

Abstract
Purpose
[131I]-meta-iodobenzylguanidine ([131I]-MIBG) is the most commonly employed treatment for metastatic pheochromocytoma and paraganglioma; however, its success is limited. Its efficacy depends on the [131I]-MIBG concentration reached within the tumor through its uptake via the norepinephrine transporter and retention in neurosecretory granules. Purpose is to enhance [123I]-MIBG uptake in cells and liver pheochromocytoma tumors.
Experimental Design
We report the in vitro effects of two histone deacetylase (HDAC) inhibitors, romidepsin and trichostatin A, on increased uptake of [3H]-norepinephrine and [123I]-MIBG in mouse pheochromocytoma (MPC) cells, and the effect of romidepsin on [18F]-fluorodopamine and [123I]-MIBG uptake in a mouse model of metastatic pheochromocytoma. The effects of both inhibitors on norepinephrine transporter activity were assessed in MPC cells by [123I]-MIBG uptake studies with and without the transporter blocking agent desipramine and the vesicular blocking agent reserpine.
Results
Both HDAC inhibitors increased [3H]-norepinephrine, [123I]-MIBG, and [18F]-fluorodopamine uptake through the norepinephrine transporter in MPC cells. In vivo, inhibitor treatment resulted in increased uptake of [18F]-fluorodopamine and in pheochromocytoma liver metastases as measured by maximal standardized uptake values on PET imaging (p < 0.001). Analysis of biodistribution after inhibitor treatment confirmed the PET results in that uptake of [123I]-MIBG was significantly increased in liver metastases (p < 0.05). Therefore, HDAC inhibitor treatment increased radioisotope uptake in MPC cells in vitro and in liver metastases in vivo, through increased norepinephrine transporter activity.
Conclusion
These results suggest that HDAC inhibitors could enhance the therapeutic efficacy of [131I]-MIBG treatment in patients with malignant pheochromocytoma.
Keywords: histone deacetylase, meta-iodobenzylguanidine, pheochromocytoma, romidepsin, trichostatin A
Introduction
Up to 36% of patients with pheochromocytoma will develop metastatic disease and have a 5-year survival rate of approximately 50% after diagnosis (1-5). Moreover, patients with metastatic pheochromocytoma exhibit excessive levels of circulating catecholamines. This results in an increased risk of hypertensive complications, including strokes and cardiac arrhythmias. Current treatments for malignant pheochromocytoma include targeted radiation using [131I]-meta-iodobenzylguanidine ([131I]-MIBG), cytotoxic chemotherapy, octreotide, tumor chemoembolization and, less frequently, radiofrequency or cryoablation (6-12). The success of these treatments is limited and varies based on the sites and growth rates of metastatic lesions.
[131I]-MIBG is one of the most effective therapies because it specifically targets chromaffin and pheochromocytoma cells. MIBG, a sympathomimetic amine analogue of guanethidine, is avidly taken up by chromaffin cells via the transmembrane norepinephrine transporter (13-15). Similar to normal chromaffin cells, pheochromocytoma cells also express the norepinephrine transporter. Uptake of [131I]-MIBG results in accumulation of 131I in the tumor cells and their destruction by beta irradiation. Unfortunately, only 30% of pheochromocytoma patients show a response to [131I]-MIBG (6). This disappointing response rate is most likely related to reduced expression of the norepinephrine transporter and a reduced number of catecholamine storage granules, which results in a suboptimal [131I]-MIBG concentration within the tumor cells. In addition, the elevated circulating catecholamines in pheochromocytoma patients compete with [131I]-MIBG for transporter-mediated entry into the tumor cell, further reducing [131I]-MIBG uptake. Kolby et al. indicated the importance of secretory granules containing vesicular monoamine transporters for the uptake and retention of [123/131I]-MIBG (16).
A number of histone deacetylase (HDAC) inhibitors have shown promise as anticancer agents (17). They can arrest growth, induce differentiation and apoptosis in various cancer cell lines, and inhibit the growth of tumors in animal models (18). Interestingly, low concentrations of the HDAC inhibitor romidepsin (depsipeptide, FK228) have been found to increase expression of the Na+/I- symporter in poorly differentiated thyroid carcinoma cells, thereby enhancing intracellular iodine accumulation (19). HDAC inhibitors might also be effective in the treatment of pheochromocytoma by virtue of their ability to induce growth inhibition and/or apoptosis in pheochromocytoma cells, increase entry and retention of radioisotopes (e.g. [131I]-MIBG), and stimulate the differentiation of malignant pheochromocytoma cells-with partial or total restoration of the phenotypic traits of the normal chromaffin cell (20).
We examined the effects of two structurally unrelated HDAC inhibitors, romidepsin and trichostatin A, in mouse pheochromocytoma (MPC) cells in vitro and in a mouse model of metastatic pheochromocytoma. MPC cells closely resemble normal and neoplastic human chromaffin cells in that they express phenylethanolamine-N-methyltransferase, which converts norepinephrine to epinephrine (21). We examined the effects of HDAC inhibition on proliferation, uptake of [3H]-norepinephrine, [123I]-MIBG, and [18F]-fluorodopamine, and norepinephrine transporter expression in MPC cells. Using positron emission tomography (PET) and biodistribution studies, we assessed the effect of a single dose of romidepsin on [18F]-fluorodopamine uptake by pheochromocytoma liver metastases in nude mice. In addition, we demonstrated the effect of trichostatin A on [123I]-MIBG uptake by liver lesions in a mouse model of metastatic pheochromocytoma (22, 23). Uptake of [18F]-fluorodopamine, an investigational imaging tracer that has been successfully used for localization of human pheochromocytoma (24), was similar to that of [131I]-MIBG in pheochromocytoma cells (25).
Materials and Methods
Reagents
RPMI 1640, Dulbecco's modified Eagle medium (DMEM), trypsin-EDTA, penicillin/streptomycin 100×, and N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES) buffer (1M) were purchased from Invitrogen-Life Technologies (Carlsbad, CA). Fetal bovine serum (FBS) and donor horse serum (DHS) were obtained from Gemini Bio-Products (Woodland, CA). Levo-2, 5, 6-3H-norepinephrine was purchased from NEN Life Science Products (Boston, MA). Bovine serum albumin (BSA), dimethyl sulfoxide (DMSO) and all the components of the Krebs Ringes Glucose (H-KRG) buffer were purchased from Sigma Chemical Co. (St. Louis, MO). Phosphate buffered saline was obtained from Biosource International (Camarillo, CA), and Triton X-100 was obtained from Fisher Scientific (Suwajanee, GA). Dr. A. T. Fojo (Developmental Therapeutics Program, National Cancer Institute, Bethesda, MD, USA) kindly provided romidepsin. Trichostatin A was purchased from Sigma Chemical Co. (St. Louis, MO).
In vitro studies
Cell culture
MPC cells (cell line 4/30/PRR) were established from heterozygous neurofibromatosis knockout mice (21). Cells were cultured in RPMI 1640 medium, supplemented with 10% DHS, 5% FBS, and penicillin/streptomycin, and maintained at 37°C in 5% CO2. Cells from passages 26-38 were used in the experiments.
Cell proliferation assay
The cytotoxic effects of romidepsin and trichostatin A were examined using the XTT-assay (Cell Proliferation Kit II, Roche Applied Science, Indianapolis, IN). MPC cells were seeded in 96-well flat-bottom plates at 50,000 cells/well and incubated with increasing concentrations of romidepsin or trichostatin A diluted in medium at 37°C for 48 or 72 hr. The XTT mixture was then added, and the cells were incubated for 18 hr. After incubation, spectrophotometric absorbance was measured using a microplate reader (Bio-Rad Laboratories, Philadelphia, PA). All experiments were performed in octuplicate.
Uptake of [3H]-norepinephrine
The uptake of norepinephrine by MPC cells was determined using [3H]-norepinephrine and a modification of the protocol described by Jaques et al. (26). MPC cells (100,000 cells/well) in 24-well plates were treated with increasing concentrations of romidepsin (0.001-10 ng/mL) or trichostatin A (6.25-100 ng/mL) and diluted in medium for 48 or 72 hr at 37°C. After treatment, cells were washed three times with 0.5 mL H-KRG buffer (H-KRG: 125 mM NaCl, 4.8 mM KCl, 2.6 mM CaCl2, 1.2 mM MgSO4, 5.6 mM glucose, 25 mM HEPES, 1 mM ascorbic acid, pH 7.35), followed by a 10 min preincubation in H-KRG buffer (0.5 mL/well) at 37°C. Next, [3H]-norepinephrine (25 nM/well) was added and the cells were incubated at 37°C for 10 min. [3H]-norepinephrine uptake was stopped by rapidly chilling the plates on ice, and the cells were washed twice with 0.1% albumin in PBS at 4°C. Cells were lysed with 0.5 mL Triton-X100 0.1% and aliquots of the cell lysates were transferred into scintillation vials. After addition of the Biosafe-II scintillation cocktail (Research Products International, Mount Prospect, IL), cell-associated beta radiation was counted in a beta-counter (Beckman Coulter LS 60000 IC).
Uptake of [123I]-MIBG and [18F]-fluorodopamine
[123I]-MIBG and [18F]-fluorodopamine uptake studies, treatment of cells, washing steps and incubations were performed following the same protocol used for [3H]-norepinephrine. MPC cells were treated with romidepsin (0.5 ng/mL) or trichostatin A (12.5 ng/mL) and incubated for 48 or 72 hr at 37°C. [123I]-MIBG (0.6-0.7 μCi/ml,, specific activity of 2mCi/0.08mg), [18F]-DA (0.6-0.7 μCi/ml, specific activity of 20mCi/3.23mg, or 17.31mCi/2.49mg) was added to the plates, which were incubated at 37°C for 10 min to asses uptake and 120 min to asses accumulation in cells. After incubation, [123I]-MIBG uptake was stopped by rapidly chilling the plates on ice, and the cells washed twice with 0.1% albumin in PBS at 4°C. The incubation medium was collected, and the cells were washed with cold PBS, trypsinized, and the cell-associated radioactivity was measured using a gamma-counter (1480 Wizard 3, Automatic Gamma Counter, Perkin-Elmer, Waltham, MA, USA).
In a subset of experiments, uptake studies with romidepsin and trichostatin A treatment were carried out in the absence or presence of 1 μM desipramine to block neuronal catecholamine uptake (27), or 10 μM reserpine to block vesicular catecholamine translocation (28, 29). All experiments were performed in quadruplicate.
Protein collection and analysis
For these experiments, cells were incubated in 25 mL flasks with 0, 0.25, 0.5 or 0.75 ng/mL of romidepsin or 0, 6.25, 12.5 or 25 ng/mL of trichostatin A for 72 hr. After two washes with PBS, cells were pelleted and lysed. Cell lysates were added to the loading buffer (1:1 GTC/PBS, Invitrogen/Cellgro). The samples were serially diluted 1:2 and loaded into a 96-well vacuum manifold (Bio-Rad, Hercules, CA) with two sheets Protran membrane (Schleicher & Schuell, Keene, NH) and a vacuum slowly applied. The membrane was removed and washed twice in TBS before being probed for GAPDH at 1:500 (American Research Products, City, State). After antibody binding, the membrane was washed three times in TTBS. The blot was then incubated with horseradish peroxidase linked anti-mouse Ig whole antibody at 1:1000 (Amersham) and imaged using ECL western blotting kit (Amersham). Next, the membrane was stripped using Re-Blot Plus Mild (Chemicon, Temecula, CA) for 10 min. Subsequently, it was probed for anti-acetyl histone H3 1:2200 and again washed three times in TTBS, followed by horseradish peroxidase linked anti-rabbit Ig whole antibody at 1:1000 (Amersham) and imaged as before. Densitometry was performed on films using the IPLab gel software.
Ultrastructural studies
The effect of treatment with either 0.5 ng/mL romidepsin or 12.5 ng/mL trichostatin A on MPC cell morphology and ultrastructure was examined by electron microscopy. After 3 washes with PBS, cells were double-fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated, embedded into Spurr's epoxy resin, and coded to insure unbiased assessment. Ultrathin sections (90 nm) were made and double-stained with uranyl acetate and lead citrate and viewed in a Philips CM10 transmission electron microscope. The percentage of cells containing neurosecretory granules and the number of granules per cell were counted under the microscope in a minimum of 100 randomly selected cells per treatment. The influence of the treatments on the occurrence and frequency of apoptotic features was also evaluated. Two investigators blinded to treatment performed the microscopic assessment. Experiments were done in duplicate.
In vivo studies
Animal model
The metastatic mouse pheochromocytoma model used in this study was previously described (22, 30). Briefly, female 6-8 week old (20-22 g) athymic nude mice (NCr-nu; Taconic, Germantown, NY) were injected by tail vein with 1×106 MPC (MPC 4/30 PRR) cells. MPC cells were mixed with 100 μL of PBS and held at room temperature prior to injection. Mice were anesthetized using isoflurane/O2 (1.5-5% v/v) before all imaging and treatment procedures. All animal studies were conducted according to the National Institutes of Health Guide for the Care and Use of Animals under an approved protocol from the Institutional Animal Care and Use Committee.
In vivo magnetic resonance imaging (MRI)
Magnetic resonance images were obtained with a 3 Tesla MRI scanner (Intera, Philips Medical System, Best, Netherlands) using a dedicated 40 mm inner diameter solenoid coil (Philips, Best, Netherlands). Localization and monitoring of liver lesions were carried out as previously described (30). Briefly, with anesthetized animals in the prone position, respiratory triggered T2-weighted spectra were acquired with the following parameters: FOV 8.0 × 8.0 × 2.0 cm3, data matrix 512 × 512, 40 slices, TE/TR 65/4500 ms, flip angle 90°, slice thickness 0.5 mm, 0.156 × 0.156 mm2 in-plane resolution, scan time of 5-7 min for two signal averages depending on the respiratory rate. No contrast agent was used for MRI. Animals were scanned 4-5 weeks after the injection of MPC cells to determine liver tumor size.
[123I]-MIBG and [18F]-fluorodopamine biodistribution studies
HDAC inhibitor treatment was carried out using the protocol described by Goldsmith et al. (31) with a slight modification. Briefly, female athymic nude mice were treated with romidepsin at a dose of 3.6 mg/kg; however, mice with liver metastases did not tolerate that dose. Thus, a single lower dose of romidepsin (2.5 mg/kg) was administered to evaluate its effect on isotope accumulation in the tumors. The same concentration was used for trichostatin A. Both romidepsin and trichostatin A were resuspended in saline and intravenously administered by injection of 150 μL at a rate of 10 μL per min. [123I]-MIBG and [18F]-fluorodopamine biodistribution studies began 24 hr after drug administration.
For [123I]-MIBG biodistribution studies, anesthetized mice (n = 3) were injected through the lateral tail vein with a dose of 150 μL saline solution containing 2.5 mg/kg of trichostatin A over 15 min. Trichostatin A was injected twice, 24 hr and 2 hr before administration of 25-27 μCi of [123I]-MIBG. Untreated control mice (n = 4) were injected with a total volume of 150 μL saline solution and vehicle, followed at the same time points with the same dose of [123I]-MIBG. Both groups were sacrificed by cervical dislocation at approximately 120 min post injection of [123I]-MIBG. Samples of the liver lesions and normal liver were collected and weighed.
For [18F]-fluorodopamine biodistribution studies, a single dose of romidepsin 2.5 mg/kg was injected into mice (n = 7) 24 hr before administration of 50-60 μCi of [18F]-fluorodopamine. Control mice (n = 7) received the same dose [18F]-fluorodopamine. Both groups were sacrificed 60 min post-injection. Samples of the liver lesions and normal liver were collected and weighed. The concentrations of [123I]-MIBG and [18F]-fluorodopamine in the liver tumors and normal liver tissue were assayed using an automatic gamma-counter (Model 1480 Wallac Wizard, Perkin Elmer, Shelton, CT City, State). Standards of 1:10 of the injected dose were prepared and counted along with all samples. Background counts were subtracted from the reported 123I and 18F counts per minute. The injected counts were determined from the standard counts and the quantitative data, expressed as standardized uptake value, was determined as described previously (32).
[18F]-fluorodopamine positron emission tomography imaging
PET is a noninvasive imaging technique that allows the longitudinal monitoring of tumors. The procedure for romidepsin pretreatment evaluation consisted of two PET scans, where mice were injected with 50-60 μCi of [18F]-fluorodopamine in the same mice. Mice were treated with a single dose of romidepsin 2.5 mg/kg; n = 4). The first scan was performed before treatment as a baseline measurement and one week later the second scan, started 24 h hours after treatment with romidepsin, was performed on the same mice.
PET scans were performed using the Advanced Technology Laboratory Animal Scanner (ATLAS; (33) which has a transverse field-of-view of 6.8 cm and an axial FOV of 2 cm. PET images were reconstructed by a 2D-ordered-subset expectation maximization (2D OSEM) algorithm (5 iterations and 16 subsets), achieving a 1.5-mm full width at half maximum (FWHM) resolution at the center (34). The reconstructed voxel size was 0.56 × 0.56 × 1.125 mm3. No correction was applied for attenuation or scatter. Dynamic data acquisition determining the pharmacokinetics of [18F]-fluorodopamine in liver tumors and liver parenchyma started about 1 min after injection. Scanning parameters were set for one frame/10 min, up to 6 frames. Whole body data acquisitions (2 bed positions, each 10 min) started consecutively after the dynamic acquisition. Whole body acquisitions (achieving 2 × 2 cm = 4 cm of the field of view), including images of the lungs through the kidneys, were acquired after administration of 50-60 μCi of [18F]-fluorodopamine.
Each liver lesion was analyzed individually for the maximal uptake based on the most active voxel cluster located within the region of interest. This is equivalent to the maximum standardized uptake value used in clinical PET studies (35). If liver tumors are smaller (in any dimension) than approximately 2.5 times the FWHM of the spatial resolution of the PET scanner, a distortion called a partial volume effect occurs (36). Therefore, liver lesions were analyzed only if they were larger than 4 mm in diameter as obtained by MRI. This is the minimum size required to exclude partial volume effects in small animal PET when monitoring lesions for potential physiological changes (37). The 18F activity concentration of the radionuclide was expressed as the tumor-to-liver ratio (TLR), which was obtained by comparing uptake in the liver lesions with that in the liver parenchyma.
SYBR Green real-time PCR for quantification of norepinephrine transporter expression in liver tumors
Norepinephrine transporter expression was quantified by quantitative real-time PCR. Total RNA was extracted from the liver metastases of untreated control mice and from trichostatin A-treated mice using the RNeasy Midi Kit (Qiagen, City, State) according to the manufacturer's instructions. After reverse transcribing mRNA, real-time PCR was performed in triplicate using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in a 7500 Real-time PCR system (Applied Biosystems) as previously described (38). Use of forward primer 5′ AGAGCAGTGGGATCCATGAC 3′ and reverse primer 5′ CCAGGAGCACAAACAAGACA 3′ yielded a 168 base pair product. Obtained CT (threshold cycle) values of the norepinephrine transporter gene (in liver lesion samples after trichostatin A treatment, n = 7) were normalized to those of beta-actin, and their relative mRNA expression was expressed as fold induction over the baseline (control untreated liver lesions, n = 6). The dissociation curves of the primer pairs showed a single peak, and PCR reactions produced a single expected DNA band in an agarose gel analysis. Statistical analyses were carried out by unpaired t-test with the two-tailed p value.
Data analyses
Results are presented as the mean ±SEM from a minimum of three experiments for both in vitro and in vivo experiments. Before performing any statistical test, all data were tested for normal distribution and equal variance. Statistical differences between groups of data were assessed by ANOVA, followed by the Student-Neuman-Keuls test for group comparison. The level of statistical significance was set at p < 0.05.
Results
In vitro experiments
Dose- and time-dependent inhibition of MPC cell proliferation by romidepsin and trichostatin A
Treatment with romidepsin or trichostatin A induced a dose-dependent decrease in MPC cell proliferation. Growth inhibition curves after 48 and 72 hr of exposure to the drugs are shown in Fig. 1A and 1B. After a 72 hr exposure, the 50% inhibitory concentrations (IC50) were determined to be 1.56 ng/mL for romidepsin and 50 ng/mL for trichostatin A.
Figure 1.
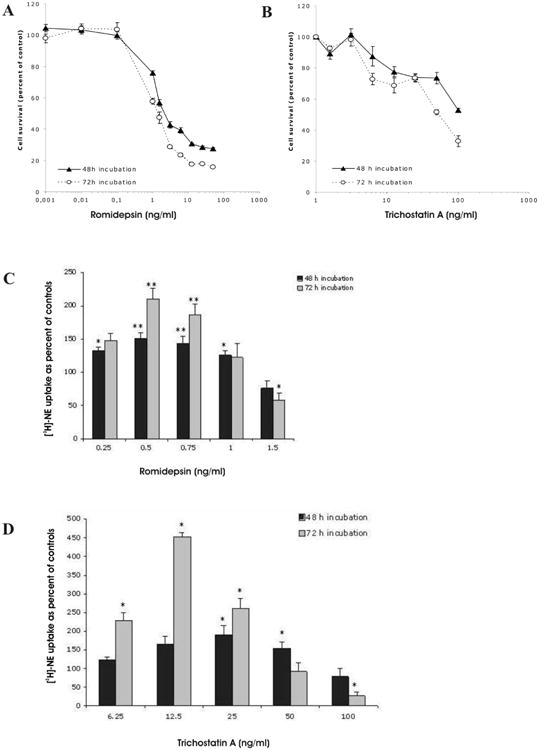
Dose- and time-dependent effect of romidepsin (A) and trichostatin A (B) on MPC cell growth. Results are presented as mean ±SEM. Increased [3H]-norepinephrine ([3H]-NE) uptake in MPC after treatment with romidepsin (C) and trichostatin A (D). (* = p < 0.05; ** = p < 0.005, compared to control, untreated cells).
Increased specific [3H]-norepinephrine uptake in MPC cells after treatment with romidepsin and trichostatin A
A significant dose- and time-dependent increase in the specific [3H]-norepinephrine uptake was observed in MPC cells treated with romidepsin concentrations of 0.25-0.5 ng/mL, and trichostatin A concentrations of 6.25-12.5 ng/mL. This increase was followed by a decrease at the highest concentrations of both romidepsin and trichostatin A (Fig. 1C and 1D). The maximal increase in [3H]-norepinephrine uptake occurred in MPC cells after 72 hr of treatment with 0.5 ng/mL of romidepsin and 12.5 ng/mL of trichostatin A. In subsequent experiments, concentrations of 0.5 ng/mL romidepsin and 12.5 ng/mL trichostatin A were used.
Increased [123I]-MIBG and [18F]-fluorodopamine uptake in MPC cells after treatment with romidepsin or trichostatin A
The maximal amount of [123I]-MIBG uptake in MPC cells occurred 72 hr after treatment with romidepsin (0.5 ng/mL), in both short-term (Fig. 2A) and long-term (Fig. 2B) contexts. Trichostatin A (12.5 ng/mL) promoted approximately the same levels of uptake of [123I]-MIBG after 48 hr as after 72 hr (Fig. 2A and 2B). The uptake of [123I]-MIBG in MPC cells was approximately 3.6 times higher than in the control group at 10 min, and 4.4 times higher at 120 min after 72 hr treatment with trichostatin A. Similarly, for romidepsin the uptake of [123I]-MIBG was approximately 1.5 times that in the control group at 10 min, and 2.2 times higher at 120 min.
Figure 2.
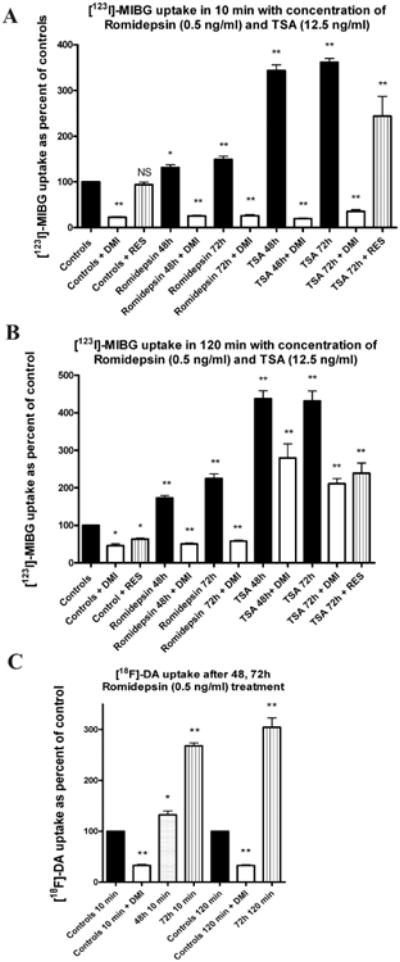
Increased [123I]-MIBG uptake in MPC cells after treatment with romidepsin and trichostatin A. (A) The effect of the norepinephrine transporter blockers desipramine (DMI) and reserpine (RES) on 10 min of uptake time of [123I]-MIBG after 48 and 72 hr of treatment with romidepsin and trichostatin A. Data are shown as percentage of [123I]-MIBG uptake in untreated control cells (baseline). (B) The effect of desipramine (DMI) and reserpine (RES) on 120 min of uptake time of [123I]-MIBG after 48 and 72 h of treatment with romidepsin and trichostatin A. (C) The uptake of [18F]-fluorodopamine in MPC cells in 10 and 120 min. The effect on [18F]-fluorodopamine uptake was studied after 48 and 72 hr of romidepsin treatment, and the association of specific transporters with uptake of [18F]-fluorodopamine was confirmed with desipramine (DMI). Desipramine and reserpine were added 30 min prior to [123I]-MIBG and [18F]-fluorodopamine dosing. Results are presented as mean ±SEM. (NS = not significant, * = p < 0.05; ** = p < 0.005)
In order to find out whether the increase of [123I]-MIBG uptake was due to inhibition of transport over the cellular membrane, or due to impaired granular storage, we examined the effect of desipramine on cellular [123I]-MIBG uptake and retention and also the effect of reserpine on storage of [123I]-MIBG in vesicles. Desipramine inhibited the entry of [123I]-MIBG into both untreated and treated MPC cells; the inhibitory effect was more pronounced at 10 min than at 120 min (Fig. 2 A and B). A similar, though less pronounced, inhibitory effect was observed with reserpine at 120 min in untreated cells, and at 10 and 120 min in cells treated with trichostatin A (Fig. 2 A, and B). Similarly, increased accumulation of [18F]-fluorodopamine was observed in MPC cells after treatment with romidepsin for 48 and 72 hr (Fig. 2C). Desipramine significantly inhibited the entry of [18F]-fluorodopamine into MPC cells (Fig. 2C).
Treatment of MPC cells with romidepsin or trichostatin A increased acetylation of histone H3
As expected, blot analysis showed increased acetylation of histone H3 in MPC cells at the same doses of romidepsin or trichostatin A that had been proven to influence proliferation and [3H]-norepinephrine and [123I]-MIBG uptake (Fig. 3A). The neurosecretory granules were present in control and treated cells, but no significant changes were visible (Fig. 3B).
Figure 3.
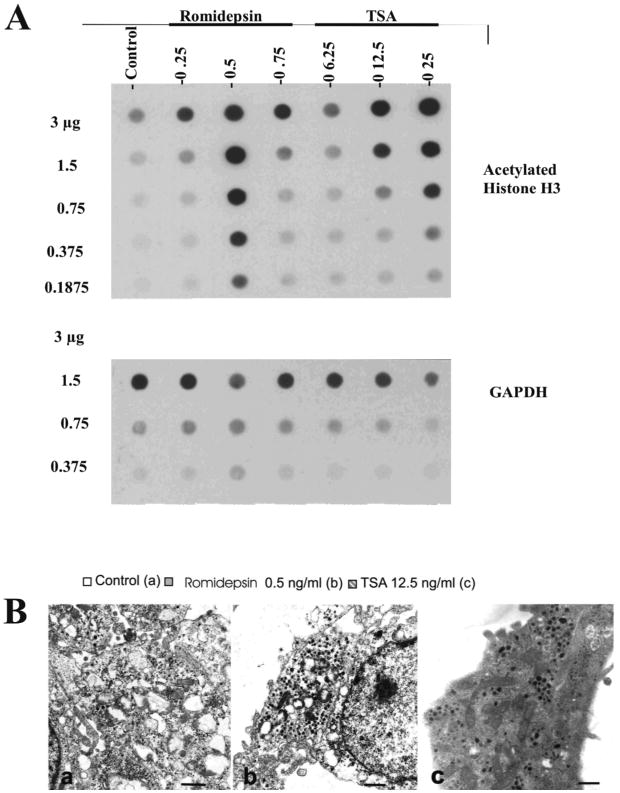
(A) Expression of acetylated histone H3 for at various concentrations of romidepsin and trichostatin A as examined by dot blot analysis. GAPDH was a loading [?] control. (B) EM photomicrographs of MPC cells showing the presence of granules in treated cells: (a) untreated cells, (b) romidepsin 0.5 ng/mL, (c) trichostatin A 12.5 ng/mL. Bar = 1 micron.
In vivo experiments
MRI imaging
MRI detected multiple liver lesions in our in vivo model. Liver lesions 4 mm in diameter were found five weeks after tail vein injection of MPC cells. Lesions at this size were suitable for PET imaging. Also, we were able to determine the growth of individual lesions and correlate radionuclide uptake with the size of the lesions.
Trichostatin A and romidepsin increased [18F]-fluorodopamine and [123I]-MIBG uptake in metastatic lesions
Tumor bearing mice were sacrificed 120 min post [123I]-MIBG injection, and various sized liver lesions (2 mm to >8 mm diameter) were dissected to determine the relationship between [123I]-MIBG uptake and tumor size. The average concentration of [123I]-MIBG in all liver metastases after treatment with trichostatin A was 2.08 ±0.287 standardized uptake value units compared to 0.87 ±0.123 in untreated control liver metastases (p < 0.001). The corresponding TLR values were 3.09 after treatment with trichostatin A and 1.82 in the untreated control mice. Similar results were observed for [18F]-fluorodopamine biodistribution. Mice treated with romidepsin demonstrated standardized uptake values of 3.522 ±0.329 vs. 0.926 ±0.464 (p < 0.001), and the TLR value was significantly higher in the treated group, 4.402 vs. 1.12. Figure 4 demonstrates no relationship between tumor size and uptake of either [18F]-fluorodopamine or [123I]-MIBG in both the control and HDAC inhibitor-treated groups. If the increased uptake of [18F]-fluorodopamine and [123I]-MIBG by liver tumors is independent of tumor size, it must be an effect of increased expression of the norepinephrine transporter.
Figure 4.
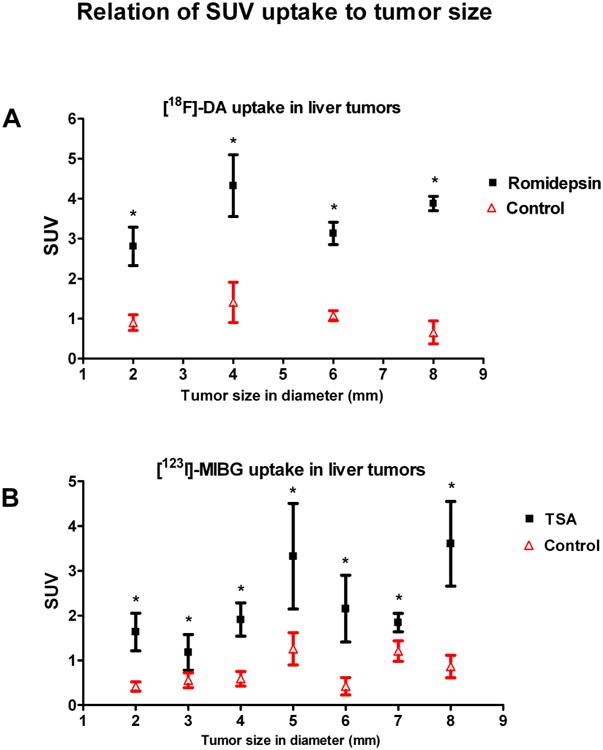
Summary of biodistribution of both tracers after romidepsin and trichostatin A treatment. The biodistribution of (A) [18F]-fluorodopamine and (B) [123I]-MIBG has no relationship to tumor size in either the control or the treated group. [18F]-Fluorodopamine uptake was evaluated from 14 control liver tumor samples and 15 treated liver tumor samples. [123I]-MIBG uptake was evaluated from 30 control tumor samples and 29 treated tumor samples. Every tumor size is derived from the mean ±SEM of at least three tumor samples. Increased uptake of [123I]-MIBG was significant in liver metastases after treatment compared to uptake in control liver lesions (p < 0.05).
Increased [18F]-fluorodopamine uptake in metastatic pheochromocytoma liver lesions in mice treated with romidepsin
The effect of romidepsin was evaluated by longitudinal PET imaging. The size of liver lesions was measured by MRI one day before [18F]-fluorodopamine PET imaging both pre- and post-romidepsin treatment. Representative dynamic PET images, acquired over 60 min after injection of [18F]-fluorodopamine, showed increased radionuclide accumulation in post-treatment scans compared to pretreatment scans of large and small tumors (Fig. 5A). Whole body PET images confirmed the increased accumulation of [18F]-fluorodopamine in pheochromocytoma liver metastases after romidepsin treatment compared to the baseline scan. Uptake of [18F]-fluorodopamine was significantly higher in liver lesions after romidepsin treatment (SUVmax 3.8 ±1.44) compared to liver lesions in pretreatment scans (SUVmax 1.259 ±0.357; p < 0.001; Fig. 5B). TLR values were 1.9 pretreatment and 6.3 for post-treatment. These results were consistent with the increased [18F]-fluorodopamine concentration detected in liver lesions. Representative pre- and post-treatment PET/MRI images of the same mouse are presented in Fig. 5C. The color scale indicates the maximum standardized uptake values in the PET images.
Figure 5.
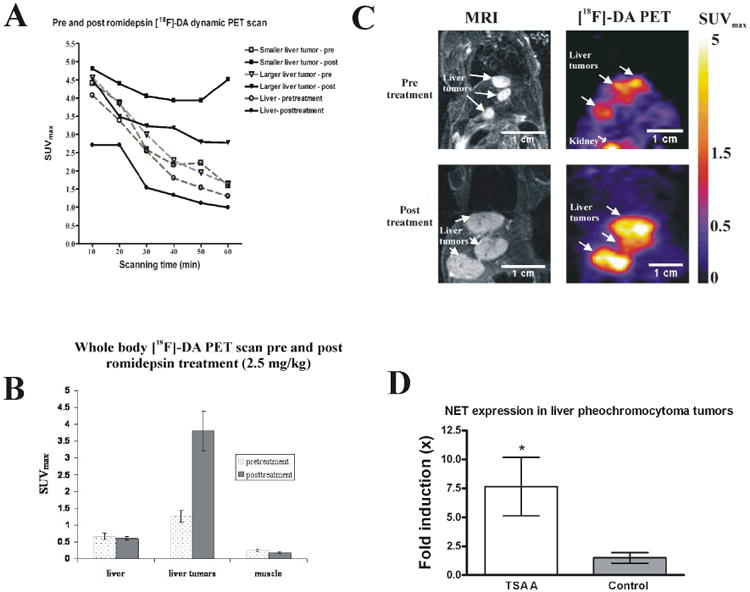
(A) Dynamic PET acquisition captured the pharmacokinetics of [18F]-fluorodopamine in liver tumors, and in liver, both pre- and post-treatment, with a 60-minute imaging period. (B) PET imaging showing [18F]-fluorodopamine uptake as mean (±SEM) maximum standardized uptake values (SUVmax) 6 days before and 24 hr after treatment with a single intravenous dose of romidepsin (2.5mg/kg), in liver, pheochromocytoma liver metastases, and muscle. [18F]-fluorodopamine uptake in the metastatic liver lesions is significantly increased after administration of romidepsin compared to the uptake in the same lesions one week earlier on the pretreatment scan (p < 0.001). (C) Representative pre- and post-treatment PET/MRI images of the same mouse. There was one week between pre- and post-treatment scans. Tumor growth over that one week is presented in MRI (left panels), and the corresponding PET of [18F]-fluorodopamine uptake (right panel) was imaged 24 hours after MRI. (D) Relative mRNA expression of the norepinephrine transporter in liver lesion samples after trichostatin A treatment (n = 7) is higher than in control, untreated liver lesions (n = 6). RNA concentrations were normalized to beta-actin.
Increased genomic expression of the norepinephrine transporter in liver tumors treated with trichostatin A compared to untreated liver tumors
Quantitative PCR demonstrated a significant increase in the expression of the norepinephrine transporter in the liver metastases of mice treated with trichostatin A in comparison to untreated animals' tumors (Fig. 5D).
Discussion
HDAC inhibitors can induce growth arrest, differentiation and/or apoptosis in various tumor cell lines. Additionally, they can inhibit tumor growth in vivo (39). In view of these anti-tumor effects, several HDAC inhibitors are now in clinical trials or have been approved for the treatment of selected cancers, either as monotherapies or in combination with other agents. The inhibitor voronistat (Zolinza®) is approved for the treatment of T-cell lymphoma as a single agent (40). HDAC inhibitors have also been combined with cytotoxic chemotherapy, differentiation agents, or radiotherapy (41, 42).
Current treatment options for malignant pheochromocytoma are limited and responses are often transient (6-9, 12, 25). The success of [131I]-MIBG, the most frequently applied therapeutic modality, depends on the dose and interval used (43, 44). Response rates are increased with higher doses of [131I]-MIBG (45), but higher doses are associated with greater toxicity (43, 44). These side-effects are the results of systemic beta-irradiation from the nonspecific uptake of [131I]-MIBG by normal tissues (46, 47). High doses of [131I]-MIBG increase the long term risk of a second malignant neoplasm (48, 49).
One way of increasing the efficacy of [131I]-MIBG, while at the same time decreasing nonspecific uptake in non-target tissues, is to increase the expression of the norepinephrine transporter in the tumor cells. This transporter carries the [131I]-MIBG across the plasma membrane (13, 15, 50), and increased expression results in increased [131I]-MIBG uptake (13). Conversely, drugs that block NET activity, such as tricyclic antidepressants, reduce [131I]-MIBG uptake (50). These findings have led to the use of the norepinephrine transporter as a reporter gene (51). In our experiments, we showed that nanomolar concentrations of two structurally unrelated HDAC inhibitors increase [3H]-norepinephrine uptake in MPC cells in vitro. The increase is dose-dependent and at its greatest 72 hours after treatment with either 0.5 ng/mL of romidepsin or 12.5 ng/mL of trichostatin A. Moreover, increased uptake of [123I]-MIBG in MPC cells after romidepsin or trichostatin A in vivo, supports the potential use of HDAC inhibitors to increase [131I]-MIBG uptake in clinical pheochromocytomas. Examining the effect of desipramine and reserpine in MPC cells suggested that both immediate uptake via the norepinephrine transporter and longer-term retention in vesicles are responsible for the increased [123I]-MIBG activity in MPC cells after romidepsin and trichostatin A treatment. However, other effects like permeability of the cell membrane and osmotic activity should be taken into consideration and should be explored in future work.
In a phase I trial of romidepsin, patients with various refractory neoplasms were administered doses of romidepsin without significant toxicity. This indicates that the concentrations used in the present study can be easily achieved in the clinic (52). At higher concentrations, a decrease in norepinephrine uptake was observed. This effect may result from the decreased number of cells available for uptake because of the growth inhibition by HDAC inhibitors at these concentrations.
The biodistribution studies were performed for confirmation of the HDAC inhibitors' effect on [18F]-fluorodopamine and [123I]-MIBG uptake in hepatic pheochromocytoma lesions. We wanted to test both romidepsin and trichostatin A on mice having a similar amount of liver involvement, and similar tumor growth rates. Increased uptake of both [18F]-fluorodopamine and [123I]-MIBG in inhibitor-treated liver tumors compared to that in control liver tumors in vivo confirmed our in vitro findings. These results could potentially be translated into the clinic for improvement of [123I]-MIBG scintigraphy as well as enhancement of radiotreatment with [131I]-MIBG for metastatic pheochromocytoma and paraganglioma patients.
Promising results from in vitro studies prompted us to investigate the effects of a single dose of romidepsin on [18F]-fluorodopamine uptake in liver metastases by PET. Post-treatment [18F]-fluorodopamine uptake in liver metastases was significantly increased 24 hours after administration of romidepsin, compared to the [18F]-fluorodopamine uptake in the same lesions on the pre-treatment PET scan. This increased uptake was not related to the volume of the liver lesions. [18F]-fluorodopamine uptake in the same liver lesion as it grows either remained the same or decreased over the time.
Of concern, HDAC inhibitors can cause toxicity in mice. Mice treated with romidepsin with dose of 3.6 mg/kg were dying during dosing. Toxic effects were seen as a result of anesthesia intolerance and liver toxicity due to the romidepsin treatment. Due to numerous liver metastases in this animal model and the low percentage of healthy liver parenchyma, anesthesia itself can be toxic (53). Anesthesia will not be used in human patients during romidepsin administration, thus we do not expect similar complications.
For biodistribution studies, trichostatin A was administered twice, 24 hours and 2 hours before [18F]-fluorodopamine injection. The reason for this was the rapid and extensive metabolism of trichostatin A. It was demonstrated that following intraperitoneal administration to mice (54), trichostatin A was rapidly absorbed from the peritoneum and was detectable in plasma within 6-9 minutes. To translate these results into humans, the half-life of the HDAC inhibitors should be taken into consideration. From these studies, we conclude that romidepsin would be more suitable for translational human study than trichostatin A due to its longer biological half-life.
Our results raise the possibility of using romidepsin to increase the sensitivity of metastatic pheochromocytoma lesions to [131I]-MIBG treatment; however, several issues remain to be addressed before a clinical trial with a combination therapy can be initiated in malignant pheochromocytoma. First, our results need to be confirmed with [123\131I]-MIBG scintigraphy coupled with dosimetry measurements in patients with metastatic pheochromocytoma. Second, [18F]-fluorodopamine uptake is overall superior to [123\131I]-MIBG in patients with malignant pheochromocytoma (55). Whether the results of similar experiments with [131I]-MIBG would be less dramatic than observed in the present study with [18F]-fluorodopamine is unknown. However, there is also a possibility that such results could also be more impressive, as [131I]-MIBG may exhibit greater uptake due to increased expression of the norepinephrine transporter. Third, future studies will focus on determining the optimal timing and dose of romidepsin. Ideally, synergy between romidepsin and [131I]-MIBG would allow both drugs to be used at concentrations lower than employed as single agent therapy. This may be especially important for studies involving patients with metastatic pheochromocytoma. Since romidepsin will not be used as a monotherapy, but as a sensitizer, and only a few doses well below that associated with cardiac toxicity will be administered before [131I]-MIBG treatment, we expect that the dosage and treatment intervals required will be associated with minimal risk of cardiotoxicity (42, 56).
In summary, treatment with HDAC inhibitors increased radioisotope uptake in MPC cells in vitro and in vivo, through both increased expression of the norepinephrine transporter and enhanced retention in neurosecretory granules. These results suggest that HDAC inhibitors might be used to increase the response of patients with malignant pheochromocytoma to [131I]-MIBG treatment. The same approach could be used also to improve diagnostic localization of metastatic lesions. Pretreatment with romidepsin could be beneficial to the patients whose tumor do not take up [123I]-MIBG for scintigraphy and thus diagnostic approach could be increased. It was also confirmed, that loss of NET with chemo therapy which is similar to what happens with 131I treatment of thyroid cancer described by Beierwaltes (57, 58). Further studies are in progress to investigate this possibility.
Acknowledgments
We thank Andrea Limpuangthip for her technical assistance. We thank Dr. Nancy Nader for her guidance and support. This research was supported in part by NIH grant R01-NS 37685 and by a grant from the Pheo Para Alliance (to A.S.T.) and APVV-0148-06 (to R.K.). This work was supported by the intramural program of the National Institute of Child Health and Human Development, the National Cancer Institute, the National Institute of Biomedical Imaging and Bioengineering, and the National Institute of Mental Health at the National Institutes of Health. Melissa Stauffer, PhD, of Scientific Editing Solutions, provided editorial assistance.
References
- 1.Mundschenk J, Lehnert H. Malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 1998;106(5):373–6. doi: 10.1055/s-0029-1212001. [DOI] [PubMed] [Google Scholar]
- 2.Plouin PF, Gimenez-Roqueplo AP, La Batide Alanore A, Salenave S, Duclos JM. Recent progress in the diagnosis, prognostic evaluation and treatment of pheochromocytomas. Rev Med Interne. 2000;21(12):1075–85. doi: 10.1016/s0248-8663(00)00268-x. [DOI] [PubMed] [Google Scholar]
- 3.John H, Ziegler WH, Hauri D, Jaeger P. Pheochromocytomas: can malignant potential be predicted? Urology. 1999;53(4):679–83. doi: 10.1016/s0090-4295(98)00612-8. [DOI] [PubMed] [Google Scholar]
- 4.O'Riordain DS, Young WF, Jr, Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg. 1996;20(7):916–21. doi: 10.1007/s002689900139. [DOI] [PubMed] [Google Scholar]
- 5.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665–75. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 6.Loh KC, Fitzgerald PA, Matthay KK, Yeo PP, Price DC. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest. 1997;20(11):648–58. doi: 10.1007/BF03348026. [DOI] [PubMed] [Google Scholar]
- 7.Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–73. doi: 10.7326/0003-4819-109-4-267. [DOI] [PubMed] [Google Scholar]
- 8.Sisson JC, Shapiro B, Shulkin BL, Urba S, Zempel S, Spaulding S. Treatment of malignant pheochromocytomas with 131-I metaiodobenzylguanidine and chemotherapy. Am J Clin Oncol. 1999;22(4):364–70. doi: 10.1097/00000421-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Kopf D, Bockisch A, Steinert H, Hahn K, Beyer J, Neumann HP, et al. Octreotide scintigraphy and catecholamine response to an octreotide challenge in malignant phaeochromocytoma. Clin Endocrinol (Oxf) 1997;46(1):39–44. doi: 10.1046/j.1365-2265.1997.d01-1738.x. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, Ashizawa N, Minami T, Suzuki S, Sakamoto I, Hayashi K, et al. Malignant pheochromocytoma with multiple hepatic metastases treated by chemotherapy and transcatheter arterial embolization. Intern Med. 1999;38(4):349–54. doi: 10.2169/internalmedicine.38.349. [DOI] [PubMed] [Google Scholar]
- 11.Pacak K, Chrousos GP, Koch CA, Lenders JW, Eisenhofer G. Pheochromocytoma: progress in diagnosis, therapy, and genetics. In: Margioris A, Chrousos GP, editors. Adrenal Disorders. 1. Totowa: Humana Press; 2001. pp. 479–523. [Google Scholar]
- 12.Munver R, Del Pizzo JJ, Sosa RE. Adrenal-preserving minimally invasive surgery: the role of laparoscopic partial adrenalectomy, cryosurgery, and radiofrequency ablation of the adrenal gland. Curr Urol Rep. 2003;4(1):87–92. doi: 10.1007/s11934-003-0065-4. [DOI] [PubMed] [Google Scholar]
- 13.Sisson JC, Wieland DM. Radiolabeled meta-iodobenzylguanidine: pharmacology and clinical studies. Am J Physiol Imaging. 1986;1(2):96–103. [PubMed] [Google Scholar]
- 14.Sisson JC, Shapiro B, Meyers L, Mallette S, Mangner TJ, Wieland DM, et al. Metaiodobenzylguanidine to map scintigraphically the adrenergic nervous system in man. J Nucl Med. 1987;28(10):1625–36. [PubMed] [Google Scholar]
- 15.Glowniak JV, Kilty JE, Amara SG, Hoffman BJ, Turner FE. Evaluation of metaiodobenzylguanidine uptake by the norepinephrine, dopamine and serotonin transporters. J Nucl Med. 1993;34:1140–1146. [PubMed] [Google Scholar]
- 16.Kolby L, Bernhardt P, Levin-Jakobsen AM, Johanson V, Wangberg B, Ahlman H, et al. Uptake of meta-iodobenzylguanidine in neuroendocrine tumours is mediated by vesicular monoamine transporters. Br J Cancer. 2003;89(7):1383–8. doi: 10.1038/sj.bjc.6601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marks PA, Rifkind RA, Richon VM, Breslow R. Inhibitors of histone deacetylase are potentially effective anticancer agents. Clin Cancer Res. 2001;7(4):759–60. [PubMed] [Google Scholar]
- 18.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92(15):1210–6. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 19.Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(-) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86(7):3430–5. doi: 10.1210/jcem.86.7.7621. [DOI] [PubMed] [Google Scholar]
- 20.Li QY, Jones PL, Lafferty RP, Safer D, Levy RJ. Thymosin beta4 regulation, expression and function in aortic valve interstitial cells. J Heart Valve Dis. 2002;11(5):726–35. [PubMed] [Google Scholar]
- 21.Powers JF, Evinger MJ, Tsokas P, Bedri S, Alroy J, Shahsavari M, et al. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res. 2000;302(3):309–20. doi: 10.1007/s004410000290. [DOI] [PubMed] [Google Scholar]
- 22.Martiniova L, Lai EW, Elkahloun AG, Abu-Asab M, Wickremasinghe A, Solis DC, et al. Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin Exp Metastasis. 2009;26(3):239–50. doi: 10.1007/s10585-009-9236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohta S, Lai EW, Morris JC, Pang AL, Watanabe M, Yazawa H, et al. Metastasis-associated gene expression profile of liver and subcutaneous lesions derived from mouse pheochromocytoma cells. Mol Carcinog. 2008;47(4):245–51. doi: 10.1002/mc.20388. [DOI] [PubMed] [Google Scholar]
- 24.Pacak K, Eisenhofer G, Carrasquillo JA, Chen CC, Li ST, Goldstein DS. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension. 2001;38(1):6–8. doi: 10.1161/01.hyp.38.1.6. [DOI] [PubMed] [Google Scholar]
- 25.Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134(4):315–29. doi: 10.7326/0003-4819-134-4-200102200-00016. [DOI] [PubMed] [Google Scholar]
- 26.Jaques S, Jr, Tobes MC, Sisson JC, Baker JA, Wieland DM. Comparison of the sodium dependency of uptake of meta-lodobenzylguanidine and norepinephrine into cultured bovine adrenomedullary cells. Mol Pharmacol. 1984;26(3):539–46. [PubMed] [Google Scholar]
- 27.Lingen B, Bruss M, Bonisch H. Cloning and expression of the bovine sodium-and chloride-dependent noradrenaline transporter. FEBS Lett. 1994;342(3):235–8. doi: 10.1016/0014-5793(94)80508-3. [DOI] [PubMed] [Google Scholar]
- 28.Cornelissen J, Tytgat GA, van den Brug M, van Kuilenburg AB, Voute PA, van Gennip AH. Menadione inhibits MIBG uptake in two neuroendocrine cell lines. J Neurooncol. 1997;31(1-2):147–51. doi: 10.1023/a:1005718421774. [DOI] [PubMed] [Google Scholar]
- 29.Eisenhofer G, Hovevey-Sion D, Kopin IJ, Miletich R, Kirk KL, Finn R, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther. 1989;248(1):419–27. [PubMed] [Google Scholar]
- 30.Martiniova L, Kotys MS, Thomasson D, Schimel D, Lai EW, Bernardo M, et al. Noninvasive monitoring of a murine model of metastatic pheochromocytoma: a comparison of contrast-enhanced microCT and nonenhanced MRI. J Magn Reson Imaging. 2009;29(3):685–91. doi: 10.1002/jmri.21654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldsmith ME, Aguila A, Steadman K, Martinez A, Steinberg SM, Alley MC, et al. The histone deacetylase inhibitor FK228 given prior to adenovirus infection can boost infection in melanoma xenograft model systems. Mol Cancer Ther. 2007;6(2):496–505. doi: 10.1158/1535-7163.MCT-06-0431. [DOI] [PubMed] [Google Scholar]
- 32.Green MV, Seidel J, Vaquero JJ, Jagoda E, Lee I, Eckelman WC. High resolution PET, SPECT and projection imaging in small animals. Comput Med Imaging Graph. 2001;25(2):79–86. doi: 10.1016/s0895-6111(00)00057-4. [DOI] [PubMed] [Google Scholar]
- 33.Seidel J. Resolution uniformity and sensitivity of the NIH ATLAS small animal PET scanner: comparison to simulated LSO scanners without depth-of-interaction capability. IEEE Trans Nuc Sci. 2003;50:1347–1350. [Google Scholar]
- 34.Toyama H, Ichise M, Liow JS, Modell KJ, Vines DC, Esaki T, et al. Absolute quantification of regional cerebral glucose utilization in mice by 18F-FDG small animal PET scanning and 2-14C-DG autoradiography. J Nucl Med. 2004;45(8):1398–405. [PubMed] [Google Scholar]
- 35.Aliaga A, Rousseau JA, Cadorette J, Croteau E, van Lier JE, Lecomte R, et al. A small animal positron emission tomography study of the effect of chemotherapy and hormonal therapy on the uptake of 2-deoxy-2-[F-18]fluoro-D-glucose in murine models of breast cancer. Mol Imaging Biol. 2007;9(3):144–50. doi: 10.1007/s11307-007-0091-6. [DOI] [PubMed] [Google Scholar]
- 36.Hutchins GD. Small animal PET imaging. Astroparticle, particle and space physics, detectors and medical physics applications. 2003:381–390. [Google Scholar]
- 37.Keyes JW., Jr SUV: standard uptake or silly useless value? J Nucl Med. 1995;36(10):1836–9. [PubMed] [Google Scholar]
- 38.Ichijo T, Voutetakis A, Cotrim AP, Bhattachryya N, Fujii M, Chrousos GP, et al. The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorticoid receptor: potential clinical implications. J Biol Chem. 2005;280(51):42067–77. doi: 10.1074/jbc.M509338200. [DOI] [PubMed] [Google Scholar]
- 39.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 40.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 41.Piekarz R, Luchenko V, Draper D, Wright J, Figg W, Fojo A, et al. Phase I trial of romidepsin, a histone deacetylase inhibitor, given on days one, three and five in patients with thyroid and other advanced cancers. J Clin Oncol. 2008 J Clin Oncol(26):abstr 3571. [Google Scholar]
- 42.Piekarz RL, Frye AR, Wright JJ, Steinberg SM, Liewehr DJ, Rosing DR, et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin Cancer Res. 2006;12(12):3762–73. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- 43.Rose B, Matthay KK, Price D, Huberty J, Klencke B, Norton JA, et al. High-dose 131I-metaiodobenzylguanidine therapy for 12 patients with malignant pheochromocytoma. Cancer. 2003;98(2):239–48. doi: 10.1002/cncr.11518. [DOI] [PubMed] [Google Scholar]
- 44.Safford SD, Coleman RE, Gockerman JP, Moore J, Feldman JM, Leight GS, Jr, et al. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery. 2003;134(6):956–62. doi: 10.1016/s0039-6060(03)00426-4. discussion 962-3. [DOI] [PubMed] [Google Scholar]
- 45.Fitzgerald PA, Goldsby RE, Huberty JP, Price DC, Hawkins RA, Veatch JJ, et al. Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG) Ann N Y Acad Sci. 2006;1073:465–90. doi: 10.1196/annals.1353.050. [DOI] [PubMed] [Google Scholar]
- 46.Tristam M, Alaamer AS, Fleming JS, Lewington VJ, Zivanovic MA. Iodine-131-metaiodobenzylguanidine dosimetry in cancer therapy: risk versus benefit. J Nucl Med. 1996;37(6):1058–63. [PubMed] [Google Scholar]
- 47.Blottner A, Bratouss A, Ertl S, Wollny P. 131-I-MIBG--pharmacokinetics and dosimetry. Radiobiol Radiother. 1988;29(2):202–5. [PubMed] [Google Scholar]
- 48.Weiss B, Vora A, Huberty J, Hawkins RA, Matthay KK. Secondary myelodysplastic syndrome and leukemia following 131I-metaiodobenzylguanidine therapy for relapsed neuroblastoma. J Pediatr Hematol Oncol. 2003;25(7):543–7. doi: 10.1097/00043426-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 49.Garaventa A, Gambini C, Villavecchia G, Di Cataldo A, Bertolazzi L, Pizzitola MR, et al. Second malignancies in children with neuroblastoma after combined treatment with 131I-metaiodobenzylguanidine. Cancer. 2003;97(5):1332–8. doi: 10.1002/cncr.11167. [DOI] [PubMed] [Google Scholar]
- 50.Bomanji J, Levison DA, Flatman WD, Horne T, Bouloux PM, Ross G, et al. Uptake of iodine-123 MIBG by pheochromocytomas, paragangliomas, and neuroblastomas: a histopathological comparison. J Nucl Med. 1987;28(6):973–8. [PubMed] [Google Scholar]
- 51.Morozumi T, Fukuchi K, Uehara T, Kusuoka H, Hori M, Nishimura T. Abnormal iodine-123-MIBG images in healthy volunteers. J Nucl Med. 1996;37:1686–1688. [PubMed] [Google Scholar]
- 52.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8(3):718–28. [PubMed] [Google Scholar]
- 53.Martiniova L, Schimel D, Lai EW, Limpuangthip A, Kvetnansky R, Pacak K. In vivo microCT imaging of liver lesions in small animal models. Methods. 2009 doi: 10.1016/j.ymeth.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanderson L, Taylor GW, Aboagye EO, Alao JP, Latigo JR, Coombes RC, et al. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab Dispos. 2004;32(10):1132–8. doi: 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- 55.Ilias I, Yu J, Carrasquillo JA, Chen CC, Eisenhofer G, Whatley M, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab. 2003;88(9):4083–7. doi: 10.1210/jc.2003-030235. [DOI] [PubMed] [Google Scholar]
- 56.Molife R, Fong P, Scurr M, Judson I, Kaye S, de Bono J. HDAC inhibitors and cardiac safety. Clin Cancer Res. 2007;13(3):1068. doi: 10.1158/1078-0432.CCR-06-1715. author reply 1068-9. [DOI] [PubMed] [Google Scholar]
- 57.Simon D, Korber C, Krausch M, Segering J, Groth P, Gorges R, et al. Clinical impact of retinoids in redifferentiation therapy of advanced thyroid cancer: final results of a pilot study. Eur J Nucl Med Mol Imaging. 2002;29(6):775–82. doi: 10.1007/s00259-001-0737-6. [DOI] [PubMed] [Google Scholar]
- 58.Gruning T, Tiepolt C, Zophel K, Bredow J, Kropp J, Franke WG. Retinoic acid for redifferentiation of thyroid cancer--does it hold its promise? Eur J Endocrinol. 2003;148(4):395–402. doi: 10.1530/eje.0.1480395. [DOI] [PubMed] [Google Scholar]