

Summary
In this study, human embryonic stem cell-derived hepatocytes (hESC-Heps) were investigated for their ability to support hepatitis C virus (HCV) infection and replication. hESC-Heps were capable of supporting the full viral life cycle, including the release of infectious virions. Although supportive, hESC-Hep viral infection levels were not as great as those observed in Huh7 cells. We reasoned that innate immune responses in hESC-Heps may lead to the low level of infection and replication. Upon further investigation, we identified a strong type III interferon response in hESC-Heps that was triggered by HCV. Interestingly, specific inhibition of the JAK/STAT signaling pathway led to an increase in HCV infection and replication in hESC-Heps. Of note, the interferon response was not evident in Huh7 cells. In summary, we have established a robust cell-based system that allows the in-depth study of virus-host interactions in vitro.
Highlights
-
•
hESC-derived hepatocytes support hepatitis C virus (HCV) infection and replication
-
•
hESC-derived hepatocytes activate innate immunity in response to HCV infection
-
•
Inhibition of JAK/STAT signaling improves HCV infection and replication
Human embryonic stem cell-derived hepatocytes (hESC-Heps) were investigated for their ability to support hepatitis C virus (HCV) life cycle. Although supportive, hESC-Hep viral infection levels were not as great as those observed in Huh7 cells. Upon further investigation, Hay, Patel, and colleagyes identified a strong type III interferon response in hESC-Heps that was not evident in Huh7 cells.
Introduction
Hepatitis C virus (HCV) infects an estimated 2%–3% of the world population and is a major cause of liver disease and cancer. It is estimated that more than 350,000 people die of the HCV-related liver disease each year (Te and Jensen, 2010; Yang and Roberts, 2010). Although the efficacy of current treatments has improved considerably, the high genetic variation of the virus still poses significant issues. Therefore, to develop new targets for effective therapy, it is necessary to gain greater understanding of the processes that control viral infection, replication, and ultimately pathogenesis.
The organ primarily affected by HCV is the liver. HCV entry into target cells occurs via receptor-mediated endocytosis and fusion with intracellular membranes. This process requires multiple attachment and entry factors. Among those, CD81, scavenger receptor class B type 1 (SR-B1), claudin 1, and occludin play a critical role (Evans et al., 2007; Pileri et al., 1998; Ploss et al., 2009; Scarselli et al., 2002). Postviral infection, the host innate immune system is the first line of defense. Human hepatocytes mount their initial immune response, producing interferon (IFN) (Horner and Gale, 2013; Kotenko et al., 2003; Takeuchi and Akira, 2009). IFNs are released from the infected cells and serve to reduce viral replication and spread (Dickensheets et al., 2013).
In order to limit the persistence, and therefore the pathology associated with HCV, it is imperative that we develop a better understanding of virus-host interactions. Cell-based models that support HCV propagation have provided the field with enabling technology. Although enabling, current models possess significant drawbacks, including diminished innate immunity (Foy et al., 2005). Therefore, if we are to gain a better understanding of HCV life cycle and associated pathogenesis, biologically relevant model systems, which more closely mimic human physiology, must be developed. For this reason, primary human hepatocytes (PHHs) have been employed. However, their scarcity, inconsistency, and rapid dedifferentiation in culture impede their widespread deployment.
The delivery of human hepatocytes, from a renewable source, is therefore an attractive strategy to bypass the issues associated with primary material (Sun et al., 2013; Hay, 2013). Of note, several reports have demonstrated the potential of pluripotent stem cells to deliver functional hepatocytes (Cai et al., 2007, Duan et al., 2007, Hay et al., 2008, 2011; Medine et al., 2013; Si-Tayeb et al., 2010; Sullivan et al., 2010; Szkolnicka et al., 2014; Zhou et al., 2012; Lucendo-Villarin et al., 2012). Most recently, stem cell-derived hepatocytes have been used to support HCV infection (Roelandt et al., 2012; Schwartz et al., 2012; Wu et al., 2012); however, the host innate immune response has not yet been studied in detail.
To study this in detail, we employed a robust and serum-free hepatocyte differentiation procedure (Szkolnicka et al., 2014). Human embryonic stem cells were efficiently differentiated toward the hepatocyte lineage. Importantly, those cells expressed critical viral receptors, supported the full life cycle of HCV and exhibited a “tunable” type III interferon response, which was not intact in Huh7s. Therefore, human embryonic stem cell-derived hepatocytes (hESC-Heps) represent an important, defined, and renewable model system with which to study HCV.
Results
Robust Hepatocyte Differentiation from Pluripotent Stem Cells
hESCs were cultured and differentiated using previously described conditions (Szkolnicka et al., 2014). In line with morphological changes (Figure 1A), we observed changes in gene expression confirming hepatocyte commitment. OCT4 expression was not detected in stem cell-derived hepatocytes (0%). In contrast, albumin, HNF4α and E-cadherin were expressed in 87% (±5%), 90% (±4%), and 92% (±2%) of cells, respectively (Figure 1B). Furthermore, stem cell-derived hepatocytes exhibited liver specific function. This peaked at day 19 with the greatest cytochrome P450 3A (CYP3A) and cytochrome P450 1A2 (CYP1A2) activities detected (Figure 1C). These data demonstrate the robust delivery of hESC-Heps, which were suitable in character for further modeling studies.
Figure 1.
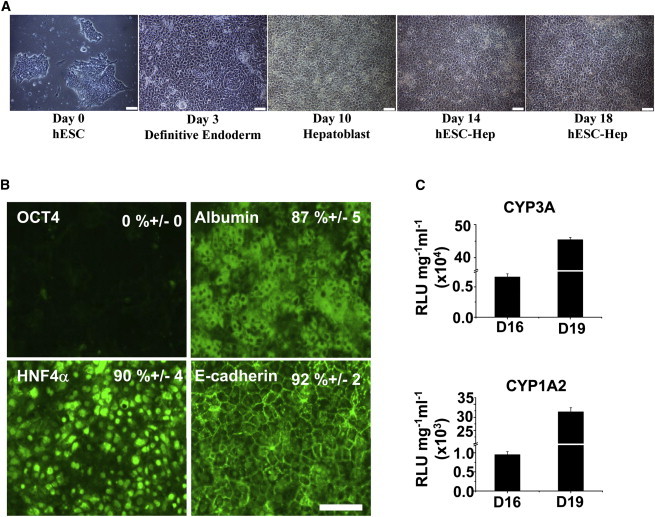
Hepatocyte Differentiation from Human Embryonic Stem Cells
(A) Morphologic change of hESCs to hESC-Heps during cellular differentiation. Scale bar, 100 μm.
(B) At day 19 hESC-Heps were fixed and immunostained for the hepatic markers Albumin/HNF4a/E-cadherin and hESC marker OCT4. Scale bar, 100 μm.
(C) Following 16 and 19 days of differentiation, hESC-Heps exhibited increasing CYP3A4 and CYP1A2 metabolic activity, which were measured using p450-Glo systems (Promega). Relative luminescence unit (RLU) values were normalized to protein (mg) and medium volume (ml), and shown as the mean ± SD. Error bars represent the SD of the mean. n = 6, biological replicates.
hESC-Heps Express the Essential HCV Entry Factors
hESCs and hESC-Heps were fixed and immunostained for the major HCV host cell entry factors; CD81, SR-B1, claudin-1, and occludin (Evans et al., 2007; Pileri et al., 1998; Ploss et al., 2009; Scarselli et al., 2002). Claudin-1 expression was not detected in hESCs, whereas it was abundant in hESC-Heps (90% ± 5%). The other viral entry factors were expressed in both hESCs and hESC-Heps, with levels increased in hESC-Heps. Expression of occludin, CD81, SR-B1, was estimated at 92% (±4%), 90% (±5%), and 84% (±4%), respectively (Figure 2A). These results were confirmed by western blotting and quantitative PCR (qPCR) (Figures 2B and 2C), suggesting that hESC-Heps would support HCV entry.
Figure 2.
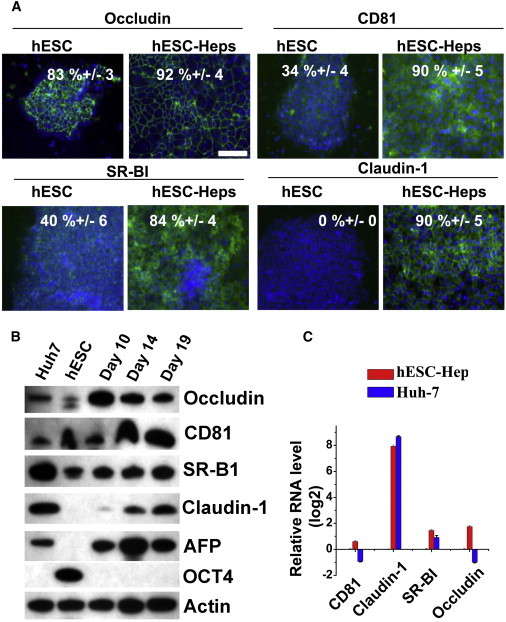
hESC-Heps Express the Essential HCV Entry Factors
(A) Immunostaining of occludin, CD81, SR-BI, and claudin-1 in hESCs and hESC-Heps. Scale bar, 100 μm.
(B) Western blotting for HCV entry factors (occludin, CD81, SR-BI, and claudin-1), stem cell (OCT4), and cell differentiation (AFP) markers in Huh7, hESCs, and hESC-Heps, respectively.
(C) The expression level of HCV entry factors in hESC-Heps and Huh7, relative to hESC, was determined by qPCR.
Error bars represent the SD of the mean. n = 3, biological replicates. See also Tables S1 and S2.
hESC-Heps Support HCV Infection and Infectious Virion Production
To test our hypothesis, HCV strain JFH-1 (Wakita et al., 2005) was selected to infect hESC-Heps. This strain replicates efficiently in Huh7 cells. At 96 hr postinfection, HCV nonstructural protein NS5A and core protein expression were detected in hESC-Heps by immunostaining demonstrating viral entry (Figure 3A). Infected foci were visualized by NS5A staining, and the total focus number per well of 12-well plate (3.8 cm2) were calculated. The size of infected foci was measured as average number of cells per focus (Calland et al., 2012). The average number of infected foci per well and the size of the infected foci were 242.5 ± 82.3 and 4.2 ± 1.8, respectively. To test for viral entry, we also examined HCV RNA levels in the presence or absence of a HCV polymerase inhibitor (2′CMA). The HCV RNA levels relative to HCV with 2′CMA group were measured by qPCR. In line with previous studies, 2′CMA inhibited HCV replication in hESC-Heps indicating assay specificity (Figure 3B). In addition to infection and viral genome replication, we were interested in studying virus assembly and secretion. To assess this, the presence and infectious titer of the virus progeny from hESC-Heps were calculated by performing a focus-forming unit (FFU) assay in Huh7 cells (Figure 3C and Figure S2 available online). hESC-Heps were capable of supporting HCV life cycle, including the release of new infectious virions. However, we noted that the level of infection and replication was markedly less than that detected in Huh7 cells. In support of this, we found that HCV replication plateaued in hESC-Heps by 48 hr, but this was not observed in Huh7 cells, even at the 72 hr time point (Figure 3D). To test if the medium of HCV-infected hESC-Heps carried antiviral activity, we incubated the reporter cell line Huh7-J20 that had been preinfected with HCVcc, with hESC-Hep supernatant collected at various times postinfection. Of note, there was a significant inhibition of HCV replication in Huh7-J20 cells when they were incubated with the medium collected from hESC-Heps (Figure 3E). These data strongly suggest the presence of key factor(s) in the infected hESC-Heps supernatants that likely accounted for the inhibition of viral replication observed in the Huh7-J20 cells.
Figure 3.
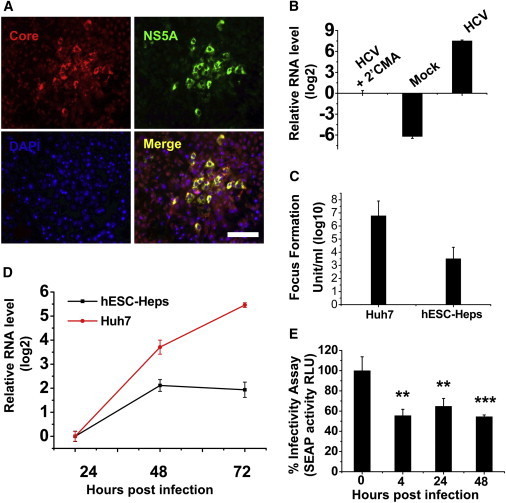
HCV Infection of hESC-Heps and Huh7
(A) Day 19 hESC-Heps were exposed to JFH-1-based HCVcc. Three days postinfection, cells were double stained for HCV core (red) and NS5A (green). Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm.
(B) hESC-Heps were either mock infected or infected with HCV in the presence or absence of 2′CMA. HCV RNA levels, relative to HCV with 2′CMA group, were detected by qPCR.
(C) At 72 hr postinfection, the infectious virus yield in the medium of infected Huh7 or hESC-Heps was determined on naive Huh7 cells by focus-forming assay, and the values presented as focus-forming units (FFU) per ml (log10).
(D) Comparison of HCV RNA levels in infected cells by qPCR. Fold change relative to that of 24 hr postinfection in hESC-Heps (black line) or Huh7 (red line) cells was calculated.
(E) Antiviral activity of conditioned medium from hESC-Heps infected by HCV. Huh7-J20 reporter cells preinfected with HCV for 3 hr were incubated for 48 hr with the supernatants from hESC-Heps collected at 0, 4, 24, or 48 hr postinfection with HCV. The effect of hESC-Hep supernatants on virus infection in Huh7-J20 cells was determined by measuring SEAP activity in the medium, which correlates directly with virus replication. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with the group of 0 hr postinfection.
Error bars represent the SD of the mean. n = 3, biological replicates. See also Figure S2 and Tables S1 and S2.
HCV Infection of hESC-Heps Activates a Type III IFN Response
Given the important role that the IFN response plays in defense against microbial and viral infection, we opted to study gene expression of key immune mediators. hESC-Heps were infected with HCVcc for 4 hr, before replacing with fresh cell medium. Seventy-two hours postinfection, total RNA was prepared and profiled using an IFN-stimulated gene (ISG) PCR array. In response to HCV infection, hESC-Hep gene expression was representative of a type III IFN response (Table 1). These findings were corroborated by qPCR (Figure S3) and strongly implicated interleukin (IL)-29, the dominant type III IFN produced by primary human and primate hepatocytes in response to hepatitis C virus infection (Park et al., 2012).
Table 1.
Increase of IFNs and ISG Expression in Stem Cell-Derived Hepatocytes after Treatment with IL-29 or HCV
| Gene Symbol | RefSeq | Fold Increase |
Gene Description | |
|---|---|---|---|---|
| IL-29 Treatment | HCV Treatment | |||
| IFIT1 | NM_001548 | 2,488.73 | 4.64 | IFN-induced protein with tetratricopeptide repeats 1 |
| IFI27 | NM_005532 | 927.55 | 5.91 | alpha-inducible protein 27 |
| IFI44 | NM_006417 | 389.86 | 6.64 | IFN-induced protein 44 |
| IFI44L | NM_006820 | 216.25 | 2.54 | IFN-induced protein 44-like |
| IFIT2 | NM_001547 | 152.75 | 3.41 | IFN-induced protein with tetratricopeptide repeats 2 |
| IFI6 | NM_002038 | 111.52 | 5.12 | IFN, alpha-inducible protein 6 |
| CXCL10 | NM_001565 | 110.63 | 4.32 | chemokine (C-X-C motif) ligand 10 |
| OAS1 | NM_002534 | 88.16 | 5.73 | 2′-5′-oligoadenylate synthetase 1, 40/46 kDa |
| ISG15 | NM_005101 | 52.36 | 4.35 | ISG15 ubiquitin-like modifier |
| IFIH1 | NM_022168 | 51.89 | 4.19 | IFN induced with helicase C domain 1 |
| IFITM1 | NM_003641 | 20.27 | 6.29 | IFN-induced transmembrane protein 1 (9–27) |
| IRF7 | NM_001572 | 18.56 | 12.51 | IFN regulatory factor 7 |
| IFNA8 | NM_002170 | 16.90 | 6.49 | IFN, alpha 8 |
| IFI35 | NM_005533 | 11.27 | 5.65 | IFN-induced protein 35 |
| MX1 | NM_002462 | 11.26 | 4.66 | myxovirus (influenza virus) resistance 1, IFN-inducible protein p78 (mouse) |
| IL-29 | NM_172140 | 10.99 | 7.10 | IL-29 (IFN, lambda 1) |
See also Table S3.
To confirm that IL-29 was eliciting a strong activation of ISGs in our model, we incubated hESC-Heps with recombinant IL-29 (Table 1). Recombinant IL-29 activated the JAK/STAT pathway in hESC-Heps (Figure 4A), leading to the phosphorylation of STAT1 and induction of ISG expression (IFIT1, MX1, OAS1, ISG15, CXCL10, IRF9, IRF7, IRF1, and IRF2) in line with the literature (Figures 4A and 4B; Table 1). In contrast to hESC-Heps, very little or no induction of RIG-I, ISG15, CXCL10, CXCL11, and IFIT1 gene expression was observed in Huh7 cells (Figure 4B).
Figure 4.
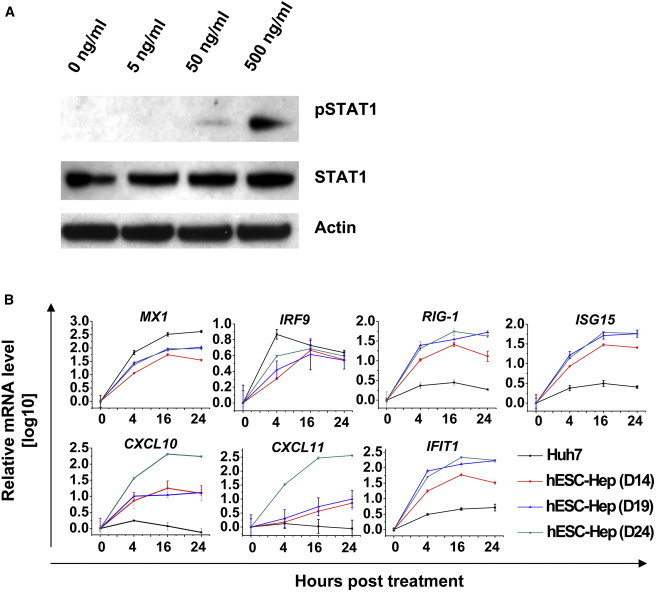
Induction of Innate Immune Response in hESC-Heps by IL-29
(A) hESC-Heps were stimulated with different concentrations of IL-29. Proteins were extracted, and phosphorylated STAT1 and loading control, β-actin, were detected by western blotting.
(B) hESC-Heps or Huh7 cells were treated with 100 ng/ml IL-29 for the different times as indicated. Total RNA was isolated and reverse transcribed, and then qPCR was performed for the named genes.
Error bars represent the SD of the mean. n = 3, biological replicates. See also Tables S1 and S2.
Poly I:C Induces IL-29 and A Type III IFN Response in hESC-Heps
To further establish the role of IFN response in our cell-based model, hESC-Heps were transfected with polyinosinic/polycytidylic acid (polyI:C) to mimic intracellular viral RNA. Twenty-four hours posttransfection, we observed upregulation of both type I (IFN-α and IFN-β) and type III IFNs (IL-28 and IL-29) by PCR or ELISA (Figures 5A and 5B). Of note, IRF9, a JAK/STAT pathway enhancer (Samuel, 2001), and RIG-1, a detector of extraneous double-stranded RNAs (dsRNAs) responsible for IFNβ production (Kato et al., 2006), were also upregulated. Additionally, inflammatory chemokines (CXCL10 and CXCL11) and antiviral ISGs (IFIT1, MX1, and ISG15) were also strongly stimulated by dsRNA (Figure 5A). To study the effect of host factors generated in hESC-Heps after polyI:C treatment on viral replication, supernatant collected from transfected hESC-Heps were transferred to the infected Huh7-J20 reporter cells. In support of our previous experiments (Figure 3E), the supernatants of polyI:C-treated hESC-Heps possessed antiviral agents, including IL-29 (Figure 5B), strongly inhibiting virus replication (Figure 5C).
Figure 5.
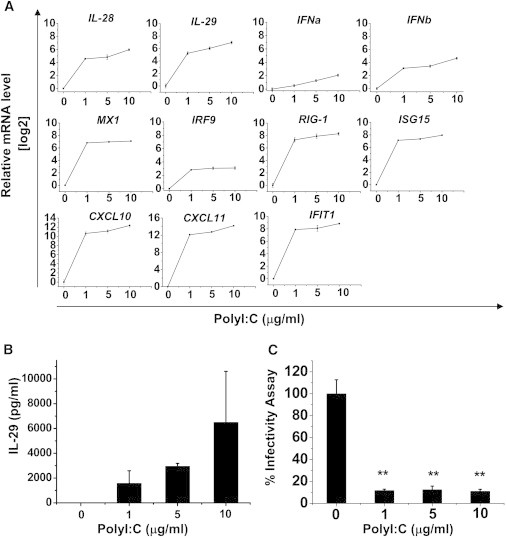
ISG Response of hESC-Heps to dsRNA
(A) IFN and ISG mRNA were determined by qPCR in polyI:C relative to mock-transfected hESC-Heps.
(B) IL29 secretion in polyI:C-treated hESC-Heps was measured by ELISA.
(C) Antiviral activity of conditioned medium (CM) from polyI:C-treated hESC-Heps cells. Huh7-J20 reporter cells were first infected with HCVcc for 3 hr and then they were incubated with CM from hESC-Heps treated with 0, 1, 5, or 10 μg/ml polyI:C. At 72 hr postincubation SEAP activity in the medium, which correlates directly with viral RNA replication, was measured. The SEAP levels are presented as relative light units (RLUs). ∗∗p < 0.01 compared with the group treated with 0 μg/ml polyI:C.
Error bars represent the SD of the mean. n = 3, biological replicates. See also Tables S1 and S2.
Inhibition of JAK/STAT Pathway Promotes HCV Replication in hESC-Heps
To improve viral infection and replication in hESC-Heps, we hypothesized that downregulation of the IFN response was necessary. To test this hypothesis, we pretreated hESC-Heps with JAK/STAT inhibitor (JAK Inhibitor I; 10 μM) for 1 hr prior to infection. HCV replication was significantly increased in JAK/STAT-inhibitor-treated hESC-Heps (Figure 6). We observed that the infected foci were larger in hESC-Heps treated with a JAK/STAT inhibitor (Figure 6A). In the hESC-Heps without inhibitor pretreatment, the total number of infected foci was 284.3 ± 94.5 per well of 12-well plate, and the average size of infected foci was 3.8 ± 3.1. In contrast, hESC-Heps pretreated with the inhibitor, the number of infected foci was 241.2 ± 77.5, and the average size of infected foci was 14.3 ± 5.8. In line with this observation, HCV RNA level in JAK/STAT-inhibitor-treated hESC-Heps was significantly increased (Figure 6B). Moreover, we also detected increases (>2-fold) in ISG expression (IFI27, IFI44, IFI44L, CXCL10, ISG15, IFIH1, IFITM1, IFNA8) in cells treated with the JAK/STAT inhibitor and following infection (Table S4).
Figure 6.
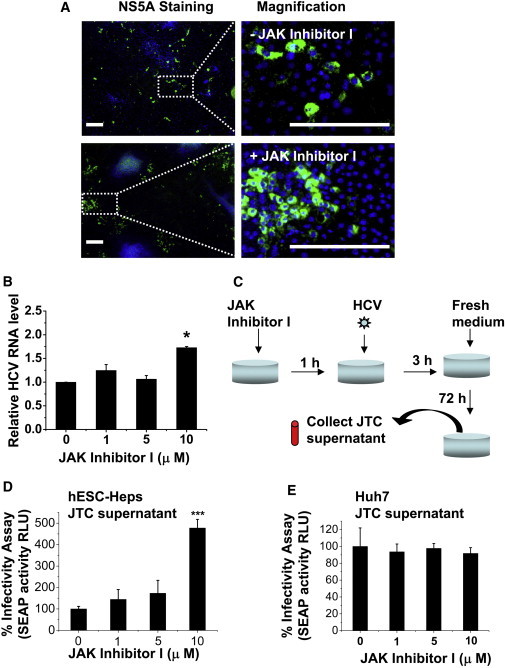
JAK Inhibitor I Improves HCV Infectivity in hESC-Heps
(A) hESC-Heps pretreated (bottom panel) or not (top panel) with 10 μM JAK inhibitor I were infected with HCVcc. At 3 days postinfection, cells were immunostained for viral NS5A antigen (green) and counterstained with DAPI (blue). Scale bar, 200 μm.
(B) hESC-Heps were pretreated with 0, 1, 5, or 10 μM JAK inhibitor I before infection. HCV RNA levels in infected hESC-Heps were measured by qPCR. The results are presented as fold change of HCV RNA level relative to that of mock-treated cells.
(C) Generation of JAK-inhibitor-I-treated conditioned medium (JTC). hESC-Heps or Huh7 were pretreated for 1 hr with 0, 1, 5, and 10 μM JAK inhibitor I, respectively, before infection with HCVcc and the JTC at 72 hr postinfection was collected.
(D) JTC of hESC-Heps improved HCV infectivity in human hepatoma cells. Huh7-J20 reporter cells were infected in advance by HCV for 3 hr, washed with PBS, and then incubated with JTC of hESC-Heps for 72 hr. The virus infectivity levels were determined by measuring SEAP activity in the medium.
(E) JTC of Huh7 has no effect on HCV infectivity in human hepatoma cells. Huh7-J20 reporter cells were infected in advance by HCV for 3 hr, washed with PBS, and then incubated with Huh7 JTC for 72 hr. The virus infectivity was determined as described in (D).
∗p < 0.05, ∗∗∗p < 0.001 compared with the group treated with 0 μM JAK inhibitor I. Error bars represent the SD of the mean. n = 3, biological replicates.
To further confirm that JAK/STAT inhibitor treatment facilitated HCV replication, we collected the supernatants from JAK/STAT-inhibitor-treated hESC-Heps, Huh7, or control cells and examined their effect on HCV replication in the Huh7-J20 reporter cell line (Figure 6C). Strikingly, only supernatants collected from JAK/STAT-inhibitor-treated hESC-Heps (Figure 6D), not Huh7s (Figure 6E), significantly increased HCV replication.
Discussion
Pluripotent stem cells are scalable and retain the ability to form every cell type in the human body. The ability to derive human soma in limitless amounts offers great possibilities for regenerative medicine and cell-based modeling. In these studies, we used hESCs to derive human hepatocytes. hESCs were differentiated using established procedures (Szkolnicka et al., 2014). The derivative cells displayed stable hepatocyte function (Figure 1), expressed the main entry factors for HCV (Da Costa et al., 2012; Evans et al., 2007) (Figure 2), and supported full virus life cycle, including the release of infectious progeny (Figures 3, 4, 5, and 6).
Although our model supported viral life cycle, there were key elements of the system that required more detailed attention. Despite robust infection, hESC-Heps and primary hepatocytes produce less infectious virions than Huh7 line (Liang et al., 2009; Ploss et al., 2010; Podevin et al., 2010; Roelandt et al., 2012; Wu et al., 2012). We hypothesized the lesser infection of hESC-Heps may be due to a robust induction of cellular immunity. Indeed, after HCV infection, hESC-Heps demonstrated strong induction of IL-29, followed by IFN-stimulated gene (ISG) expression. This was lesser in Huh7 cells and is likely attributable to defects in retinoic acid-inducible gene 1 (RIG-1) pathway (Sumpter et al., 2005). These observations were tested extensively in vitro, and further supported by studies in which extraneous RNA was introduced to the cells to stimulate the IFN response (Park et al., 2012).
Following the demonstration that the IFN response was intact, we sought to alter the dynamics of this system to see if we could improve viral infection and replication. In these studies, we chose one of the major signaling pathways effecting the IFN response. Through the use of a JAK/STAT pathway inhibitor, it was possible to attenuate the hESC-Hep innate immune response. In line with reduced JAK/STAT activity, hESC-Heps displayed enhanced HCV replication, which was most likely attributable to cell-to-cell transmission of virus. Notably, innate immunity was not intact in the Huh7 line, highlighting the need to develop new models of HCV biology, which more accurately reflect virus-host interactions.
In conclusion, although infection in hESC-Heps has been established, it is relatively low (Roelandt et al., 2012; Wu et al., 2012). Prior to these studies, the reason for this had proved elusive. We provide evidence that by modulating the JAK/STAT pathway and the downstream IFN response, hESC-Hep infection and subsequent replication is “tunable.” This, in combination with the scalable nature of our system and the defined genetics, provides the field with an important model and platform technology.
Experimental Procedures
Reagents
RPMI, 50× B27 Supplement, Knockout DMEM (KO-DMEM), Knockout Serum Replacement Medium (KO-SR), GlutaMAX, Penicillin/Streptomycin (P/S), and HepatoZYME-SFM (HZM) were purchased from Life Technologies. Recombinant Mouse Wnt3a, Human Activin A (AA), Human Hepatocyte Growth Factor (HGF), and Human Oncostatin M (OSM) were from PeproTech (Hannoun et al., 2010; Hay et al., 2011; Szkolnicka et al., 2013).
Hepatocyte Differentiation
H9 were maintained on MEF cells in MEF-CM (R&D Systems). Before differentiation, H9 was passaged onto feeder-free Matrigel-coated plates in mTeSR medium (STEMCELL Technologies). hESC identity was assessed using a number of criteria including, the absence of stage-specific embryonic antigen-1 (SSEA-1) expression and presence of stage-specific embryonic antigen-4 (SSEA-4) (Figure S1). A stepwise method for cellular differentiation was employed as described (Szkolnicka et al., 2014). Briefly, hESC (H9) differentiation to endoderm was driven by incubating cells in RPMI/B27 supplemented with 50 ng/ml Wnt3a and 100 ng/ml Activin A for 72 hr. Following which cells were maintained in 20% SR/1% DMSO/KO-DMEM for a further 4–5 days to generate hepatoblastic populations. Hepatocytes were subsequently specified in HepatoZYME-SFM (supplemented with 10 μM Hydro-cortisone, 10 ng/ml HGF, and 20 ng/ml OSM) for a further 10–15 days.
Immunofluorescence Staining for HCV Receptors and Intracellular Antigens
Cells were fixed with ice-cold methanol for 10 min. After washing with PBS-0.05% Tween 20 (PBST) and blocking in 0.5% BSA in PBS for 1 hr, cells were incubated with primary antibodies overnight at 4°C. Primary antibodies used were rabbit anti-human Oct4 (Abcam), mouse anti-human Albumin (Sigma-Aldrich), rabbit anti-human HNF4α (Santa Cruz Biotechnology), mouse anti-human E-cadherin (Abcam), mouse anti-human Occludin (Invitrogen), mouse anti-human CD81 (Santa Cruz Biotechnology), mouse anti-human claudin-1 (Invitrogen), mouse anti-human SR-BI (BD), mouse anti-NS5A (9E10, a kind gift from Charles M. Rice, Center for the Study of Hepatitis C, The Rockefeller University, New York), rabbit anti-HCV core serum (R308, a kind gift from John McLauchlan). Secondary antibodies were Alexa Fluor 488 donkey anti-mouse (Molecular Probes), Alexa Fluor 488 donkey anti-rabbit (Probe molecular), and Alexa Fluor 594 donkey anti-rabbit (Molecular Probes) conjugates. Details of working dilution of each antibody are provided in Table S1. Cells were counterstained with DAPI (Sigma-Aldrich), and the pictures were captured by microscope of Zeiss Axio Observer.
CYP3A and CYP1A2 Assay
CYP3A and CYP1A2 assay were conducted as per the manufacturer’s instructions (Promega, CYP3A P450-GloTM Assay and CYP1A2 P450-GloTM Assay). The relative light unit (RLU) of the product was determined by the GloMax-96 Microplate Luminometer (Promega) and normalized to per milligram protein.
Western Blot Analysis
The XCell II Blot Module (Invitrogen) was employed according to the manufacturer’s instructions. Total protein was extracted by RIPA buffer (Pierce). The protein concentrations were measured using the standard BCA assay (Pierce). Samples containing equal amounts of total proteins were separated by Bis-Tris Gels (Life Technologies) and electrophoretically transferred to PVDF membranes (Bio-Rad). Blots were blocked with 5% BSA in TBST (Tris-buffered saline containing 0.1% Tween 20) for 1 hr at room temperature and then probed with primary antibody at 4°C overnight under constant rotation. The secondary antibody was incubated for 1 hr at room temperature, and detected using SuperSignal West Pico substrate (Thermo Scientific). Details of antibody sources are provided in Table S1.
Preparation of Cell Culture Infectious HCV and Infection
Human hepatoma Huh7 cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (Gibco), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.1 none essential amino acids. JFH-1-based HCVcc were produced in Huh7 cells as previously described (Wakita et al., 2005). The HCVcc virus used in this study, JFH-1DSGCSL, is a JFH-1-derived cell-culture adaptive mutant, the characterization of which will be described elsewhere (A.G.N.A. and A.H.P., unpublished data). HCVcc titers were determined by infection of Huh7 cells with serial dilutions of virus, followed by indirect immunofluorescence for HCV NS5A protein, and expressed as focus-forming units (FFUs)/ml. These virus stocks at 107 FFU/ml were diluted ten times in DMEM medium and were used to inoculate hESC-Heps for 3 hr. Cultures were washed with DMEM medium and propagated in HGF and OSM containing medium. To see the specificity of HCV replication, the HCV NS5B polymerase inhibitor 2′CMA (generously supplied by Craig Gibbs [Gilead Sciences]) was added to the hESC-Heps 3 hr postinfection at final concentration of 10 μM.
Determination of Virus Yield by Focus-Forming Assay
To test if infected hESC-Heps produced infectious virions, supernatants were harvested at 5 days postinoculation and serially diluted to infect Huh7 cells. To assess viral NS5A antigen expression, the infected Huh7 cells were fixed in methanol, counterstained with DAPI (Invitrogen), and immunostained with mouse anti-NS5A (9E10) antibody followed by Donkey anti-mouse Alexa Fluor 488 (Invitrogen). NS5A foci formed in the plate were calculated and the infectious virus yield presented as focus-forming unit (FFU) per ml. Virus produced from infected Huh7 cells were used as a control throughout.
Quantification of Virus Infectivity Using a Reporter Cell Line
The Huh7-J20 reporter cell line has been described previously (Iro et al., 2009). This cell line stably express eGFP fused in-frame to secreted alkaline phosphatase (SEAP) via a recognition sequence of the viral NS3/4A serine protease. The level of SEAP activity in the culture medium directly correlates with the level of intracellular viral RNA replication. Huh7-J20 cells seeded in a 96-well tissue culture plate were first infected with the virus for 3 hr after which the inoculum was replaced with fresh medium and the cells incubated at 37°C for 3 days. The SEAP activity in the infected cell medium was determined as described (Iro et al., 2009) using the Hidex Chameleon plate reader and expressed as relative light units (RLUs).
Quantitative PCR
Quantitative PCR was performed using the ABI 7500 instrument and kit (ABI). Primers were designed separately to span at least an intron. First-strand cDNA was used as a template. The relative expression of the target genes to internal control (GAPDH) was calculated by the comparative threshold cycle (DDCt) method. Experiments were performed in triplicate.
PCR Array for IFN-Stimulated Gene Assay
The quality of RNA extracted by RNAeasy kit (QIAGEN) was analyzed using a Nanodrop. One microgram of RNA was reverse transcribed using the RT2 First Strand kit (QIAGEN). Real-time PCR was performed to evaluate the expression of 84 genes using RT2 profiler PCR array PAHS-064ZE (RT2 Profiler PCR Array Human Interferons & Receptors, SABiosciences). Relative changes in gene expression were calculated using the comparative threshold cycle (DDCt) method. The GAPDH gene in array was used to normalize to the RNA amount.
Polyinosinic/Polycytidylic Acid Transfection of hESC-heps
hESC-Heps were transfected with 1, 5, or 10 μg polyI:C (tlrl-pic, InvivoGen) lipofectamine 2000 (Invitrogen) as described (Park et al., 2012). Supernatant and RNA were collected 24 hr after infection and frozen at −20°C and −70°C, respectively.
IL-29 ELISA
IL-29 levels were measured using the human IL-29 sandwich ELISA kit (eBioscience) according to manufacturer’s instructions.
Chemical Inhibitor Studies
hESC-Heps or Huh7 cells were exposed to 1, 5, 10 μM InSolution JAK inhibitor 1 (Millipore) for 1 hr prior to infection with HCV. Cells were infected with HCVcc at 10 FFU/ml for 3 hr and changed to fresh medium. Cultures were incubated for 5 days. During the incubation, medium was renewed every 2 days. The supernatant from infected cells were collected and stored at 4°C prior to use in the infectivity influence assay described below.
Infectivity Influence Assay
Huh7-J20 reporter cells were plated in a 24-well plate at 30% confluence. Cells were first infected with HCVcc for 3 hr and washed with DMEM. To test the influence of exogenous supernatant to the HCV replication, supernatant collected from either polyI:C or JAK inhibitor I-treated cells were added to the infected Huh7-J20 cells. The cells were incubated and the medium collected after 3 days. The infectivity of HCV in collected medium was measured by SEAP assay.
Statistical Analyses
Data collected from biological replicates are expressed as mean ± SD. Differences between groups were examined for statistical significance using Student’s t test, or one-way ANOVA followed by Dunnett t tests; p values < 0.05 were regarded as significant.
Author Contributions
X.Z., P.S., A.H.P., and D.C.H. conceived and designed the experiments. P.S., X.Z., B.L.-V., and A.H.P. performed the experiments. X.Z., P.S., A.H.P., A.G.N.A., K.C., and D.C.H. analyzed the data. D.C.H., A.G.N.A., S.L.F., B.L.-V., D.S., and K.C. contributed reagents/materials/analysis tools. X.Z., P.S., A.H.P., and D.C.H. wrote the manuscript.
Acknowledgments
We are grateful to the MRC Centre for Regenerative Medicine and the MRC Centre for Virus Research for hosting these studies. We thank Joyce Mitchell for the virus training, Olivia Rodrigues for help with flow cytometry, and Valeria Berno for imaging assistance. We also thank Charles M. Rice, Takaji Wakita, and John McLauchlan for kind gift of reagents used in this study. This work was supported by the MRC (Ref. MR/K008757/1) and NSFC (Ref. 81261130312) Stem Cell Partnership scheme, partly by the Natural Science Foundation of Guangdong Province, China (Ref. S2012010009414, S2012010009605), and University Young Scholars Overseas Visiting Foundation of Guangdong Province. D.C.H. was supported by a RCUK fellowship. B.L.-V. and D.S. were supported by MRC PhD studentships. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. D.C.H. is a founder, CSO, shareholder, and director of FibromEd Products Ltd.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Arvind H. Patel, Email: arvind.patel@glasgow.ac.uk.
David C. Hay, Email: davehay@talktalk.net.
Supplemental Information
References
- Cai J., Zhao Y., Liu Y., Ye F., Song Z., Qin H., Meng S., Chen Y., Zhou R., Song X. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239. doi: 10.1002/hep.21582. [DOI] [PubMed] [Google Scholar]
- Calland N., Albecka A., Belouzard S., Wychowski C., Duverlie G., Descamps V., Hober D., Dubuisson J., Rouillé Y., Séron K. (-)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology. 2012;55:720–729. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- Da Costa D., Turek M., Felmlee D.J., Girardi E., Pfeffer S., Long G., Bartenschlager R., Zeisel M.B., Baumert T.F. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J. Virol. 2012;86:11919–11925. doi: 10.1128/JVI.01066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickensheets H., Sheikh F., Park O., Gao B., Donnelly R.P. Interferon-lambda (IFN-λ) induces signal transduction and gene expression in human hepatocytes, but not in lymphocytes or monocytes. J. Leukoc. Biol. 2013;93:377–385. doi: 10.1189/jlb.0812395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y., Catana A., Meng Y., Yamamoto N., He S., Gupta S., Gambhir S.S., Zern M.A. Differentiation and enrichment of hepatocyte-like cells from human embryonic stem cells in vitro and in vivo. Stem Cells. 2007;25:3058–3068. doi: 10.1634/stemcells.2007-0291. [DOI] [PubMed] [Google Scholar]
- Evans M.J., von Hahn T., Tscherne D.M., Syder A.J., Panis M., Wölk B., Hatziioannou T., McKeating J.A., Bieniasz P.D., Rice C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- Foy E., Li K., Sumpter R., Jr., Loo Y.M., Johnson C.L., Wang C., Fish P.M., Yoneyama M., Fujita T., Lemon S.M., Gale M., Jr. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannoun Z., Fletcher J., Greenhough S., Medine C., Samuel K., Sharma R., Pryde A., Black J.R., Ross J.A., Wilmut I. The comparison between conditioned media and serum-free media in human embryonic stem cell culture and differentiation. Cell Reprogram. 2010;12:133–140. doi: 10.1089/cell.2009.0099. [DOI] [PubMed] [Google Scholar]
- Hay D.C. Rapid and scalable human stem cell differentiation: now in 3D. Stem Cells Dev. 2013;22:2691–2692. doi: 10.1089/scd.2013.1500. [DOI] [PubMed] [Google Scholar]
- Hay D.C., Fletcher J., Payne C., Terrace J.D., Gallagher R.C., Snoeys J., Black J.R., Wojtacha D., Samuel K., Hannoun Z. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA. 2008;105:12301–12306. doi: 10.1073/pnas.0806522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D.C., Pernagallo S., Diaz-Mochon J.J., Medine C.N., Greenhough S., Hannoun Z., Schrader J., Black J.R., Fletcher J., Dalgetty D. Unbiased screening of polymer libraries to define novel substrates for functional hepatocytes with inducible drug metabolism. Stem Cell Res. (Amst.) 2011;6:92–102. doi: 10.1016/j.scr.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Horner S.M., Gale M., Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013;19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iro M., Witteveldt J., Angus A.G., Woerz I., Kaul A., Bartenschlager R., Patel A.H. A reporter cell line for rapid and sensitive evaluation of hepatitis C virus infectivity and replication. Antiviral Res. 2009;83:148–155. doi: 10.1016/j.antiviral.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kotenko S.V., Gallagher G., Baurin V.V., Lewis-Antes A., Shen M., Shah N.K., Langer J.A., Sheikh F., Dickensheets H., Donnelly R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Liang Y., Shilagard T., Xiao S.Y., Snyder N., Lau D., Cicalese L., Weiss H., Vargas G., Lemon S.M. Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology. 2009;137:1448–1458. doi: 10.1053/j.gastro.2009.07.050. [DOI] [PubMed] [Google Scholar]
- Lucendo-Villarin B., Khan F., Pernagallo S., Bradley M., Iredale J.P., Hay D.C. The effect of biological and synthetic matrices on hepatic stem cell gene expression. BioResearch Open Access. 2012;1:50–53. doi: 10.1089/biores.2012.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medine C.N., Lucendo-Villarin B., Storck C., Wang F., Szkolnicka D., Khan F., Pernagallo S., Black J.R., Marriage H.M., Ross J.A. Developing high-fidelity hepatotoxicity models from pluripotent stem cells. Stem Cells Transl. Med. 2013;2:505–509. doi: 10.5966/sctm.2012-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H., Serti E., Eke O., Muchmore B., Prokunina-Olsson L., Capone S., Folgori A., Rehermann B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology. 2012;56:2060–2070. doi: 10.1002/hep.25897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A.J., Houghton M., Rosa D., Grandi G., Abrignani S. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- Ploss A., Evans M.J., Gaysinskaya V.A., Panis M., You H., de Jong Y.P., Rice C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploss A., Khetani S.R., Jones C.T., Syder A.J., Trehan K., Gaysinskaya V.A., Mu K., Ritola K., Rice C.M., Bhatia S.N. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. USA. 2010;107:3141–3145. doi: 10.1073/pnas.0915130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podevin P., Carpentier A., Pène V., Aoudjehane L., Carrière M., Zaïdi S., Hernandez C., Calle V., Méritet J.F., Scatton O. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology. 2010;139:1355–1364. doi: 10.1053/j.gastro.2010.06.058. [DOI] [PubMed] [Google Scholar]
- Roelandt P., Obeid S., Paeshuyse J., Vanhove J., Van Lommel A., Nahmias Y., Nevens F., Neyts J., Verfaillie C.M. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J. Hepatol. 2012;57:246–251. doi: 10.1016/j.jhep.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Samuel C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R.M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R.E., Trehan K., Andrus L., Sheahan T.P., Ploss A., Duncan S.A., Rice C.M., Bhatia S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012;109:2544–2548. doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tayeb K., Noto F.K., Nagaoka M., Li J., Battle M.A., Duris C., North P.E., Dalton S., Duncan S.A. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan G.J., Hay D.C., Park I.H., Fletcher J., Hannoun Z., Payne C.M., Dalgetty D., Black J.R., Ross J.A., Samuel K. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter R., Jr., Loo Y.M., Foy E., Li K., Yoneyama M., Fujita T., Lemon S.M., Gale M., Jr. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005;79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P., Zhou X., Farnworth S.L., Patel A.H., Hay D.C. Modeling human liver biology using stem cell-derived hepatocytes. Int. J. Mol. Sci. 2013;14:22011–22021. doi: 10.3390/ijms141122011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkolnicka D., Zhou W., Lucendo-Villarin B., Hay D.C. Pluripotent stem cell-derived hepatocytes: potential and challenges in pharmacology. Annu. Rev. Pharmacol. Toxicol. 2013;53:147–159. doi: 10.1146/annurev-pharmtox-011112-140306. [DOI] [PubMed] [Google Scholar]
- Szkolnicka D., Farnworth S.L., Lucendo-Villarin B., Storck C., Zhou W., Iredale J.P., Flint O., Hay D.C. Accurate prediction of drug-induced liver injury using stem cell-derived populations. Stem Cells Transl Med. 2014;3:141–148. doi: 10.5966/sctm.2013-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Akira S. Innate immunity to virus infection. Immunol. Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te H.S., Jensen D.M. Epidemiology of hepatitis B and C viruses: a global overview. Clin. Liver Dis. 2010;14:1–21. doi: 10.1016/j.cld.2009.11.009. vii. [DOI] [PubMed] [Google Scholar]
- Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H.G., Mizokami M. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Robotham J.M., Lee E., Dalton S., Kneteman N.M., Gilbert D.M., Tang H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.D., Roberts L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Hannoun Z., Jaffray E., Medine C.N., Black J.R., Greenhough S., Zhu L., Ross J.A., Forbes S., Wilmut I. SUMOylation of HNF4α regulates protein stability and hepatocyte function. J. Cell Sci. 2012;125:3630–3635. doi: 10.1242/jcs.102889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.