

Summary
Conversion of EpiSCs to naive ESCs is a rare event that is driven by the reestablishment of the naive transcription factor network. In mice, STAT3 activation is sufficient to drive conversion of EpiSCs to the naive pluripotent stem cell (PSC) state. However, the lack of responsiveness of EpiSCs to LIF presents a bottleneck in this conversion process. Here, we demonstrate that local accumulation of BMP-SMAD1 signaling, in cooperation with GP130 ligands, enhances the recovery of LIF responsiveness by directly controlling transcription of the LIF receptor (Lif-r). Addition of BMP and LIF to EpiSCs increases both LIF responsiveness and conversion frequencies to naive PSCs. Mechanistically, we show that the transcriptional cofactor P300 plays a critical role by mediating complex formation between STAT3 and SMAD1. This demonstration of how the local microenvironment or stem cell niche reactivates dormant signaling responsiveness and developmental potential may be applicable to other stem cell niche-containing systems.
Graphical Abstract

Highlights
-
•
Addition of BMP increases mouse EpiSC-to-ESC conversion frequency
-
•
Accumulation of BMP and gp130 ligands resuscitates JAK-STAT signaling in EpiSCs
-
•
BMP and gp130 ligands control LIF-R transcription
-
•
p300 mediates STAT3-SMAD1 complex to control LIF responsiveness
Mouse EpiSC-to-ESC conversion is a rare event that is limited by the insufficient activation of STAT3 in EpiSCs. Engineering the microenvironment overcomes this limitation through local accumulation of endogenous BMP and GP130 ligands. This drives a P300-mediated, feed-forward mechanism that revives dormant JAK-STAT signaling. Zandstra and colleagues describe the role of the in vitro niche in influencing early stem cell fate decisions.
Introduction
Mouse embryonic stem cells (mESCs) and epiblast stem cells (EpiSCs) are stable, pluripotent cells derived from mouse blastocysts and early postimplantation embryo, respectively. Embryonic stem cells demonstrate characteristics of naive pluripotency, while, in contrast, EpiSCs are in a primed pluripotent state (Nichols and Smith, 2009). These cells, despite their temporal proximity in vivo, exhibit marked differences in their response to signals to maintain their pluripotency; mESC stability is governed by leukemia inhibitory factor (LIF) and bone morphogenic protein 4 (BMP4) signaling, while Activin A and basic fibroblast growth factor (bFGF) maintain EpiSCs. Conversion of mESCs to EpiSCs is primarily driven by culture in EpiSC-supportive conditions, specifically, by addition of the cytokines Activin A and bFGF (Guo et al., 2009). This conversion robustly yields culture-derived EpiSCs (CDEs). Similarly, conversion of EpiSCs to mESCs can be driven by activation of STAT3 (Bao et al., 2009; Onishi et al., 2012; van Oosten et al., 2012; Yang et al., 2010), the transcription factor directly downstream of mESC-supportive LIF signaling. Alternative methods of EpiSC conversion involve overexpression of transgenes (Festuccia et al., 2012; Gillich et al., 2012; Guo and Smith, 2010; Guo et al., 2009; Hall et al., 2009; Hanna et al., 2009; Yang et al., 2010) or culture on feeders (Bao et al., 2009; Bernemann et al., 2011; Greber et al., 2010; Han et al., 2010a; Sasaki et al., 2011; Zhou et al., 2010). Frequency of conversion remains low in most cases, with optimal conversion occurring at ∼10% efficiency when potent conversion-driving transcription factors, NANOG or KLF4, are overexpressed in combination with either optimal STAT3 activation (Yang et al., 2010) or orphan nuclear receptor, LRH-1 (Guo and Smith, 2010). Not surprisingly, STAT3-driven conversion of EpiSCs is critically limited by the inability of EpiSCs to robustly respond to LIF. We have previously demonstrated that EpiSCs regain LIF responsiveness upon control of the cellular microenvironment and local cell density using micropatterning (μP) (Onishi et al., 2012). This recovery of LIF responsiveness facilitates feeder-free conversion of EpiSCs at frequencies comparable to transgenic systems. We have previously demonstrated the importance of endogenous GP130 ligands in gaining LIF responsiveness (Davey et al., 2007). However, the observation that supplementing exogenous LIF to EpiSCs does not recover LIF responsiveness suggests the presence of additional signaling conversion barriers (Onishi et al., 2012).
BMP signaling in the presence of LIF functions to maintain mESC pluripotency. However, when EpiSCs are exposed to BMP, SMAD1 activation induces their differentiation to both primitive endoderm and trophectoderm (Brons et al., 2007). Additionally, at higher concentrations (50 to 500 ng/ml), BMP drives the differentiation of EpiSCs to germ cells (Hayashi and Surani, 2009; Tesar et al., 2007). This behavior is consistent with that of human embryonic stem cells (hESCs); an observation further corroborated in other differentiation protocols involving BMP signaling (Vallier et al., 2009). The role of BMP in differentiation of EpiSCs is extensive and well defined. In contrast to its role in differentiation, here we demonstrate a role for BMP signaling in the upregulation of LIF signaling responsiveness and subsequent enhancement of the reprogramming efficiency of EpiSCs. The increase in LIF responsiveness of EpiSCs upon μP (Onishi et al., 2012) can be explained by an increase in local accumulation of BMP and GP130 (i.e., IL-6 family) ligands. Specifically, inhibition of endogenous BMP and GP130 signaling on μP results in the loss of LIF responsiveness. Additionally, in standard, nonpatterned culture, supplementation with both LIF and BMP4 increases LIF responsiveness by as much as 3-fold over addition of either LIF or BMP4 alone. We demonstrate that STAT3 and SMAD1, the respective downstream targets of LIF and BMP4, cooperate by directly binding and regulating transcription of the promoter region the gene encoding JAK-STAT pathway receptor LIF receptor (Lif-r). As is observed in neural precursors (Nakashima et al., 1999), the interaction between SMAD1 and STAT3 in EpiSCs is bridged by P300. We demonstrate that manipulation of P300 can tune the reinitiation of dormant JAK-STAT signaling in response to LIF and BMP, thereby affecting frequency of conversion. This demonstration of how signaling crosstalk changes the context and cell fate effects of BMP signaling may be indicative of conserved mechanisms in other systems and an important consideration for harnessing the full potential of pluripotent and multipotent stem cells.
Results
SMAD1 Activation by BMP Signaling Is Enhanced upon Micropatterning
We have previously demonstrated niche-mediated control of endogenous BMP signaling in hESC colonies by μP (Peerani et al., 2007). We reasoned that the μP of EpiSCs, in addition to its effects on JAK-STAT signaling, may impact BMP signaling and provide insight into how LIF responsiveness is regained. Upon μP, the transcription of several BMP ligands and the SMAD1 targets, Id1 and Id3, increased (Figure 1A; Figure S1A available online). To address whether SMAD1 was being activated upon μP, we stained for activated SMAD1 (i.e., phospho-SMAD1 [pSMAD1]). Levels of pSMAD1 in μP EpiSCs increased significantly over nonpatterned (non-μP) EpiSCs, both in the absence or presence of exogenous BMP (Figure 1B). Addition of LIF did not significantly increase SMAD1 activity over μP-AF cells (Figure 1B) or in non-μP cells (Figure S1B), suggesting that these pathways act independently with respect to activation of SMAD1. A time course reveals that μP induced gene expression changes within 24 hr postseeding (Figure 1C), whereby Nanog transcript is elevated at 6 hr over the nonpatterned control, suggesting that endogenous signals begin concentrating locally within a few hours after seeding onto patterns. Taken together, we observe an increase in overall BMP signaling in EpiSCs upon μP and that these signals begin to concentrate early into the patterning process.
Figure 1.

BMP Signaling Rapidly Increases in EpiSCs on μP
(A) qRT-PCR on (i) culture-derived EpiSCs (CDEs) and (ii) an embryo-derived EpiSC line (129S2C1a) demonstrates an overall increase in transcription of BMP-signaling-related genes upon μP. Genes are categorized as follows: pluripotency related (Plurip.), BMP related (BMP), and JAK-STAT related (JAK-STAT).
(B) Representative images of phosphorylated SMAD1 (pSMAD1) staining on EpiSCs that are cultured in Activin A and FGF media (AF) either not patterned (non-μP) or patterned (μP) in the presence of exogenous BMP4 (AF +BMP4) or LIF (AF+LIF). Quantification of images is shown in the right panel.
(C) Time course of gene expression changes upon μP of EpiSCs. All time points were compared to their respective non-μP controls. ∗p < 0.05 as tested by a two-sided, two-sample t test.
All data are presented as mean ± SD (n = 3, technical replicates, independent wells) except (B) (n = 6, technical replicates, independent wells). (A) and (C) are representative plots.
Addition of BMP4 Enhances the Conversion Frequency of EpiSCs
We next asked if addition of BMP4 for 1 day prior to reseeding in stringent mESC media (2iL) would increase conversion frequency. We observed a dramatic 8-fold increase in alkaline phosphatase (ALP)-positive colonies upon BMP4 treatment (Figure 2A) and BMP2 (Figure S2Bi) at both 10 ng/ml and 50 ng/ml (Figure S2Bii). To exclude the possibility of the existence of ALP-positive EpiSCs in 2iL, we costained ALP with KLF4 and SOX2 in EpiSCs (CDE), serum-cultured mESCs (R1), and revertant EpiSCs in 2iL (rCDEs) (Figure S1A). Cells staining positive for both KLF4 and SOX2 (double positive [DP]) mark cells in the naive pluripotent state (Figure S2Ai). Additionally, ALP-positive cells exist only in the DP subpopulation in both mESCs and in revertant EpiSCs (Figures S2Aii and S2Aiii), demonstrating that ALP serves as a faithful reporter of the naive state. BMP4 signaling is critical during primordial germ cell (PGC) derivation from EpiSCs (Hayashi and Surani, 2009; Tesar et al., 2007). These EpiSC-derived PGCs are then able to form embryonic germ cells (EGCs) that closely resemble mESCs. To exclude the possibility that EpiSCs are undergoing differentiation through PGCs in response to BMP4, we assayed transcript levels of known PGC markers (Blimp1, Nanos3, Prdm14, Fragilis, Dnd1, Hoxa1, and Stella) in the presence of BMP2 and BMP4 at 10 ng/ml and 50 ng/ml (Figures S2Ci and S2Cii). All markers demonstrated no observable increase in the presence of BMP signaling at the two concentrations. Additionally, immunostaining of BLIMP1, a marker of early PGC formation, demonstrated no significant increases with 10 ng/ml or 50 ng/ml of BMP4 treatment for 1 day (Figure S2D). These observations, along with the requirement for 500 ng/ml of BMP4 exposure for 4–7 days to induce low-frequency PGC formation from EpiSCs (Hayashi et al., 2011; Hayashi and Surani, 2009), strongly suggest that cells are not converting to EGCs. BMP signaling, both endogenous and exogenous, therefore improves yield of ALP-positive colonies from EpiSCs by reversion, not by differentiation to PGCs. The embryo-derived EpiSC line 129S2C1a increased conversion frequency by about 13-fold upon supplementation with BMP4 (Figure 2Bi). We next assayed for its ability to form chimeras upon blastocyst injection. As EpiSCs were reverted in the absence of transgenes, the converted, mESC-like cells were transfected with a constitutively expressed β-geo (β-galactosidase + neomycin resistance) transgene to distinguish between donor and host cells. Importantly, these BMP4-treated, reverted cells contribute to all three germ layers of the developing embryo, as assayed by β-galactosidase staining, demonstrating full conversion to naive pluripotency (Figure 2Bii).
Figure 2.
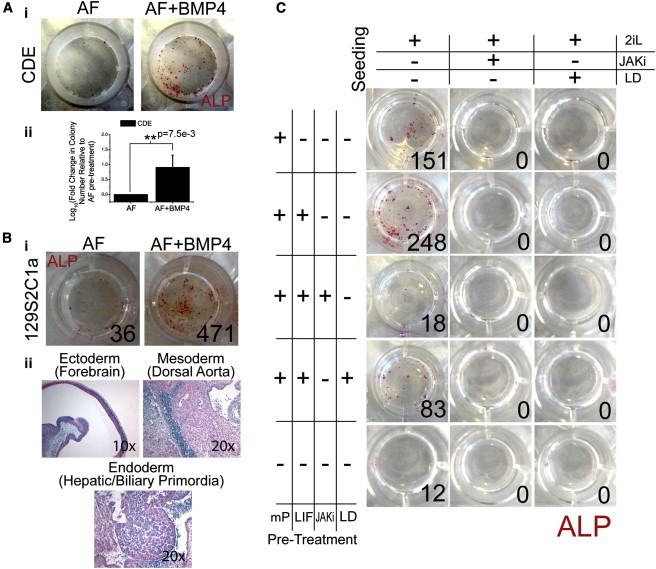
BMP4 Improves Frequency of Conversion to Naive Pluripotency, while Inhibition of BMP Signaling Decreases Frequency
(A) (i) Addition of BMP4 to AF media in the absence of patterning increases conversion frequency in CDEs as measured by alkaline phosphatase (ALP)-positive colonies in 2i + LIF (2iL) media, and (ii) quantification of conversion frequency. (n = 3 biological replicates, independent experiments).
(B) (i) Similar results are observed in embryo-derived 129S2C1a cells upon addition of BMP4. Representative picture: (ii) these revertant cells (after transfection with a constitutive β-geo construct) generate all three germ layers (endoderm, primordial hepatic biliary; ectoderm, forebrain; mesoderm, dorsal aorta) after blastocyst aggregation.
(C) Representative image of factorial experiment to determine the role of both STAT3 and SMAD1 activity before and during conversion. ALP-positive colonies were counted and reported in the lower corner of each panel. μP CDEs were pretreated for 1 day in LIF, JAK inhibitor (JAKi, 2μM), or LDN-193189 (LD, 3μM) (rows). Cells were then seeded in 2iL media containing either JAKi or LD (columns). ∗p < 0.05 as tested by a two-sided, two-sample t test.
All data are presented as mean ± SD (n = 3).
With BMP signaling emerging as a potent enhancer of conversion in EpiSCs, we next asked whether it, along with JAK-STAT signaling, is a requirement for conversion. To test this, we first micropatterned EpiSCs in Activin A/bFGF (AF) media supplemented with LIF (10 ng/ml), a JAK inhibitor (JAKi; 2 μM), a BMP signaling inhibitor (targeting the BMP receptors ALK2 and ALK3), or LDN193189 (LDN) (3 μM) or without any supplementation. This pretreatment of EpiSCs on μP was performed for 1 day. Nonpatterned CDEs cultured in AF were used as controls. Test cells were then dissociated and seeded in 2iL media supplemented with JAKi (2 μM) or LDN (3 μM). Colonies were stained for ALP and counted after 7 days (Figure 2C). No colonies were observed in 2iL supplemented with either JAKi or LDN, suggesting that continuous activation of both STAT3 and SMAD1 during conversion is essential. CDEs cultured in JAKi or LDN supplemented AF for 15 days maintain OCT4 expression, demonstrating that cytotoxic effects of the small molecules alone are negligible (Figure S2Ei). Similar results were observed with mESCs treated with LDN (Figure S2Eii) for 9 days and JAKi for 10 days (Onishi et al., 2012) in 2iL. Pretreatment with either JAKi or LDN did not abolish colony formation in 2iL but resulted in a 13- and 3-fold decrease in colony number, respectively, suggesting an important, but not critical, role of JAK-STAT and BMP signaling in priming EpiSCs for conversion. Taken together, the continuous activation of STAT3 and SMAD1 is required during the onset of conversion (i.e., upon addition of 2iL media) and for the duration of conversion, but not during the pretreatment phase.
Increase in LIF Responsiveness upon μP Can Be Explained by Signaling through LIF and BMP4
As JAK-STAT and BMP signaling are both present during μP-mediated priming of EpiSCs prior to conversion, and as both have beneficial roles in conversion, we next asked whether these signaling pathways were involved in the resuscitation of LIF responsiveness seen on patterns. JAK-STAT signaling is autoregulated in a positive feed-forward loop (He et al., 2005) and buffers mESCs from undergoing differentiation in the absence of exogenous LIF (Davey et al., 2007). Despite this, addition of LIF for 9 days was able to only subtly resuscitate LIF responsiveness in EpiSCs (Onishi et al., 2012). The role of BMP signaling in the context of LIF responsiveness remains undefined. To this end, EpiSCs (both CDEs and EpiB3s) supplemented with LIF and BMP4 (both at 10ng/ml) for 1 day prior demonstrated significantly increased levels of pSTAT3 when compared to cells supplemented with LIF or BMP4 alone after a 15 min stimulation with LIF (Figure 3A). Interestingly, addition of BMP4 at concentrations below 10 ng/ml (i.e., 5 ng/ml, 1 ng/ml, 0.1 ng/ml) did not allow EpiSCs to recover LIF responsiveness even in the presence of elevated LIF (10 ng/ml) (Figure S3A), consistent with previous observations of the lack of responsiveness in EpiSCs upon addition of LIF alone (Onishi et al., 2012). To address whether BMP4 signaling is central to μP-mediated recovery of LIF responsiveness, we treated EpiSCs with NOGGIN (a BMP receptor antagonist) (Figure 3Bi), LDN (Figure 3Bii), or small interfering RNA (siRNA) sequences targeting Smad1 (Figure 3Biii) prior to or during μP. NOGGIN (300 ng/ml) was added to cells at day 0 (d0) as they were seeded onto μP, or at day 1 after μP (d1). LDN (100 nM) was added at d1. Cells were assayed after 2 days (d2) on μP. Inhibition of BMP signaling by all three methods prevented EpiSCs from regaining LIF responsiveness on μP (Figure 3B). Consistent with our observation that μP leads to transcriptional changes within the span of 1 day (Figure 1C), we observed that addition of NOGGIN at the time of seeding (i.e., d0) was more effective in preventing recovery of LIF responsiveness than when it was added at d1 (Figure 3Bi). Inhibition of JAK-STAT signaling with JAKi also abolishes LIF responsiveness on μP (Onishi et al., 2012), and similar to NOGGIN, we observed a more potent effect of JAKi when added at d0 as compared to JAKi added at d1 (Figure S3B). Importantly, addition of both LIF and BMP4 increased conversion frequency of nonpatterned EpiSCs by 5-fold over untreated EpiSCs, 3.25-fold over LIF only, and 1.5-fold over BMP only (Figure 3C). These data suggest that accumulation of local GP130 and BMP ligands soon after μP serves to increase LIF responsiveness of EpiSCs and function to promote conversion frequencies.
Figure 3.

LIF and BMP4 Increase LIF Responsiveness in EpiSCs On and Off μP
(A) Immunostaining for pSTAT3 and OCT4 in (i) CDEs and (ii) embryo-derived EpiB3 treated with LIF and/or BMP4 then pulsed with LIF to assay LIF responsiveness. (iii) Quantification of images seen in (i) and (ii).
(B) Immunostaining for pSTAT3 and OCT4 after inhibition of BMP signaling on μP by (i) NOGGIN (Nog. 300 ng/ml), (ii) LDN-193189 (LDN, 0.1 μM), or (iii) siRNA against Smad1 (Smad1i) followed by a LIF pulse to assay LIF responsiveness.
(C) ALP staining of revertant CDEs after treatment with any combination of LIF and BMP4 (both at 10 ng/ml). Quantification on bottom panel, ∗p < 0.05, as tested by a two-sided, two-sample t test.
All data are presented as mean ± SD (A: n = 4 technical replicates, independent wells; B: n = 6 for Bi, n = 4 for Bii, all technical replicates, independent wells; C: n = 3, biological replicates across independent experiments).
LIF and BMP4 Increase LIF Responsiveness by Directly Targeting and Regulating Lif-r Transcription in EpiSCs
We next asked how LIF and BMP4 synergized to increase LIF responsiveness in EpiSCs. The respective downstream transcription factor targets of LIF and BMP4, STAT3 and SMAD1, are known to form a complex, bridged by P300, in neural precursors to promote differentiation to astrocytes (Nakashima et al., 1999). Additionally, in mESCs, STAT3 and SMAD1 coimmunoprecipitate (Ying et al., 2003) and colocalize with P300 to the same regions of the genome (Chen et al., 2008). We examined chromatin immunoprecipitation sequencing (ChIP-seq) data generated by Chen et al. (Chen et al., 2008) and found that pull downs of SMAD1, P300, and STAT3 all enriched for DNA fragments mapping upstream of the Lif-r gene. We therefore hypothesized that STAT3 and SMAD1 form a complex in EpiSCs and regulate transcription of genes mediating LIF responsiveness, specifically Lif-r. A time course of BMP4-treated EpiSCs reveals a steady increase of Lif-r expression over 24 hr (Figure 4A). With the addition of LIF and BMP4 in combination, after 3 hr, transcription of Lif-r reaches the level observed at 24 hr with BMP4 alone. Furthermore, the peak transcription level of Lif-r in LIF and BMP4-treated EpiSCs is greater than that observed in EpiSCs treated with only LIF for 24 hr (Figure S4) and 48 hr (Figure 4A). Oct4 and Nanog levels remain high throughout the time course, demonstrating that EpiSCs do not undergo significant differentiation when in the presence of exogenous BMP4 for 24 hr. Importantly, the effects of LIF and BMP4 are observed even in the presence of the translation inhibitor, cycloheximide (chx), added for 3 hr at 30 ng/ml, demonstrating that this is a direct effect (Figure 4B). We next asked whether STAT3, SMAD1, and coregulator, P300, bind to the locus upstream of Lif-r as identified by Chen and colleagues (Chen et al., 2008) (Lif-r-enh) or to regions within promoters as defined by the Database of Transcriptional Start Sites (Lif-r-pro1, Lif-r-pro2) (Yamashita et al., 2010). We also included the possibility of binding to the Gp130 promoter (Gp130-pro), as ChIP-seq from Chen et al. also identified binding of SMAD1, STAT3, and P300 to this locus. To this end, we performed ChIP on STAT3, P300, and SMAD1 and amplified ∼500 bp regions within the respective sites. No regions of the Gp130 promoter were pulled down (data not shown). The ∼500 bp region within Lif-r-enh was determined by mapping the ChIP-seq data set generated by Chen et al. to the mouse genome (build GRCm38/mm10). To determine the regions within Lif-r-pro1 and Lif-r-pro2, quantitative PCR with primers specific to five regions within Lif-r-pro1 and four within Lif-r-pro2 (Table S1) was performed (data not shown). Together, we proceeded with three specific ∼500 bp regions, one for each of Lif-r-enh, Lif-r-pro1, and Lif-r-pro2 (Figure 4C, red boxes). Subsequent PCR on these three regions demonstrated enrichment only in the first promoter, Lif-r-pro1 (Figure 4C). To determine if the region enriched for STAT3, P300, and SMAD1 binding is functional and to confirm that the regions absent in binding are not functional, we cloned ∼2 kb sections encompassing the pulled down regions into luciferase constructs (pGL3-promoter for Lif-r-enh and pGL3-basic for Lif-r-pro1, Lif-r-pro2, and Gp130-pro) (Figure 4D, yellow boxes). CDEs were transfected and treated with nothing (AF), LIF, BMP4, or LIF and BMP4. Upon treatment with LIF and BMP4, we observed a significant increase in luciferase expression in Lif-r-pro1, but not Lif-r-enh, Lif-r-pro2, or Gp130-pro (Gp130 not shown), supporting the ChIP data. Additionally, 24 hr of LIF treatment alone did not increase luciferase activity of the construct, supporting quantitative RT-PCR (qRT-PCR) data of Lif-r transcript levels after equivalent treatment. Taken together, activation of STAT3 and SMAD1 in EpiSCs, either endogenously or exogenously, recovers dormant LIF responsiveness by directly regulating transcription of the JAK-STAT pathway receptor, Lif-r, but not Gp130.
Figure 4.
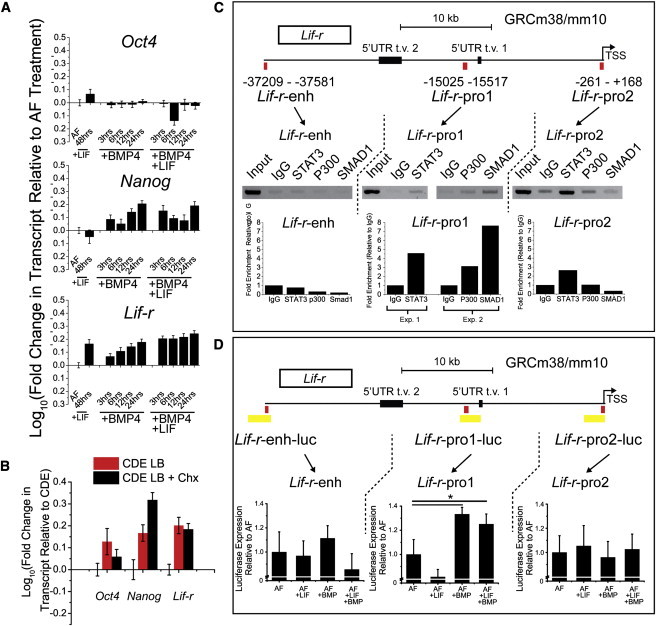
LIF and BMP4 Directly Regulate Transcription of Lif-r and Gp130
(A) Time-course qRT-PCR spanning 48 hr of Lif-r along with pluripotency markers Oct4 and Nanog after addition of either BMP4 alone or LIF and BMP4. Controls were addition of nothing (AF) or addition of LIF for 48 hr.
(B) Transcription of Oct4, Nanog, and Lif-r was assayed in the presence of LIF and BMP4 and cycloheximide (chx, 30ng/ml) for 3 hr.
(C) Chromatin immunoprecipitation (ChIP) was performed on three regions upstream of the Lif-r gene, Lif-r-enh, Lif-r-pro1, and Lif-r-pro2. These regions were determined based on data from Chen et al. (Lif-r-enh) and Yamashita et al. (Lif-r-pro1 and 2) (Chen et al., 2008; Yamashita et al., 2010). The amplified regions are denoted by the red box. Gels were run after ChIP-PCR on DNA samples collected from total cell input (Input), a negative control with just addition of rabbit immunoglobulin G (IgG), and pull-downs of total STAT3, P300, and SMAD1. Quantification of band intensities is shown below gel images.
(D) Approximately 2 kb regions flanking the binding sites (yellow boxes) were cloned upstream of a luciferase reporter, assayed for activity and relative activity quantified.
Error bars represent SD n = 3 technical replicates for (A) and (B), SE n = 4 (two replicates per two independent experiments) for (D). ∗p < 0.05, one sample two-tailed t test.
Modulation of P300 Affects LIF Responsiveness in EpiSCs and Subsequent Conversion Frequencies
We next asked if decreasing P300 protein levels in EpiSCs would result in a decrease in LIF responsiveness and conversion frequency. siRNA knockdown of P300 using a pool of four siRNA results in the decrease of only LIF receptor transcription in standard EpiSC culture relative to nontransfected controls (Figure 5Ai). On μP, we also observe a decrease in Lif-r transcription to the baseline nonpatterned EpiSCs (Figure 5Aii). Gp130 levels decrease but remain above baseline levels, supporting our earlier observation that LIF and BMP signaling do not fully contribute to transcription of Gp130, thus mitigating the effects of the loss of P300 and suggesting a direct effect of P300 on transcription of Lif-r. Stat3 levels remain higher than baseline levels when in the presence of LIF. These data suggest the maintenance of population-level LIF responsiveness even upon P300 knockdown and subsequent loss of Lif-r transcription. We postulated that could arise for two reasons: incomplete P300 knockdown or the retention of LIF responsiveness in spite of P300 knockdown. To resolve this, we used high-content imaging to costain for P300 and LIF responsiveness (i.e., pSTAT3 after LIF pulse). After P300 knockdown (P300i), cells demonstrated a marked decrease in LIF responsiveness even in the presence of LIF and BMP4 (Figure 5B). Importantly, the cells that retained the capacity to recover LIF responsiveness were those that still retained P300 (Figure 5C), suggesting incomplete knockdown was responsible for cells still remaining LIF responsive. P300-low cells did not exhibit LIF responsiveness in EpiSCs regardless of P300 knockdown (Figure 5C). Conversion frequencies as measured by ALP-positive colonies in 2iL reflected this loss of LIF responsiveness in P300 knockdown cells. Even in LIF+BMP4, a condition that robustly drives conversion, very few colonies emerged (Figure 5D). Taken together, P300 bridges SMAD1 and STAT3 in EpiSCs and acts as a gatekeeper for the recovery of dormant LIF signaling by regulating the transcription of LIF receptor. Regions of high local cell density experience a higher local concentration of BMP and GP130 ligands, thereby increasing LIF responsiveness (Figure 5E). Modulation of P300 levels tunes the responsiveness of cells to their immediate environment and their subsequent decision to undergo reprogramming.
Figure 5.
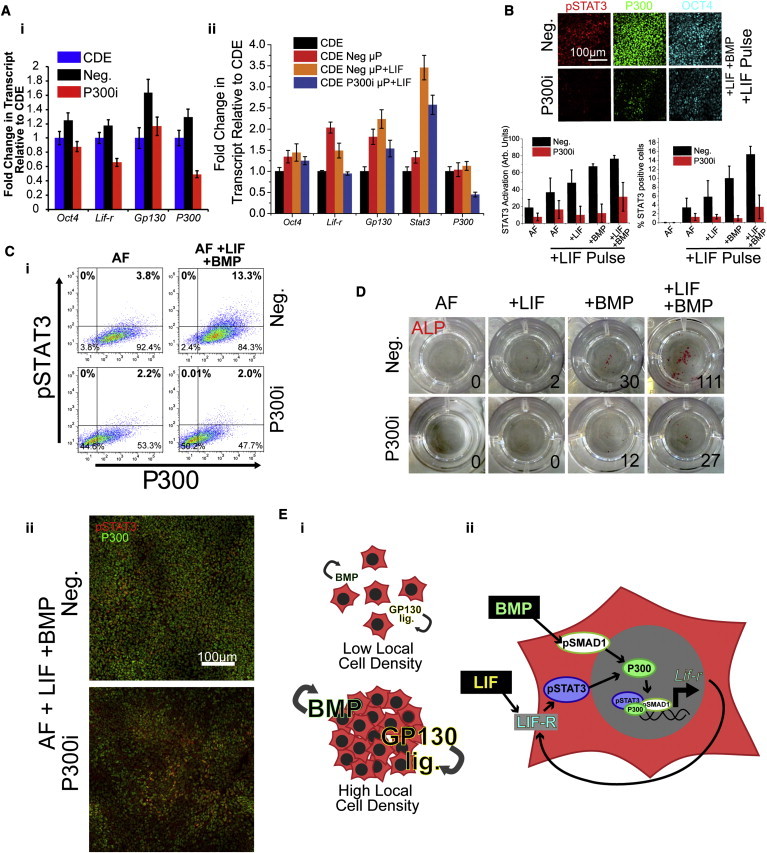
P300 Levels Tune Response to LIF and BMP4 and Adjust LIF Responsiveness and Conversion Frequencies
(A) QRT-PCR on Lif-r, Gp130, and Stat3 after siRNA knockdown with scramble control (Neg.) or a pool of siRNAs specific for P300 (P300i) on (i) non-μP CDEs and (ii) μP CDEs. Transcript levels normalized to siRNA-free CDEs.
(B) LIF responsiveness in EpiSCs was assayed in the presence of LIF and/or BMP after siRNA knockdown of P300 (P300i). Scrambled siRNA was used as the control (Neg.)
(C) (i) Scatterplots of P300i cells versus Neg. cells after treatment with nothing (AF) or LIF and BMP4 followed by a 15 min pulse of LIF to assay for LIF responsiveness. (ii) Representative images of P300i and Neg. cells treated with LIF and BMP4 prior to pulsing with LIF. Cells were costained for pSTAT3 (red) and P300 (green).
(D) ALP colony counts (insets) of knockdown cells in 2iL media following treatment in nothing (AF), LIF, BMP4, or LIF + BMP4.
(E) Schematic of model. (i) Briefly, EpiSCs residing in an area of low local cell density secrete low levels of endogenous GP130 ligands and BMP4. (ii) In regions of high local cell density, whether by μP, MEFs, or in high-confluence culture, endogenous GP130 ligands and BMP4 accumulate to regulate transcription of Lif-r through binding to P300. Endogenous signaling may be recapitulated by addition of LIF and BMP4, and results in increased LIF responsiveness and subsequent increased conversion frequency.
Error bars represent SDs n = 3 for (A) and n = 4 for (B) (both technical replicates, independent wells) all images are representative.
Discussion
An important insight provided by this study is the dominant role of signaling pathway reactivation in driving and stabilizing normally inaccessible transcriptional network changes in pluripotent cells. While mESCs and EpiSCs share a core transcriptional network (CTN), including OCT4, NANOG, and SOX2 (Brons et al., 2007; Tesar et al., 2007), mESCs differ from EpiSCs in their heightened expression of pluripotency-inducing genes. These genes include Klf4 (Takahashi and Yamanaka, 2006), Klf2, Klf5 (Nakagawa et al., 2008), Esrrb (Buganim et al., 2012), Gbx2 (Tai and Ying, 2013), and Tbx3 (Han et al., 2010b), which form components of a peripheral naive network. Combined with the CTN, these transcription factors result in a highly interconnected network robustly stabilizing naive pluripotency (Festuccia et al., 2012; Jiang et al., 2008; Loh et al., 2006; Niwa et al., 2009; van den Berg et al., 2008; Zhang et al., 2008). Importantly, LIF signaling directly regulates many members of the peripheral naive network: Klf4, Tbx3 (Niwa et al., 2009), Klf5 (Bourillot et al., 2009), and Gbx2 (Tai and Ying, 2013). It follows, then, that reactivation of STAT3 via a LIF-independent chimeric receptor is sufficient and dominant in driving reversion of EpiSCs to naive pluripotency (van Oosten et al., 2012; Yang et al., 2010). We have demonstrated that in the absence of genetic modification and in defined conditions, reestablishment of the peripheral transcriptional network and naive pluripotency is limited by responsiveness to LIF signaling pathway components. By engineering the local microenvironment, we have identified a role for BMP signaling in priming LIF signaling responsiveness, ultimately reestablishing a naive pluripotency transcriptional network by exogenous niche engineering.
We observed a role for BMP signaling in increasing EpiSC-to-ESC conversion frequency. BMP has been demonstrated to play a role in somatic cell reprogramming to induced pluripotent stem cells (iPSCs), primarily during the initial postinduction stages (Hamasaki et al., 2012; Samavarchi-Tehrani et al., 2010). Analyses support a role for BMP in accelerating/enhancing the mesenchymal-to-epithelial transition (MET) in these studies. Related to this, EpiSCs are generally thought to be epithelial, as mesendodermal progenitors are the first cells postimplantation to undergo an epithelial-to-mesenchymal transition (EMT) event during gastrulation (Thiery et al., 2009). However, as MET/EMT events involve a gradual cascade of changes in phenotype, we cannot exclude aspects of an MET event occurring (i.e., regulation of MET/EMT markers) in EpiSCs upon treatment with BMP. While it has been postulated that EpiSCs can undergo an MET event (Zhou et al., 2010), it remains an open question if specific events reminiscent of BMP-induced MET play a role in EpiSC conversion.
An important observation from this work is the presence of heterogeneity in signaling responsiveness. Even in the presence of saturating, exogenous LIF and BMP4, EpiSCs demonstrate a distributed response to LIF. At best (i.e., in LIF and BMP4), we observe ∼15% of the total EpiSC population responding to LIF. The subsequent conversion frequency of ∼0.2% in a population that does not fully activate STAT3 is consistent with previously reported conversion frequencies of ∼1%–2% in EpiSCs that fully activate STAT3 (Yang et al., 2010). Specifically, assuming a relatively homogeneous and robust activation of STAT3 in these cells, we expect to observe ∼0.3% conversion in our conditions (i.e., 15% cells responding to LIF × 2% of these cells reprogramming). The relatively small (i.e., ∼1.2- to 1.5-fold) increase in transcription and luciferase activity observed in the total population of EpiSCs is likely due to this heterogeneous response. It remains unclear if this heterogeneity is a product of the oscillatory behavior of signaling proteins/transcription factors or if it is a product of lateral inhibition, whereby a subset of permissive cells respond to LIF and BMP4 to increase LIF responsiveness while simultaneously suppressing the responsiveness of neighboring cells. Additionally, there may exist a threshold of STAT3 activation that is required for EpiSC to mESC conversion. By uniformly increasing LIF responsiveness, the frequency of cells in the population reaching this threshold would concomitantly increase, thereby resulting in a higher frequency of conversion.
While we have excluded PGC formation from EpiSCs in response to BMP induction, suggesting a direct conversion to ESCs, we cannot exclude the possibility that JAK-STAT signaling recovery is a conserved mechanism in both EpiSC conversion and PGC specification from the posterior epiblast. BMP signaling provided by both the developing definitive endoderm and primitive ectoderm creates a niche for emerging PGCs (Ying and Zhao, 2001). Due to many parallels between PGCs and ESCs (Leitch et al., 2013), JAK-STAT recovery may play an important role in PGC specification. We propose that the microenvironment for early PGC cell formation is recapitulated in vitro in EpiSCs; an observation consistent with recent reports of EpiSCs resembling cells of the anterior primitive streak (Kojima et al., 2014). More broadly, we propose that upregulation of JAK-STAT signaling potential by LIF and BMP4 may be a parallel mechanism, in addition to the ascribed role of ID proteins (Ying et al., 2003) and MAPK inhibition (Qi et al., 2004), in maintenance of naive pluripotency. This remains an area of active investigation.
Broadly, this study demonstrates two important things. The first is that downregulation of signaling responsiveness, likely via internalization of cell surface receptors, precedes the formation of normally irreversible (epigenetic) barriers to cell-fate transitions. The second is that engineering the local microenvironment, and the provision of appropriate stem cell niche signals, can lead to conditions that reactivate downregulated signaling pathways (and their target transcriptional networks) to revert cell-fate transitions. Early evidence suggests that these concepts may be generalizable to stem cell-fate-control mechanisms in other niche-containing systems (Brawley and Matunis, 2004; Ritsma et al., 2014; Rompolas et al., 2013).
Experimental Procedures
Cell Culture
EpiB3 EpiSCs were a generous gift from Drs. Janet Rossant and Peter Rugg-Gunn, and 129S2C1a EpiSCs were from Dr. Ludovic Vallier (Brons et al., 2007). EpiSC and CDE lines were cultured in X-vivo base media (Lonza) supplemented with 10 μM β-mercaptoethanol (Sigma), 1 mM minimum essential medium/nonessential amino acids (Invitrogen), 2 mM Glutamax (Invitrogen), 20 ng/ml Activin A (R&D Systems), and 20 ng/ml bFGF (R&D Systems) (AF media). Cells were passaged every 3 days as single cells in TrypLE (Invitrogen) in the presence of the Rho-associated kinase (ROCK) inhibitor (Y27632) (Sigma) and seeded on wells precoated with Matrigel (BD bioscience). LIF (Millipore) and BMP4 (R&D Systems) were used at 10 ng/ml unless otherwise stated.
Micropatterninng and Conversion of EpiSCs
μP was performed as described previously (Peerani et al., 2007) on patterns that were 200 μm in diameter and either 320 or 500 μm in pitch. LIF responsiveness was assayed as described previously (Onishi et al., 2012). Briefly, cells were patterned, treated in respective conditions for 1 or 2 days and starved of cytokines for 4 hr prior to pulsing with 10 ng/ml of LIF for 15 min. Cells were immediately fixed and stained. NOGGIN (R&D Systems) was used at 300 ng/ml, and LDN193189 was used at 3 μM (low solubility) or 0.1 μM (newer, DMSO soluble) (Stemgent).
EpiSCs treated in the various ways were then dissociated using TrypLE (Invitrogen) and plated in 2i media (N2B27 base media with 3 μM CHIR99021 and 1 μM PD0325901 [Reagents Direct]) supplemented with 10 ng/ml of LIF at a density of 2 × 105 cells per one well in a 0.2% gelatin-coated 12-well plate (Corning) (approx. 1.78 × 103 cells/cm3 for non-12-well plates). Cells were allowed to form colonies for ALP staining and quantifying and for picking of clones. Colonies were further dissociated only for expansion and were not dissociated for quantification of frequencies. JAK signaling was inhibited using 2 μM JAK Inhibitor I (Calbiochem).
Alkaline Phosphatase
Alkaline phosphatase staining was performed using the instructions provided in the kit (Vector Laboratories). Cells were fixed in 10% formalin (Sigma) prior to staining.
siRNA Knockdown
SMARTpool siRNA against Smad1 and P300 (Thermo) was transfected into EpiSCs using Dharmafect (Thermo) transfection reagent and mixed in OptiMEM basal media (Life Technologies) (10% dilution of Dharmafect in OptiMEM). Final concentrations of siRNA exposed to cells ranged from 25 to 100 nM.
Generation of Chimeras
Chimeras were produced through aggregation of cell clumps (8–15 cells) of reverted EpiSCs with diploid 2.5 days postcoitum Hsd:ICR (CD-1) embryos, as described previously (Woltjen et al., 2009). Aggregations were made with reverted embryo-derived EpiSC lines, 129S2C1a and EpiB3 cells (129S2C1a-μP and EpiB3-μP) that were treated with LIF and BMP4 (10 ng/ml each) prior to conversion. We transfected these cells with a constitutively expressed β-geo cassette to facilitate visualization of developmental contribution by Lac-Z staining. Embryonic day 10.5 embryos were fixed with 0.25% glutaraldehyde for 30 min, washed in permeabilizing solution (2 mM MgCl2, 0.01% sodium deoxycholate, and 0.02% Nonidet-P40 in PBS) and then incubated with X-gal staining solution (20 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6 and 1 mg/ml X-gal in PBS) for 4 hr at 37 degrees. The lacZ-stained embryos were then paraffin embedded and sectioned.
Chromatin Immunoprecipitation
EpiSCs were grown to ∼85% confluence in a 10 cm cell-culture dish (∼3.5 × 107 cells) and fixed for 10 min in 1% formalin. Cells were lysed in 1% SDS buffer supplemented with protease inhibitors (Roche). The sample was sonicated using a Branson 450 Sonifier at 50% output, 100% duty cycle, for 10 s intervals to shear genomic DNA. Antibodies against total STAT3 (Cell Signaling Technology cat. 4904), total SMAD1 (Cell Signaling Technology, cat. 6944), and P300 (Santa Cruz Biotechnology, cat. sc-585) or normal rabbit immunoglobulin G (Cell Signaling Technology, cat. 2729) were used to pull down DNA. Sheep anti-rabbit immunoglobulin G Dynabeads (Life Technologies) were used to isolate bound DNA. Crosslinks were reversed and DNA was purified using phenol-chloroform extraction. PCR was performed using the primers listed in Table S1.
Luciferase
Regions of interest were PCR amplified using LongAMP Taq Polymerase (New England Biolabs) using primers with restriction sites flanking the 5′ ends. These were then cloned into either a pGL3-Basic vector (Promega) or pGL3-promoter vector (Promega) (only for Lif-r-enh). EpiSCs were transfected with both pGL3-derived constructs and Renilla luciferase (pRL) (Promega) using Lipofectamine 2000 (Life Technologies). The dual-luciferase reporter kit (Promega) was used to assay luciferase activation and was measured using the PheraStar Plus plate reader (BMG Technologies). All results were normalized to Renilla luciferase to account for differences in cell number and transfection efficiencies.
qRT-PCR
qRT-PCR was performed as described before (Onishi et al., 2012). Briefly, RNA was isolated using the QIAquick Miniprep kit (QIAGEN) and reverse transcribed with Superscript III (Life Technologies). PCR was performed on the ABI 7000 using SYBR Master mix (Roche).
Immunocytochemistry
Cells were fixed and stained and immunofluorescence intensity was measured as described previously (Onishi et al., 2012) using the Cellomics HCS platform (Thermo Scientific). Antibodies used were as follows: P300 (IF) (Santa Cruz, cat. sc-585; or Life Technologies, cat. 33-7600), pSTAT3 (IF) (Cell Signaling Technology, #9131), pSMAD1 (IF) (Cell Signaling Technology, #9516), OCT4 (IF) (BD Biosciences, cat. #611203), KLF4 (IF) (R&D Systems, AF3158), SOX2 (IF) (R&D Systems, MAB2018), Alkaline Phosphatase (IF) (R&D Systems MAB29091), STAT3 (ChIP) (Cell Signaling Technology, cat. #4904), SMAD1 (ChIP) (Cell Signaling Technology, #6944), and P300 (ChIP) (Santa Cruz, cat. sc-585).
Author Contributions
K.O. and P.W.Z. conceived the experiments and analyzed the data. K.O. and P.D.T. performed the experiments. K.O., P.D.T., A.N., and P.W.Z. wrote the paper.
Acknowledgments
We are grateful to Drs. Peter Rugg-gunn, Janet Rossant, and Ludovic Vallier for cell lines. We acknowledge the Canadian Institutes of Health Research (grant #MOP-57885) and the Natural Sciences and Engineering Research Council of Canada (PWZ Discovery) for funding of this work. P.W.Z. is the Canada Research Chair in Stem Cell Bioengineering.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Bao S., Tang F., Li X., Hayashi K., Gillich A., Lao K., Surani M.A. Epigenetic reversion of post-implantation epiblast to pluripotent embryonic stem cells. Nature. 2009;461:1292–1295. doi: 10.1038/nature08534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernemann C., Greber B., Ko K., Sterneckert J., Han D.W., Araúzo-Bravo M.J., Schöler H.R. Distinct developmental ground states of epiblast stem cell lines determine different pluripotency features. Stem Cells. 2011;29:1496–1503. doi: 10.1002/stem.709. [DOI] [PubMed] [Google Scholar]
- Bourillot P.Y., Aksoy I., Schreiber V., Wianny F., Schulz H., Hummel O., Hubner N., Savatier P. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells. 2009;27:1760–1771. doi: 10.1002/stem.110. [DOI] [PubMed] [Google Scholar]
- Brawley C., Matunis E. Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science. 2004;304:1331–1334. doi: 10.1126/science.1097676. [DOI] [PubMed] [Google Scholar]
- Brons I.G.M., Smithers L.E., Trotter M.W.B., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S.M., Howlett S.K., Clarkson A., Ahrlund-Richter L., Pedersen R.A., Vallier L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Cheng A.W., Itskovich E., Markoulaki S., Ganz K., Klemm S.L., van Oudenaarden A., Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Davey R.E., Onishi K., Mahdavi A., Zandstra P.W. LIF-mediated control of embryonic stem cell self-renewal emerges due to an autoregulatory loop. FASEB J. 2007;21:2020–2032. doi: 10.1096/fj.06-7852com. [DOI] [PubMed] [Google Scholar]
- Festuccia N., Osorno R., Halbritter F., Karwacki-Neisius V., Navarro P., Colby D., Wong F., Yates A., Tomlinson S.R., Chambers I. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell. 2012;11:477–490. doi: 10.1016/j.stem.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillich A., Bao S., Grabole N., Hayashi K., Trotter M.W., Pasque V., Magnúsdóttir E., Surani M.A. Epiblast stem cell-based system reveals reprogramming synergy of germline factors. Cell Stem Cell. 2012;10:425–439. doi: 10.1016/j.stem.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber B., Wu G., Bernemann C., Joo J.Y., Han D.W., Ko K., Tapia N., Sabour D., Sterneckert J., Tesar P., Schöler H.R. Conserved and divergent roles of FGF signaling in mouse epiblast stem cells and human embryonic stem cells. Cell Stem Cell. 2010;6:215–226. doi: 10.1016/j.stem.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Guo G., Smith A. A genome-wide screen in EpiSCs identifies Nr5a nuclear receptors as potent inducers of ground state pluripotency. Development. 2010;137:3185–3192. doi: 10.1242/dev.052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., Yang J., Nichols J., Hall J.S., Eyres I., Mansfield W., Smith A. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development. 2009;136:1063–1069. doi: 10.1242/dev.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J., Guo G., Wray J., Eyres I., Nichols J., Grotewold L., Morfopoulou S., Humphreys P., Mansfield W., Walker R. Oct4 and LIF/Stat3 additively induce Krüppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell. 2009;5:597–609. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Hamasaki M., Hashizume Y., Yamada Y., Katayama T., Hohjoh H., Fusaki N., Nakashima Y., Furuya H., Haga N., Takami Y., Era T. Pathogenic mutation of ALK2 inhibits induced pluripotent stem cell reprogramming and maintenance: mechanisms of reprogramming and strategy for drug identification. Stem Cells. 2012;30:2437–2449. doi: 10.1002/stem.1221. [DOI] [PubMed] [Google Scholar]
- Han D.W., Tapia N., Joo J.Y., Greber B., Araúzo-Bravo M.J., Bernemann C., Ko K., Wu G., Stehling M., Do J.T., Schöler H.R. Epiblast stem cell subpopulations represent mouse embryos of distinct pregastrulation stages. Cell. 2010;143:617–627. doi: 10.1016/j.cell.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Han J., Yuan P., Yang H., Zhang J., Soh B.S., Li P., Lim S.L., Cao S., Tay J., Orlov Y.L. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature. 2010;463:1096–1100. doi: 10.1038/nature08735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J., Markoulaki S., Mitalipova M., Cheng A.W., Cassady J.P., Staerk J., Carey B.W., Lengner C.J., Foreman R., Love J. Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell. 2009;4:513–524. doi: 10.1016/j.stem.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Surani M.A. Self-renewing epiblast stem cells exhibit continual delineation of germ cells with epigenetic reprogramming in vitro. Development. 2009;136:3549–3556. doi: 10.1242/dev.037747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Ohta H., Kurimoto K., Aramaki S., Saitou M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011;146:519–532. doi: 10.1016/j.cell.2011.06.052. [DOI] [PubMed] [Google Scholar]
- He F., Ge W., Martinowich K., Becker-Catania S., Coskun V., Zhu W., Wu H., Castro D., Guillemot F., Fan G. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Chan Y.S., Loh Y.H., Cai J., Tong G.Q., Lim C.A., Robson P., Zhong S., Ng H.H. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- Kojima Y., Kaufman-Francis K., Studdert J.B., Steiner K.A., Power M.D., Loebel D.A., Jones V., Hor A., de Alencastro G., Logan G.J. The transcriptional and functional properties of mouse epiblast stem cells resemble the anterior primitive streak. Cell Stem Cell. 2014;14:107–120. doi: 10.1016/j.stem.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Leitch H.G., Nichols J., Humphreys P., Mulas C., Martello G., Lee C., Jones K., Surani M.A., Smith A. Rebuilding pluripotency from primordial germ cells. Stem Cell Reports. 2013;1:66–78. doi: 10.1016/j.stemcr.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Nakagawa M., Koyanagi M., Tanabe K., Takahashi K., Ichisaka T., Aoi T., Okita K., Mochiduki Y., Takizawa N., Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Yanagisawa M., Arakawa H., Kimura N., Hisatsune T., Kawabata M., Miyazono K., Taga T. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284:479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Niwa H., Ogawa K., Shimosato D., Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- Onishi K., Tonge P.D., Nagy A., Zandstra P.W. Microenvironment-mediated reversion of epiblast stem cells by reactivation of repressed JAK-STAT signaling. Integr Biol (Camb) 2012;4:1367–1376. doi: 10.1039/c2ib20098h. [DOI] [PubMed] [Google Scholar]
- Peerani R., Rao B.M., Bauwens C., Yin T., Wood G.A., Nagy A., Kumacheva E., Zandstra P.W. Niche-mediated control of human embryonic stem cell self-renewal and differentiation. EMBO J. 2007;26:4744–4755. doi: 10.1038/sj.emboj.7601896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Li T.G., Hao J., Hu J., Wang J., Simmons H., Miura S., Mishina Y., Zhao G.Q. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA. 2004;101:6027–6032. doi: 10.1073/pnas.0401367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritsma L., Ellenbroek S.I., Zomer A., Snippert H.J., de Sauvage F.J., Simons B.D., Clevers H., van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature. 2014;507:362–365. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompolas P., Mesa K.R., Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502:513–518. doi: 10.1038/nature12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P., Golipour A., David L., Sung H.K., Beyer T.A., Datti A., Woltjen K., Nagy A., Wrana J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Sasaki N., Shinomi M., Hirano K., Ui-Tei K., Nishihara S. LacdiNAc (GalNAcβ1-4GlcNAc) contributes to self-renewal of mouse embryonic stem cells by regulating leukemia inhibitory factor/STAT3 signaling. Stem Cells. 2011;29:641–650. doi: 10.1002/stem.615. [DOI] [PubMed] [Google Scholar]
- Tai C.I., Ying Q.L. Gbx2, a LIF/Stat3 target, promotes reprogramming to and retention of the pluripotent ground state. J. Cell Sci. 2013;126:1093–1098. doi: 10.1242/jcs.118273. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tesar P.J., Chenoweth J.G., Brook F.A., Davies T.J., Evans E.P., Mack D.L., Gardner R.L., McKay R.D.G. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Vallier L., Touboul T., Chng Z., Brimpari M., Hannan N., Millan E., Smithers L.E., Trotter M., Rugg-Gunn P., Weber A., Pedersen R.A. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS ONE. 2009;4:e6082. doi: 10.1371/journal.pone.0006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg D.L., Zhang W., Yates A., Engelen E., Takacs K., Bezstarosti K., Demmers J., Chambers I., Poot R.A. Estrogen-related receptor beta interacts with Oct4 to positively regulate Nanog gene expression. Mol. Cell. Biol. 2008;28:5986–5995. doi: 10.1128/MCB.00301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten A.L., Costa Y., Smith A., Silva J.C. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nat Commun. 2012;3:817. doi: 10.1038/ncomms1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woltjen K., Michael I.P., Mohseni P., Desai R., Mileikovsky M., Hämäläinen R., Cowling R., Wang W., Liu P., Gertsenstein M. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R., Wakaguri H., Sugano S., Suzuki Y., Nakai K. DBTSS provides a tissue specific dynamic view of Transcription Start Sites. Nucleic Acids Res. 2010;38(Database issue):D98–D104. doi: 10.1093/nar/gkp1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., van Oosten A.L., Theunissen T.W., Guo G., Silva J.C., Smith A. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell Stem Cell. 2010;7:319–328. doi: 10.1016/j.stem.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y., Zhao G.Q. Cooperation of endoderm-derived BMP2 and extraembryonic ectoderm-derived BMP4 in primordial germ cell generation in the mouse. Dev. Biol. 2001;232:484–492. doi: 10.1006/dbio.2001.0173. [DOI] [PubMed] [Google Scholar]
- Ying Q.L., Nichols J., Chambers I., Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhang J., Wang T., Esteban M.A., Pei D. Esrrb activates Oct4 transcription and sustains self-renewal and pluripotency in embryonic stem cells. J. Biol. Chem. 2008;283:35825–35833. doi: 10.1074/jbc.M803481200. [DOI] [PubMed] [Google Scholar]
- Zhou H., Li W., Zhu S., Joo J.Y., Do J.T., Xiong W., Kim J.B., Zhang K., Schöler H.R., Ding S. Conversion of mouse epiblast stem cells to an earlier pluripotency state by small molecules. J. Biol. Chem. 2010;285:29676–29680. doi: 10.1074/jbc.C110.150599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.