

Abstract
Objectives
The LPA SNP rs10455872 has been associated with low density lipoprotein cholesterol (LDLc) lowering response to statins in several randomised control trials (RCTs) and is a known coronary artery disease (CAD) marker. However it is unclear what residual risk of CAD this marker may have during statin treatment.
Methods
Using electronic medical records linked to the GoDARTS genotyped population we identified over 8000 patients on statins in Tayside, Scotland.
Results
We replicated the findings of the RCTs, with the G allele of rs10455872 being associated with a 0.10mmol/l per allele poorer reduction in LDLc in response to statin treatment, and conducted a meta-analysis with previously published RCTs (P=1.46×10−29, n=30,467). We demonstrate an association between rs10455872 and CAD in statin treated individuals and have replicated this finding in the Utrecht Cardiovascular Pharmacogenetics Study (combined odds ratio 1·41, 95% CI 1.17-1.68, P=4.5×10−5, n=8822) suggesting that statin treatment does not abrogate this well-established genetic risk for CAD. Furthermore in a Cox proportional hazards model with LDLc measured time dependently we demonstrated the relationship between CAD and rs10455872 was independent of LDLc during statin treatment.
Conclusion
Individuals with the G allele of rs10455872, which represents approximately 1 in 7 patients, have a higher risk of CAD than the majority of the population even after treatment with statins, and therefore represent a vulnerable group requiring an alternative medication in addition to statin treatment.
Keywords: Lipoproteins, drug response, pharmacogenetics, HMG CoA Reductase inhibitors, coronary artery disease
Introduction
Statins act by lowering plasma cholesterol and are the most commonly used drugs for the prevention of coronary artery disease (CAD)1. There is broad inter-individual variability in low density lipoprotein cholesterol (LDLc) lowering response to statins2, and variants in APOE are the most robustly established genetic determinants of this3-6. Recent studies have also demonstrated a role for the LPA locus. The JUPITER study7, the combined CARDS and ASCOT studies with replication from the PROSPER study8 and the Heart Protection Study (HPS)9 have all demonstrated reduced LDLc lowering by statins with rs10455872, a known marker of LP(a) levels10.
The Global Lipid Consortium (GLC)11 demonstrated a genome wide association of rs10455872 with serum LDLc levels, and interestingly it is also a well-established marker of CAD risk in large populations where the use of statins is not documented10, 12. The HPS investigated the association of this variant with vascular risk response to statins, and concluded that although carriers were at a higher absolute risk of events than non-carriers, there was no statistically significant difference in absolute or relative risk reduction with statin therapy by genotype. The CARDS study also inferred this finding though it was underpowered in this respect. These findings suggest that statin treatment does not abrogate the CAD risk associated with this variant. However neither of these studies provided a direct estimate of the risk conferred by the LPA genotype in either the statin treated or the non-statin treated arms and therefore it is unclear what residual risk of CAD this marker may have during statin treatment.
The aims of the present study were; 1. To replicate the findings of the GLC regarding rs10455872 and LDLc levels; 2. To replicate the findings of the RCTs regarding rs10455872 and LDLc lowering efficacy of statins in our observational study; 3. To assess the role of rs10455872 in determining CAD outcomes in statin treated individuals; 4. To assess the relationship between LDLc response to statins and the risk of CAD associated with rs10455872.
Methods
We performed an observational study using data from the Genetics of Diabetes Audit and Research (GoDARTS) database, which has been described previously13, 14. This includes detailed clinical information on ~15,000 patients with and without diabetes who have provided consent to link their genetic information with their healthcare record in Tayside Scotland from 1990 to present, including demographic data, all prescriptions dispensed from Tayside pharmacies, all biochemistry data from the region-wide clinical laboratory, Scottish Morbidity Records (SMR), detailing International Classification of Disease (ICD) coding for hospital admissions and data from the General Registrars Office detailing date and cause of death. This continuously accruing data is deterministically linked to genetic information through a patient-unique healthcare identifier number known as the Community Health Index (CHI) which is used for all healthcare related activity in the region. The biochemistry laboratories in Tayside use the Friedewald equation to calculate LDLc which is not directly measured therefore all references to measured LDLc in this study are calculated values.15 This study was approved by the Tayside Regional Ethics Committee.
Genotyping
Direct typing of rs10455872 was performed using Taqman allelic discrimination assays as supplied by Applied Biosystems (Carlsbad, CA). All typing was performed in 384 well format using 10-20ng of DNA in 2ul reaction volumes using Universal Taqman mastermix (Applied Biosystems, Carlsbad, CA). Assays were plated using a DEERAC Equator GX microdispenser (Labcyte, Sunnyvale, CA), and thermal cycling was performed in a H2OBIT high throughput thermal cycler (KBiosystems, Basildon, Essex). End point fluorescence was measured and genotypes were called using an ABI 7900HT sequence detection system (Applied Biosystems, Carlsbad, CA).
Study Population
All patients taking part in GoDARTS who were resident in Tayside during the study period 1st January 1990 to 1st March 2011, genotyped for rs10455872, were initially considered for the study. There are four analyses presented with each comprising a different phenotype definition and study population (detailed in statistical methods and figure 1).
Figure 1.

Flow chart of study populations: this shows the study criteria and the number of patients who were eligible for each model
Definition of study period
Duration of statin therapy was defined as the period between first and last statin prescription. However, if patients were on a lipid-regulating drug other than statins (defined as all other drugs in BNF chapter 2.12 16) at first prescription and this drug was subsequently stopped then the study was censored at this point, or if a lipid-regulating drug was initiated during statin exposure then the study was also censored at this point.
Statistical methods
1. Association of rs10455872 with LDLc levels in the absence of lipid-regulating drug treatment was analysed using a linear regression model with the outcome measure defined as the mean of all such available LDLc measures.
2. Association of rs10455872 with LDLc lowering response to statins was analysed using a linear regression model. The outcome was on-treatment LDLc adjusted for pre-treatment LDLc. Where pre-treatment LDLc was defined as closest measure before statin initiation and on-treatment LDLc was defined as the lowest LDLc achieved whilst on statins. This model has been previously published 6, 17, 18 and is mathematically equivalent to modelling change in LDLc (i.e., the difference in on-treatment and pre-treatment LDL) adjusted for pre-treatment LDLc.
.As approximately 37% of our study population had no pre-treatment statin measure available, we imputed missing values using the multiple imputation procedure (PROC MI) in SAS (further details are provided in supplementary material). To assess the impact of the imputation on our results we performed a sensitivity analysis excluding patients with imputed measures.
3. Association of rs10455872 with CAD risk on statins was analysed as a case-control study using unconditional logistic regression. A case was defined as a CAD hospitalisation or death event (myocardial infarction (MI), unstable angina or coronary-artery revascularisation) after statin initiation with any of the following codes: ICD10: I21, I20.0; OPCS-3:3043, 8841; OPCS-4: K75.9, K49.1, K49.2, K49.3, K49.8, K49.9, K50.2, K50.3,K50.4, K50.8, K40.1, K40.2, K40.3, K41.1, K41.3,K43.3, K43.9,K44.1, K44.2,K44.9. The rest of the eligible study population were defined as controls.
4. Association of rs10455872 with CAD risk on statins with adjustment for LDLc was analysed using Cox’s proportional hazards regression. Patients were modelled from statin initiation to first CAD event (as defined in model 3) or censor (defined as earliest of date of death, end of statin therapy or 1st March 2011). To assess the impact of LDLc in the model all available on-treatment measures were modelled time dependently used the counting process format in SAS.
Covariates
All models were adjusted for age, sex and diabetes status. Due to the observational nature of the study and based on clinical judgment, models 2, 3 and 4 included additional covariates. These differed by model but included pre-treatment LDLc, statin dose (calculated as the maximum dose prescribed during the study period, and where other statins were prescribed expressed as the Simvastatin equivalent dose (SED)19), statin adherence (our adherence calculation has been previously validated20, and additional information is provided in the supplementary material), study duration, binary variables indicating a prior CAD event, whether patient was on additional lipid-regulating drug therapy, history of smoking and whether pre-treatment LDLc was imputed. The results for the full models are provided in supplementary tables to show the individual covariate effects.
We performed two meta-analyses:
1. Association of rs10455872 with LDLc lowering response to statins
We combined our result with the CARDS/ASCOT/PROSPER meta-analysis, JUPITER and HPS studies. The effect size in CARDS/ASCOT/PROSPER was scaled so that the residuals had unit variance, thereby allowing studies using different transformations to be combined. We therefore similarly scaled the GoDARTS, JUPITER and HPS studies. The response definition differed by study with CARDS/ASCOT/PROSPER using a similar model to ours, and JUPITER and HPS using absolute reduction (i.e. no adjustment for pre-treatment LDLc).
2. Association of rs10455872 with CAD risk on statins
We aimed to replicate our result (model 3) in statin treated individuals from the Utrecht Cardiovascular Pharmacogenetics (UCP) Study (details of the cohort are provided in the supplementary material)21.
Rs10455872 genotype was analysed as an additive genetic model. Meta-analyses were performed in PLINK (v 1.07) with the results of the fixed-effects model presented. All other statistical analyses were performed in SAS (v 9.2). All p-values are two-sided, with P<0.05 considered statistically significant.
Results
A flowchart of the sub-populations used in the analyses is provided in figure 1. Of the 14,635 individuals with DNA available for genotyping, 13,839 were successfully genotyped for rs10455872 giving a call rate of 95%. The minor allele frequency was 0.07 and the SNP was in HWE (P>0.05).
Association of rs10455872 with LDLc levels in the absence of lipid-regulating drug treatment
A total of 10,300 patients were eligible for this analysis, with a mean(sd) LDLc of 3.22(0.97)mmol/l. The model is presented in supplementary table 1. There was an association (P=1.36×10−5) of each copy of the G allele with a 0.11mmol/l increase in LDLc. There was no difference by diabetes status.
Association of rs10455872 with LDLc lowering response to statins
A total of 7233 patients were eligible for this analysis. Supplementary table 2 provides a breakdown of patient characteristics by genotype. Overall, statin treatment resulted in a median (IQR) 56.2(20.1)% reduction in LDLc corresponding to a mean(sd) 1.98(0.9)mmol/l absolute reduction. Clinical predictors of lower response to statins included higher pre-treatment LDLc, primary prevention, additional lipid-regulating drug, poorer adherence and lower dose (supplementary table 3). In addition, each copy of the G allele was associated with a 0.10mmol/l higher on-treatment LDLc (P=1.35×10−7). Despite patients with diabetes having a better statin response (P=4.38×10−60), the effect size for rs10455872 was similar in the individuals with and without diabetes (beta(se) 0.09(0.02) vs. 0.11(0.05) respectively, data not shown). As an additional check, regarding the high frequency of imputed pre-treatment LDLc levels, we performed a sensitivity analysis to assess the impact on our results. After removal of individuals with imputed pre-treatment LDLc the effect size for rs10455872 was still consistent with the overall study population (0.11(0.02), data not shown). We performed a meta-analysis of our result with previously published studies. The combined population size was 30,467 individuals with a combined scaled beta(se) of 0.17(0.02), P=1.46×10−29 (figure 2). In terms of the comparison of effect sizes in absolute LDLc concentrations, the GoDARTS, JUPITER and HPS study effect estimates were 0.10, 0.16 and 0.07mmol/l respectively in response, per copy of the G allele. The result for the CARDS/ASCOT/PROSPER meta-analysis was not directly interpretable due to the transformation used.
Figure 2.
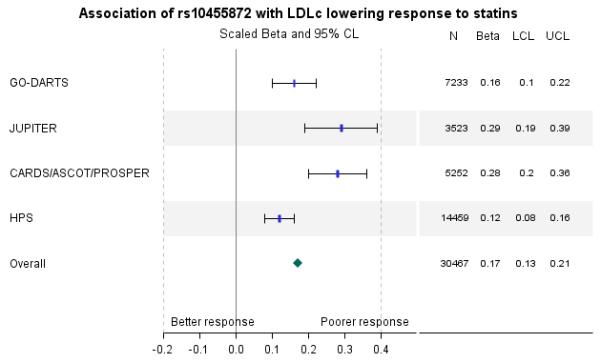
Meta-analysis of published results for association of rs10455872 with LDLc lowering response to statins in 30,467 individuals. Results for the fixed effect model are presented with an overall p-value of 1.46×10−29. Data are scaled beta (95% confidence limits) where positive beta means the minor allele is associated with a higher on-treatment LDLc and, therefore, a lower response to statins
Association of rs10455872 with CAD risk on statins
Of the 8110 patients eligible for this analysis, 932 (11.5%) had a CAD event after statin initiation (defined as cases in the analysis). This consisted of 400 MIs, 281 unstable anginas and 279 coronary-revascularisation procedures (28 patients had two of these outcomes). There was a significant difference in frequency of cases by genotype using a chi-square test: 11% A/A, 14.3% A/G, and 18% G/G, P=0.0023.
Clinical predictors of increased CAD risk included secondary prevention, diabetes, poorer adherence, lower dose, male sex and smoking (supplementary table 4). In addition, each copy of the G allele was associated with an increased risk of CAD (OR (95% CI) 1·39(1.15-1.67), P=6.76×10−4).
Replication of association of rs10455872 with CAD risk on statins
As the association of rs10455872 with CAD outcome in a statin treated population had not previously been established, we sought replication for our findings from the UCP study. The results for the meta-analysis are presented in figure 3. The combined population size for the two studies was 8822, with 1137 cases. The combined OR (95% CI) was 1·41(1.17-1.68), P=4.5×10−5.
Figure 3.
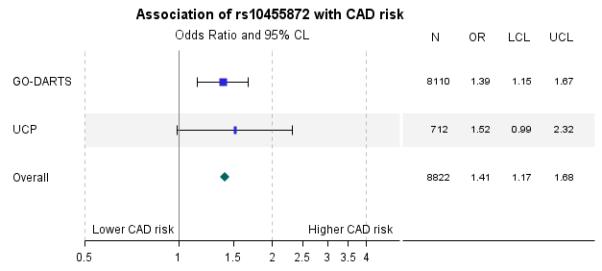
Replication of association of rs10455872 with CAD risk in statin treated individuals in the UCP Study. Results for the fixed effect model are presented with an overall p-value of 4.5×10−5. Data are odds ratio (95% CL) where an odds ratio greater than 1 means the variable is associated with an increased risk of CAD
Association of rs10455872 with CAD risk on statins with adjustment for LDLc
To assess whether the association of rs10455872 with CAD was independent of the LDLc levels achieved during statin therapy, we measured LDLc time dependently using Cox’s proportional hazards regression. A total of 6848 patients were eligible for this analysis. The median(IQR) number of LDLc measures in the model per patient was 4(5). Overall, there were 630 cases and the median (IQR) study duration was 6.5(4.7) years. The model for primary prevention is presented in supplementary table 5. Clinical predictors of CAD risk were similar to the case-control model, but importantly increasing LDLc was also a strong predictor, with each 1mmol/l increase associated with a 19% increased CAD risk. Despite adjustment for these clinical parameters each copy of the G allele was associated with a 36% increased risk of CAD.
A summary of the results for rs10455872 genotype with CAD risk, overall and in primary prevention, is presented in figure 4. To allow direct comparison the models are presented with and without LDLc adjustment. Adjusting for LDLc only reduced the HR by 2% in the overall model and 4% in primary prevention. Our results suggest that although the G allele of rs10455872 is associated with a blunted LDLc lowering response to statins, the association of rs10455872 with CAD is independent of LDLc levels achieved during statin therapy.
Figure 4.
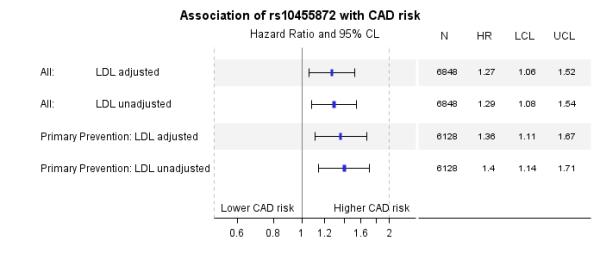
Association of rs10455872 in a Cox proportional hazards model of time to first CAD event following statin initiation. The association of rs10455872 is compared in the model adjusted and unadjusted for LDLc. The combined primary and secondary prevention model and model for primary prevention alone are presented. Data are hazard ratio (95% CL) where a hazard ratio greater than 1 means the minor allele is associated with a greater risk of an event.
Discussion
The present study adds to our understanding of the clinical implications of rs10455872 and LDLc lowering response to statins, by showing that this association, which has so far been demonstrated in RCTs across various statin types 7-9, is robust to the real life observational setting of individuals being routinely treated with statins. We replicated the findings of the GLC11 regarding rs10455872 and plasma LDLc in our population of over 10,000 individuals. We have also shown in a meta-analysis with the UCP study21, that the well-established association of rs10455872 with CAD10, 12, can be detected in a statin treated population and that this association appears to be largely independent of lipid lowering achieved, despite the confirmed association of this variant with LDLc levels and LDLc response.
In our meta-analysis of LDLc lowering response to statins, we observed a difference in the allelic point estimates between studies. This could be due to differences in the statin response definition, type of statin used and the RCT vs. observational study design. For instance, the HPS reported the lowest effect size9, despite it being the largest study. This may be due to the phenotype of absolute LDLc reduction used despite reporting a significant association of rs10455872 with pre-treatment LDLc. In our model, pre-treatment LDLc was the largest predictor of response, accounting for approximately 50% of the variation explained. Type of statin may also have an impact, with HPS - a Simvastatin trial, and our study, which comprised 74% Simvastatin prescriptions reporting the lowest effect sizes.
In order to determine the extent to which statins abrogate the genotype associated CAD risk we would need to compare rates both on and off statins within our population. Unfortunately, we were unable to formally test this as the observational nature of our data means we do not have a suitable reference group who are at a similar risk of CAD as the statin treated patients but who are not on statins. However, we modelled our study population off statins (details in supplementary methods and supplementary table 6) and showed a hazard ratio (95% CI) of 1.30(1.12-1.51) per copy of the G allele of rs10455872. This is comparable to the results we show for the combined primary and secondary prevention model unadjusted for LDLc (1.29(1.08-1.54), suggestive of no interaction of genotype with statin therapy. This is in line with the HPS study which investigated the risk of major vascular events in almost 19,000 statin treated patients by rs10455872 and found similar risk reductions across the genotypes, however they did not provide a direct estimate of the risk conferred by the LPA genotype in either the statin treated or the non-statin treated arms. Furthermore, in our study we have also demonstrated that CAD risk is independent of LDLc lowering achieved by statin therapy.
The Lipid Trialists Collaboration demonstrated that for every 1mmol/l reduction in LDLc there is a 21% reduction in CAD22, and given the impact of this variant on LDLc lowering is only 0.1 mmol/l, it follows that we would only expect a 2% reduction in CAD. This is in line with what we have shown, where adjusting for LDLc reduced the hazard ratio by 2%. This underscores the observations that there are multiple potential routes through which raised Lp(a) levels increase atherosclerotic risk other than by the increased levels of cholesterol it carries (measured as a component of the LDLc estimated by the Friedewald formula)23, and that these mechanisms are not affected by lipid lowering therapy. This has led to suggestions that individuals with high levels of LP(a) should be treated with additional drugs such as Niacin or Carnitine, which have been shown to reduce LP(a) through as yet undefined mechanisms 23-26, although to date alternative therapies by LPA genotype have yet to be performed.
Weaknesses of this study include that we have used an observational cohort in which we have modelled statin taking and outcome, and given the complexity of the real-life situation have only been able to account for what were considered the major clinical variables that we had available in the medical record, and also as mentioned above the epidemiological challenges of modelling both on and off statins. In spite of this we have achieved a high degree of internal consistency and external validity in terms of estimates of genotype and statin effects on cardiovascular outcomes. The ability to translate previous findings from trial data into clinical observational data in this way however also represents a major strength of this study in demonstrating the potential clinical impact of this pharmacogenetic paradigm.
The important conclusion of this study is that the individuals with the G allele of rs10455872 in LPA, which represents approximately 1 in 7 patients, have a higher risk of CAD than the majority of the population even after treatment with statins, and therefore represent a vulnerable group requiring an additional alternative treatment, rather than relying on statin alone.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to all the participants in this study, the general practitioners, the Scottish School of Primary Care for their help in recruiting the participants, and to the whole team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. The study complies with the Declaration of Helsinki. We acknowledge the support of the Health Informatics Centre, University of Dundee for managing and supplying the anonymised data and NHS Tayside, the original data owner. The Wellcome Trust provides support for Wellcome Trust United Kingdom Type 2 Diabetes Case Control Collection (GoDARTS) and informatics support is provided by the Chief Scientist Office. The Wellcome Trust funds the Scottish Health Informatics Programme. This research was specifically funded by the Wellcome Trust (084726/Z/08/Z, 085475/Z/08/Z, 085475/B/08/Z) and by the UK Medical Research Council (G0601261). KZ holds a Henry Wellcome Post-Doctoral Fellowship. The Utrecht Cardiovascular Pharmacogenetics project was funded by Veni grant Organization for Scientific Research (NWO), Grant no. 2001.064 Netherlands Heart Foundation (NHS), and TI Pharma Grant T6-101 Mondriaan.The department of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, has received unrestricted research funding from the Netherlands Organisation for Health Research and Development (ZonMW), the Dutch Health Insurance Board (CVZ), the Royal Dutch Association for the Advancement of Pharmacy (KNMP), the private-public funded Top Institute Pharma (www.tipharma.nl), includes co-funding from universities, government, and industry, the EU Innovative Medicines Initiative (IMI), EU 7th Framework Program (FP7), the Dutch Medicines Evaluation Board, the Dutch Ministry of Health and industry (including GlaxoSmithKline, Pfizer, and others)
Footnotes
Conflict of interest None declared
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–421. [PubMed] [Google Scholar]
- 2.Schmitz G, Schmitz-Madry A, Ugocsai P. Pharmacogenetics and pharmacogenomics of cholesterol-lowering therapy. Curr Opin Lipidol. 2007;18(2):164–73. doi: 10.1097/MOL.0b013e3280555083. [DOI] [PubMed] [Google Scholar]
- 3.Ordovas JM, Mooser V. The APOE locus and the pharmacogenetics of lipid response. Curr Opin Lipidol. 2002;13(2):113–7. doi: 10.1097/00041433-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA, et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 2010;5(3):e9763. doi: 10.1371/journal.pone.0009763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson JF, Hyde CL, Wood LS, Paciga SA, Hinds DA, Cox DR, et al. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ Cardiovasc Genet. 2009;2(2):173–81. doi: 10.1161/CIRCGENETICS.108.818062. [DOI] [PubMed] [Google Scholar]
- 6.Donnelly LA, Palmer CN, Whitley AL, Lang CC, Doney AS, Morris AD, et al. Apolipoprotein E genotypes are associated with lipid-lowering responses to statin treatment in diabetes: a Go-DARTS study. Pharmacogenet Genomics. 2008;18(4):279–87. doi: 10.1097/FPC.0b013e3282f60aad. [DOI] [PubMed] [Google Scholar]
- 7.Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet. 2012;5(2):257–64. doi: 10.1161/CIRCGENETICS.111.961144. [DOI] [PubMed] [Google Scholar]
- 8.Deshmukh HA, Colhoun HM, Johnson T, McKeigue PM, Betteridge DJ, Durrington PN, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: importance of Lp(a) J Lipid Res. 2012;53(5):1000–11. doi: 10.1194/jlr.P021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hopewell JC, Parish S, Offer A, Link E, Clarke R, Lathrop M, et al. Impact of common genetic variation on response to simvastatin therapy among 18 705 participants in the Heart Protection Study. Eur Heart J. 2012 doi: 10.1093/eurheartj/ehs344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 11.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopewell JC, Clarke R, Parish S, Armitage J, Lathrop M, Hager J, et al. Lipoprotein(a) genetic variants associated with coronary and peripheral vascular disease but not with stroke risk in the Heart Protection Study. Circ Cardiovasc Genet. 2011;4(1):68–73. doi: 10.1161/CIRCGENETICS.110.958371. [DOI] [PubMed] [Google Scholar]
- 13.Doney AS, Lee S, Leese GP, Morris AD, Palmer CN. Increased cardiovascular morbidity and mortality in type 2 diabetes is associated with the glutathione S transferase theta-null genotype: a Go-DARTS study. Circulation. 2005;111(22):2927–34. doi: 10.1161/CIRCULATIONAHA.104.509224. [DOI] [PubMed] [Google Scholar]
- 14.Doney AS, Fischer B, Leese G, Morris AD, Palmer CN. Cardiovascular risk in type 2 diabetes is associated with variation at the PPARG locus: a Go-DARTS study. Arterioscler Thromb Vasc Biol. 2004;24(12):2403–7. doi: 10.1161/01.ATV.0000147897.57527.e4. [DOI] [PubMed] [Google Scholar]
- 15.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low- density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. [PubMed] [Google Scholar]
- 16.Committee JF. British National Formulary 62. BMJ Group and Pharmaceutical Press; London: 2011. [Google Scholar]
- 17.Donnelly LA, Doney AS, Dannfald J, Whitley AL, Lang CC, Morris AD, et al. A paucimorphic variant in the HMG-CoA reductase gene is associated with lipid-lowering response to statin treatment in diabetes: a GoDARTS study. Pharmacogenet Genomics. 2008;18(12):1021–6. doi: 10.1097/FPC.0b013e3283106071. [DOI] [PubMed] [Google Scholar]
- 18.Donnelly LA, Doney AS, Tavendale R, Lang CC, Pearson ER, Colhoun HM, et al. Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin Pharmacol Ther. 2011;89(2):210–6. doi: 10.1038/clpt.2010.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilleman DE, Wurdeman RL, Lenz TL. Therapeutic change of HMG-CoA reductase inhibitors in patients with coronary artery disease. Pharmacotherapy. 2001;21(4):410–5. doi: 10.1592/phco.21.5.410.34491. [DOI] [PubMed] [Google Scholar]
- 20.Wei L, Wang J, Thompson P, Wong S, Struthers AD, MacDonald TM. Adherence to statin treatment and readmission of patients after myocardial infarction: a six year follow up study. Heart. 2002;88(3):229–33. doi: 10.1136/heart.88.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Wieren-de Wijer DB, Maitland-van der Zee AH, de Boer A, Stricker BH, Kroon AA, de Leeuw PW, et al. Recruitment of participants through community pharmacies for a pharmacogenetic study of antihypertensive drug treatment. Pharm World Sci. 2009;31(2):158–64. doi: 10.1007/s11096-008-9264-x. [DOI] [PubMed] [Google Scholar]
- 22.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordestgaard BG. Commentary: The Finnish success of cardiovascular risk factor reduction. Int J Epidemiol. 2010;39(2):518–9. doi: 10.1093/ije/dyq004. [DOI] [PubMed] [Google Scholar]
- 24.Rainwater DL. Lp(a) concentrations are related to plasma lipid concentrations. Atherosclerosis. 1996;127(1):13–8. doi: 10.1016/s0021-9150(96)05922-9. [DOI] [PubMed] [Google Scholar]
- 25.Chapman MJ, Redfern JS, McGovern ME, Giral P. Niacin and fibrates in atherogenic dyslipidemia: pharmacotherapy to reduce cardiovascular risk. Pharmacol Ther. 2010;126(3):314–45. doi: 10.1016/j.pharmthera.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Scanu AM, Bamba R. Niacin and lipoprotein(a): facts, uncertainties, and clinical considerations. Am J Cardiol. 2008;101(8A):44B–7B. doi: 10.1016/j.amjcard.2008.02.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.