevention of relapse to smoking. Journal of Clinical Oncology. 2003;21:914–920. doi: 10.1200/JCO.2003.08.160. [DOI] [PubMed] [Google Scholar]
- Irvin JE, Hendricks PS, Brandon TH. The increasing recalcitrance of smokers in clinical trials II: Pharmacotherapy trials. Nicotine and Tobacco Research. 2003;5:27–35. doi: 10.1080/1462220031000070534. [DOI] [PubMed] [Google Scholar]
- Jackson KJ, Mcintosh JM, BrunzeU DH, Sanjakdar SS, Damaj MI. The role of α6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. Journal of Pharmacology and Experimental Therapeutics. 2009;331:547–554. doi: 10.1124/jpet.109.155457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TB, Hogankamp DJ, Coyne L, Su J, HaUiweU RF, Tran MB, et al. Modifying quinolone antibiotics yields new anxiolytics. Nature Medicine. 2004;10:31–32. doi: 10.1038/nm967. [DOI] [PubMed] [Google Scholar]
- Jorenby DE, Hays JT, Rigotti NA, Azoulay S, Watsky EJ, Williams KE, et al. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: A randomized controlled trial . JAMA. 2006;296:56–63. doi: 10.1001/jama.296.1.56. [DOI] [PubMed] [Google Scholar]
- Kaiser SA, Soliakov L, Harvey SC, Luetje CW, Wonnacott S. Differential inhibition by α-conotoxin-MII of the nicotinic stimulation of [3H] dopamine release from rat striatal synaptosomes and slices. Journal of Neurochemistry. 1998;10:1069–1076. doi: 10.1046/j.1471-4159.1998.70031069.x. [DOI] [PubMed] [Google Scholar]
- Koelle GB. Neuromuscular blocking agents. In: Goodman LS, Gilman A, editors. The pharmacological basis of therapeutic. New York, NY: MacMillan Publishing Co; 1975. pp. 575–588. [Google Scholar]
- Kulak JM, Nguyen TA, Olivera BM, Mcintosh JM. α-Conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. Journal of Neuroscience. 1997;11:5263–5270. doi: 10.1523/JNEUROSCI.17-14-05263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Lindstrom J. Expression of functional human α6β2β3 acetylcholine receptors in Xenopus laevis oocytes achieved through subunit chimeras and con-catamers. Molecular Pharmacology. 2011;19:126–140. doi: 10.1124/mol.110.066159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Control of the reinforcing effects of nicotine by associated environmental stimuli in animals and humans. TRENDS in Pharmacological Sciences. 2005;26:287–293. doi: 10.1016/j.tips.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Handbook of experimental pharmacology. Vol. 192. New York, NY: Springer; 2009. Effects of nicotine in experimental animals and humans: An update on addictive properties; pp. 335–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. European Journal of Neuroscience. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- Lockman PR, Geldenhuys WJ, Manda V, Thomas F, Crooks PA, Dwoskin LP, et al. Carrier mediated transport at the blood-brain barrier for the quaternary ammonium nicotinic receptor antagonist, N, N-dodecyl-bis-picolinium bromide (bPiDDB) Journal of Pharmacology and Experimental Therapeutics. 2008;324:244–250. doi: 10.1124/jpet.107.130906. [DOI] [PubMed] [Google Scholar]
- Lukas RJ. Pharmacological effects of nicotine and nicotinic receptor subtype pharmacological profiles. Boca Raton, PL: CRC Press LLC; 2007. [Google Scholar]
- Lundahl LH, Henningfield JE, Lukas SE. Mecamylamine blockade of both positive and negative effects of IV nicotine in human volunteers. Pharmacology, Biochemistry, and Behavior. 2000;66:637–643. doi: 10.1016/s0091-3057(00)00252-5. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Collins AC, Paylor R, Marks MJ. Deletion of the beta2 nicotinic acetylcholine receptor subunit alters development of tolerance to nicotine and eliminates receptor upregulation. Psychopharmacology. 2006;184:314–327. doi: 10.1007/s00213-005-0076-6. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annual Review of Physiology. 1995;51:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- Mcintosh JM, Plazas PV, Watkins M, Gomez-Casati ME, Olivera BM, Elgoyhen AB. A novel alpha-conotoxin, PelA, cloned from Conus pergrandis, discriminates between rat alpha9alphal0 and alpha7 nicotinic cholinergic receptors. Journal of Biological Chemistry. 2005;280:30107–30112. doi: 10.1074/jbc.M504102200. [DOI] [PubMed] [Google Scholar]
- Mihalak KB, Carroll PI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Molecular Pharmacology. 2006;10:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- Millar NS, Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology. 2009;70:801–805. doi: 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Mills EJ, Wu P, Spurden D, Ebbert JO, Wilson K. Efficacy of pharmacotherapies for short-term smoking abstinence: A systematic review and meta-analysis. Harm Reduction Journal. 2009;6:25. doi: 10.1186/1477-7517-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Somenzi O, Picciotto MR. Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6 J mice. Neuropharmacology. 2007;52:1256–1262. doi: 10.1016/j.neuropharm.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I, et al. A comparative study of the effects of the intravenous self-administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Molecular Pharmacology. 2010;18:287–296. doi: 10.1124/mol.110.064071. [DOI] [PubMed] [Google Scholar]
- Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. Effect of a novel nicotinic receptor antagonist, N, N’-dodecane-l,12-diyl-bis-3-picolinium dibromide (bPiDDB), on nicotine self-administration and hyperactivity in rats. Psychopharmacology. 2006;184:426–434. doi: 10.1007/s00213-005-0163-8. [DOI] [PubMed] [Google Scholar]
- Papke RL, Wecker L, Stitzel JA. Activation and inhibition of mouse muscle and neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. Journal of Pharmacology and Experimental Therapeutics. 2010;333:501–518. doi: 10.1124/jpet.109.164566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Min W, Hackett A, Lowe D, Hanania T, Caldarone B, et al. The high-affinity nAChR partial agonists varenicline and sazetidine-A exhibit reinforcing properties in rats. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2010;34:1455–1464. doi: 10.1016/j.pnpbp.2010.07.037. [DOI] [PubMed] [Google Scholar]
- Perkins KA. Nicotine self-administration. Nicotine and Tobacco Research. 1999;2:133–137. doi: 10.1080/14622299050011951. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harbor Perspectives in Medicine. 2013:3. doi: 10.1101/cshperspect.a012112. http://dx.doi.org/10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed]
- Pons S, Pattore L, Cossu G, Tolu S, Porcu E, Mcintosh JM, et al. Crucial role of α4 and α6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. Journal of Neuroscience. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarta D, Ciruela F, Patkar K, Borycz J, Solinas M, Lluis C, et al. Heteromeric nicotinic acetylcholine-dopamine autoreceptor complexes modulate striatal dopamine release. Neuropsychopharmacology. 2007;32:35–42. doi: 10.1038/sj.npp.1301103. [DOI] [PubMed] [Google Scholar]
- Rahman S, Zhang Z, Papke RL, Crooks PA, Dwoskin LP, &Bardo MT. Region-specific effects of the novel nicotinic receptor antagonist N, N’-dodecane-l,12-diyl-bis-3-picolinium bromide (bPiDDB) on the nicotine-induced increase in extracellular dopamine: An in vivo reverse microdialysis study. British Journal of Pharmacology. 2008;153:792–804. doi: 10.1038/sj.bjp.0707612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau KS, Birdsall E, Hanson JE, Johnson-Davis KL, Carroll PI, Wilkins DG, et al. Bupropion increases striatal vesicular monoamine transport. Neuropharmacology. 2005;49:820–830. doi: 10.1016/j.neuropharm.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Rose JE. Nicotine and non-nicotine factors in cigarette addiction. Psycho-pharmacology. 2006;184:274–285. doi: 10.1007/s00213-005-0250-x. [DOI] [PubMed] [Google Scholar]
- Rose JE. Disrupting nicotine reinforcement from cigarette to brain. Annals of the New York Academy of Sciences. 2008;1141:233–256. doi: 10.1196/annals.1441.019. [DOI] [PubMed] [Google Scholar]
- Rose JE. New findings on addiction and treatment. In: Bevins RA, Caggiula AR, editors. 55th annual Nebraska symposium on motivation: The motivational impact of nicotine and its role in tobacco use. Vol. 55. New York, NY: Springer; 2009. pp. 230–242. [Google Scholar]
- Rose JE, Behm FM, Westman EC, Levin ED, Stein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clinical Pharmacology and Therapeutics. 1994;56:86–99. doi: 10.1038/clpt.1994.105. [DOI] [PubMed] [Google Scholar]
- Rose JE, Salley A, Behm PM, Bates JE, Westman EC. Reinforcing effects of nicotine and non-nicotine components of cigarette smoke. Psychopharmacology. 2010;1:1–12. doi: 10.1007/s00213-010-1810-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, Mcintosh JM, Collins AC, Marks MJ, Grady SR. Pharmacology of α-conotoxin Mil-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Molecular Pharmacology. 2007;11:1563–1571. doi: 10.1124/mol.106.031492. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, Mcintosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Molecular Pharmacology. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Sanofi Pipeline. http://en.sanofi-aventis.com/binaries/080212_PDF_Slides_media_tcm28-15767.pdf.
- Schuh LM, Schuh KJ, Henningfield JE. Pharmacologic determinants of tobacco dependence. American fournal of Therapy. 1996;3:335–341. doi: 10.1097/00045391-199605000-00002. [DOI] [PubMed] [Google Scholar]
- Shahan TA, Odum AL, Bickel WK. Nicotine gum as a substitute for cigarettes: A behavioral economic analysis. Behavioral Pharmacology. 2000;11:71–79. doi: 10.1097/00008877-200002000-00008. [DOI] [PubMed] [Google Scholar]
- Slemmer JE, Martin BR, Damaj MI. Bupropion is a nicotinic antagonist. Journal of Pharmacology and Experimental Therapeutics. 2000;295:321–327. [PubMed] [Google Scholar]
- Smith AM, Dhawan GK, Zhang Z, Siripurapu KB, Crooks PA, Dwoskin LP. The novel nicotinic receptor antagonist, N, N′-dodecane-l,12-diyl-bis-3-picolinium dibromide (bPiDDB), inhibits nicotine-evoked [3H]norepinephrine overflow from rat hippocampal slices. Biochemical Pharmacology. 2009;78:889–897. doi: 10.1016/j.bcp.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Pivavarchyk M, Wooters TE, Zhang Z, Mcintosh JM, Crooks PA, et al. Repeated nicotine increases bPiDDB potency to inhibit nicotine-evoked DA release from rat striatum. Biochemical Pharmacology. 2010;80:402–409. doi: 10.1016/j.bcp.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AK, Markou A. Unraveling the neurobiology of nicotine dependence using genetically engineered mice. Current Opinion in Neurobiology. 2013;23:493–499. doi: 10.1016/j.conb.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KH, Martin RM, Davies NM, Metcalfe C, Windmeijer P, Gunnel D. Smoking cessation treatment and risk of depression, suicide, and self harm in the Clinical Practice Research Datalink prospective cohort study. British Journal of Medicine. 2013;347:f5704. doi: 10.1136/bmj.f5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan TV, Zhou W, et al. AT-1001: A high affinity and selective α3β4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychophartnacology. 2012;37:1367–1376. doi: 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Kellar KJ. Nicotinic cholinergic receptors in the rat cerebellum: Multiple heteromeric subtypes. Journal of Neuroscience. 2005;25:9258–9265. doi: 10.1523/JNEUROSCI.2112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen JW, Cohen LM, Watson NL. Utilizing a multimodal assessment strategy to examine variations of impulsivity among young adults engaged in co-occurring smoking and binge drinking behaviors. Drug and Alcohol Dependence. 2012;127:150–155. doi: 10.1016/j.drugalcdep.2012.06.026. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha2, alpha3, alpha4, and beta2 neuronal nicotinic receptor subunit mPvNAs in the central nervous system: A hybridization histochemical study in the rat. Journal of Comparative Neurology. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Marks MJ, Grady SR, Lu Y, Picciotto MR, Changeux JP, et al. Pharmacological and null mutation approaches reveal nicotinic receptor diversity. European Journal of Pharmacology. 2000;393(1–3):123–135. doi: 10.1016/s0014-2999(00)00052-2. [DOI] [PubMed] [Google Scholar]
- Whiting P, Lindstrom J. Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:595–599. doi: 10.1073/pnas.84.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileyto P, Patterson F, Niaura R, Epstein L, Brown R, Audrain-McGovern J, et al. Do small lapses predict relapse to smoking behavior under bupropion treatment? Nicotine and Tobacco Research. 2004;6:357–366. doi: 10.1080/1462220042000202463. [DOI] [PubMed] [Google Scholar]
- Wooters TE, Smith AM, Pivavarchyk M, Siripurapu KB, Mcintosh JM, Zhang Z, et al. bPiDI: A novel selective α6β2 nicotinic receptor antagonist and preclinical candidate treatment for nicotine abuse. British Journal of Pharmacology. 2011;163:346–357. doi: 10.1111/j.1476-5381.2011.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Wilson K, Dimoulas P, Mills EJ. Effectiveness of smoking cessation therapies: A systematic review and meta-analysis. BMC Public Health. 2006;6:300. doi: 10.1186/1471-2458-6-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, et al. Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Molecular Pharmacology. 2006;70:1454–1460. doi: 10.1124/mol.106.027318. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Woolverton WL, Sahibzada N, Yasuda RP, Kellar KJ. Pharmacological properties of sazetidine-A, a desensitizer of alpha4beta2 nicotinic acetylcholine receptors. Society for NeuroscienceAbstracts. 2007;33:574.6.. [Google Scholar]
- Yoshimura RP, Hogenkamp DJ, Li WY, Tran MB, Belluzzi JD, Whittemore ER, et al. Negative allosteric modulation of nicotinic acetylcholine receptors blocks nicotine self-administration in rats . Journal of Pharmacology and Experimental Therapeutics. 2007;323:907–915. doi: 10.1124/jpet.107.128751. [DOI] [PubMed] [Google Scholar]
- Zatonski W, Cedzynska M, Tutka P, West R. An uncontrolled trial of cytisine (Tabex) for smoking cessation. Tobacco Control. 2006;15:481–484. doi: 10.1136/tc.2006.016097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lockman PR, Mittapalli RK, Allen DD, Dwoskin LP, Crooks PA. bis-Pyridinium cyclophanes: Novel ligands with high affinity for the blood-brain barrier choline transporter. Bioorganic and Medicinal Chemistry Letters. 2008;18:5622–5625. doi: 10.1016/j.bmcl.2008.08.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pivavarchyk M, Subramanian KL, Deaciuc AG, Dwoskin LP, Crooks PA. Novel bis-2,2,6,6-tetramethylpiperidine (bis-TMP) and bis-mecamylamine antagonists at neuronal nicotinic receptors mediating nicotine-evoked dopamine release. Bioorganic and Medicinal Chemistry Letters. 2010;20:1420–1423. doi: 10.1016/j.bmcl.2009.12.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zheng G, Pivavarchyk M, Deaciuc AG, Dwoskin LP, Crooks PA. tetrakis-Azaaromatic quaternary ammonium: Selective neuronal nicotinic receptor antagonists at subtypes that mediate nicotine-evoked dopamine release. Bioorganic and Medicinal Chemistry Letters. 2008;18:5753–5757. doi: 10.1016/j.bmcl.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zheng G, Pivavarchyk M, Deaciuc AG, Dwoskin LP, Crooks PA. Novel bis-, tris-, and tetrakis tertiary amino analogs as antagonists at neuronal nicotinic receptors that mediate nicotine-evoked dopamine release. Bioorganic and Medicinal Chemistry Letters. 2011;21:88–91. doi: 10.1016/j.bmcl.2010.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Bayram E, Sumithran SP, Ayers JT, Zhan CG, Schmitt JD, et al. QSAR modeling of mono- and bis-quaternary ammonium salts that act as antagonists at neuronal nicotinic acetylcholine receptors mediating dopamine release. Bioorganic and Medicinal Chemistry. 2006;14:3017–3037. doi: 10.1016/j.bmc.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Zheng F, McConell M, Zhan C-G, Dwoskin LP, Crooks PA. QSAR study on maximal inhibition (I max) of quaternary ammonium antagonists for S-(−)-nicotine-evoked dopamine release from dopaminergic nerve terminals in rat striatum. Bioorganic and Medicinal Chemistry. 2009;11:AA77–AA4485. doi: 10.1016/j.bmc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Sumithran SP, Deaciuc AG, Dwoskin LP, Crooks PA. tris-Azaaromatic quaternary ammonium salts: Novel templates as antagonists at nicotinic receptors mediating nicotine-evoked dopamine release. Bioorganic and Medicinal Chemistry tetters. 2007;11:6701–6706. doi: 10.1016/j.bmcl.2007.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Zhang Z, Lockman PPR, Geldenhuys WJ, Allen DD, Dwoskin LP, et al. bis-Azaaromatic quaternary ammonium salts as ligands for the blood-brain barrier choline transporter. Bioorganic and Medicinal Chemistry Letters. 2010;20:3208–3210. doi: 10.1016/j.bmcl.2010.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Zhang Z, Pivarvarchyk M, Deaciuc AG, Dwoskin LP, Crooks PA. bis-Azaaromatic quaternary ammonium salts as antagonists at nicotinic receptors mediating nicotine-evoked dopamine release: An investigation of binding conformation. Bioorganic and Medicinal Chemistry Letters. 2007;11:6734–6738. doi: 10.1016/j.bmcl.2007.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Zheng G, Deaciuc AG, Zhan C, Dwoskin LP, Crooks PA. Computational neural network analysis of the affinity of N-n-alkylnicotinium salts for the α4β2 nicotinic acetylcholine receptor. Journal of Enzyme Inhibition and Medicinal Chemistry. 2009;24:157–168. doi: 10.1080/14756360801945648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Lena C, Picciotto MR, Changeux JP. Identification of four classes of brain nicotinic receptors using beta2 mutant mice. Journal of Neuroscience. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, et al. Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Molecular Pharmacology. 2008;13:1834–1843. doi: 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]