

Abstract
Abstinence from cocaine self-administration (SA) is associated with neuroadaptations in the prefrontal cortex (PFC) and nucleus accumbens (NAc) that are implicated in cocaine-induced neuronal plasticity and relapse to drug-seeking. Alterations in cAMP-dependent protein kinase A (PKA) signaling are prominent in medium spiny neurons in the NAc after repeated cocaine exposure but it is unknown whether similar changes occur in the PFC. Because cocaine SA induces disturbances in glutamatergic transmission in the PFC-NAc pathway, we examined whether dysregulation of PKA-mediated molecular targets in PFC-NAc neurons occurs during abstinence and, if so, whether it contributes to cocaine seeking. We measured the phosphorylation of CREB (Ser133) and GluA1 (Ser845) in the dorsomedial (dm) PFC and the presynaptic marker, synapsin I (Ser9, Ser62/67, Ser603), in the NAc after 7 days of abstinence from cocaine SA with or without cue-induced cocaine-seeking. We also evaluated whether infusion of the PKA inhibitor, 8-bromo-Rp-cyclic adenosine 3′, 5′-monophosphorothioate (Rp-cAMPs), into the dmPFC after abstinence would affect cue-induced cocaine-seeking and PKA-regulated phosphoprotein levels. Seven days of forced abstinence increased the phosphorylation of CREB and GluA1 in the dmPFC and synapsin I (Ser9) in the NAc. Induction of these phosphoproteins was reversed by a cue-induced relapse test of cocaine-seeking. Bilateral intra-dmPFC Rp-cAMPs rescued abstinence-elevated PKA-mediated phosphoprotein levels in the dmPFC and NAc and suppressed cue-induced relapse. Thus, by inhibiting abstinence-induced PKA molecular targets, relapse reverses abstinence-induced neuroadaptations in the dmPFC that are responsible, in part, for the expression of cue-induced cocaine-seeking.
Keywords: CREB, AMPA receptor, nucleus accumbens, synapsin
Drug craving and relapse after withdrawal are major clinical challenges that hinder the treatment of cocaine addiction (Mendelson and Mello, 1996; O’Brien, 1997). The susceptibility to drug relapse and other addictive behaviors is thought to depend on long-term neuroadaptations in the mesocorticolimbic circuitry, including the dorsomedial prefrontal cortex (dmPFC)-nucleus accumbens (NAc) projection (Koob et al., 1997; Wang and McGinty, 1999; White and Kalivas, 1998). Although the molecular mechanisms underlying drug addiction are not fully-established, changes in protein kinase A (PKA)-mediated signaling may contribute to drug-induced neuroadaptations leading to relapse. For example, in animal models, abstinence from cocaine self-administration (SA) or repeated experimenter-administered cocaine resulted in the elevation of PKA activity and PKA substrate phosphorylation in the NAc (Boudreau et al., 2009; Hope et al., 2005; Lu et al., 2003). Further, direct intra-NAc infusion of dopamine or, conversely, a PKA inhibitor reinstated cocaine-seeking in rats (Cornish and Kalivas, 2000; Self et al., 1998). We have previously reported that abstinence from cocaine SA increased the phosphorylation of CREB (p-CREB), but not ERK (p-ERK), in the dmPFC, in a time-dependent manner (Whitfield et al., 2011), implying a gradual increase in PKA-regulated molecular activation during abstinence. Further, dopamine or cocaine microinfusions into the dmPFC have been shown to reinstate cocaine seeking (McFarland and Kalivas, 2001; Park et al., 2002). Taken together, these results suggest that increased PKA-mediated signaling in the dmPFC and NAc plays a critical role in initiating cocaine seeking after withdrawal/abstinence.
Glutamate transmission in the dmPFC-NAc projection has been implicated in drug-induced neuronal plasticity and reinstatement. For instance, the surface expression of GluA1-containing AMPA receptors is gradually increased in the NAc of rats during abstinence from cocaine self administration (Conrad et al., 2008). PKA-mediated phosphorylation of GluA1 Ser845 promotes AMPA receptor plasma membrane insertion (Esteban et al., 2003; Oh et al., 2006). Additionally, cocaine SA results in abnormalities in extracellular glutamate levels in the NAc after extinction and reinstatement that are dependent on neurons projecting from the PFC (McFarland et al., 2003). Furthermore, we have shown that a single brain-derived neurotrophic factor (BDNF) infusion into dmPFC immediately following the final cocaine SA session rescued abnormalities in glutamate transmission in the NAc and suppressed subsequent cocaine-seeking in an ERK MAP kinase-dependent manner (Berglind et al., 2007, 2009; Whitfield et al., 2011). The distinct molecular mechanisms underlying the cocaine-mediated alterations in glutamate release are still unknown; however, synapsins are potentially involved as they are key presynaptic regulators of glutamate release. In mice lacking synapsins, regulation of the reserve pool of synaptic vesicles in excitatory terminals is compromised (Gitler et al., 2004) and the ability to release dopamine in the striatum is reduced following a cocaine challenge (Venton et al., 2006). The functional roles of different phosphorylation sites in synapsin I (p-synapsin I) are particularly well-characterized (Greengard et al., 1993). For example, PKA/CaMKI-mediated p-synapsin I Ser9 activation results in synapsin dissociation from synaptic vesicles and dispersion into the cytosol. The ERK/MAPK-mediated p-synapsin I Ser62/67 promotes glutamate release in isolated nerve terminals (Jovanovic et al., 2001). In addition, the CaMKII-activated p-synapsin I Ser603 is one of the most effective sites promoting dissociation from the actin cytoskeleton and dispersion of synapsin I into axons, allowing vesicles to diffuse to the active zone and fuse with the presynaptic membrane to be released upon neuronal stimulation (Hilfiker et al., 1999). Phosphorylation of p-synapsin I Ser9 has been implicated previously in cocaine SA-induced neuroadaptations because it is increased in the NAc immediately after cessation of cocaine intake (Edwards et al., 2007).
Based on this background, the goals of the present study were twofold. First, we evaluated whether PKA-mediated molecular signaling, including the phosphorylation of CREB and GluA1 Ser845 in the dmPFC and p-synapsin I Ser9 in the NAc was changed following abstinence from cocaine SA with or without a conditioned cue-induced relapse test. Secondly, we investigated the possible effect of pharmacological PKA inhibition in the dmPFC on these phosphoproteins after abstinence and relapse to cocaine seeking in both brain regions as well as its contribution to cue-induced cocaine seeking. These studies aimed to further establish PKA and its downstream signaling targets as essential contributors to cocaine-mediated plasticity in the dmPFC-NAc pathway and their contribution to the emergence of cocaine relapse.
MATERIALS AND METHODS
Animals and surgery
Adult male Sprague-Dawley rats (Charles River Laboratories, Raleigh, NC; initial weight: 301–325 g) were housed individually in a temperature- and humidity-controlled animal facility on a reversed 12-hr light/dark cycle (lights off at 7:00 AM). Animals were given 20–25 g of standard rat chow with ad libitium access to water daily. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina and complied with National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 80–23, revised 1996). Animals were given at least a 3-day acclimation period. On the day of surgery, rats were anesthetized with a mixture of ketamine and xylazine (66 and 1.33 mg/kg i.p., respectively), followed by equithesin (0.5 ml/kg i.p.) and ketorolac (2 mg/kg i.p.). A silastic catheter, implanted into the right jugular vein, exited on the back to connect to an infusion harness (Instech Laboratories, Plymouth Meeting, PA) for intravenous infusions. Immediately after catheterization, rats were implanted with 26 gauge bilateral stainless steel guide cannulae (Plastics One) aimed 1 mm above the dmPFC infusion target. Coordinates used in the stereotaxic procedures were +3.0 mm AP, ±0.6 mm ML, and −1.6 mm DV from the dural surface (Paxinos and Watson, 2005). Guide cannulae were secured to the skull with dental cement and steel screws. Two 10 mm stylets (Plastics One) were placed into the guide cannulae following surgery to prevent blockage throughout the experiment. After surgeries, daily intravenous infusions of 0.1 ml of the antibiotics, Cefazolin (10 mg/ml) or Timentin (2.4 mg/ml), and 0.1 ml of heparinized saline (100 U/ml) were administered during surgical recovery and maintained throughout self-administration to maintain the catheter patency. Catheter patency was verified as needed with an intravenous infusion of 0.1 ml of methohexital sodium (10 mg/ml).
Cocaine self-administration, abstinence and cue-induced drug-seeking test
After 5 days of recovery, rats were subjected to 2-hr daily cocaine or yoked saline self-administration sessions under a FR1 reinforcement schedule in self-administration chambers (30 × 20 × 20 cm; MED Associates, East Fairfield, VT). Each chamber contained two retractable levers (7 cm above floor) and circular stimulus light above each lever. A Tygon infusion line within the spring leash was mounted to a balanced metal arm and attached to a liquid swivel (Instech, Polymouth Meeting, PA). Before each session, the catheter was secured to the infusion line. Presses on the active lever resulted in a 2-s cocaine hydrochloride (0.2 mg/infusion; National Institute on Drug Abuse, Research Triangle Park, NC) infusion via a computer-controlled pump. Cocaine infusions were paired with a 5-s presentation of a conditioned-stimulus complex including the illumination of a white light above the active lever and a tone (2 kHz, 15 dB above ambient noise), followed by 20-s timeout period, during which additional responses on the active lever resulted in no programmed consequences. Responses on the inactive lever were recorded but had no consequence. For each session, rats had to self-administer at least 10 infusions to fulfill the maintenance criterion. After the end of 10 self-administration sessions to criterion, rats were returned to their colony room for 7 days of abstinence. Following the abstinence period, the rats received a 30 min cue-induced cocaine-seeking test under extinction conditions. During the test, active lever responses resulted in the presentation of the conditioned-stimulus complex (light+tone) without infusion. Presses on the inactive lever had no programmed consequence.
Locomotor activity
After the cue test, locomotor activity was measured in clear acrylic chambers (40 × 40 × 30 cm) equipped with Digiscan monitors (AccuScan Instruments Inc., Columbus, OH). Each chamber was equipped with two 4 × 4 photobeam arrays for horizontal activity (total distance traveled) and vertical activity (rearing). Photobeam breaks were continuously clustered in 5 min bins for 1 hr by a Digiscan analyzer and recorded by Versamax software (version 1.82; Accuscan Instruments Inc.).
Immunoblotting
The procedure of immunoblotting was described previously (Sun et al., 2007). Briefly, rats were rapidly decapitated and brains were removed and placed in a brain matrix (Braintree Scientific, Inc., Warren, MI). Two 2 mm coronal slices at AP 4.7–2.7 and 2.7-(−0.3) from Bregma were cut and the dmPFC and NAc, respectively, were bilaterally punched and frozen at −80°C. Protein was extracted by sonication in ice-cold RIPA lysis buffer and centrifuged at 10,000 g for 20 min at 4°C. The resulting supernatants were collected and subjected to protein analysis with the Micro-Bicinchoninic Acid assay kit (Pierce, Rockford, IL). Equal amounts of protein extracts were boiled in Lammeli buffer containing 1% β-mercaptoethanol for 5 min and run on SDS-PAGE gels, then transferred to PVDF membranes. Membranes were then blocked with 5% nonfat dry milk in Tris-buffered saline with Tween-20 (TBST; pH=7.6) for 1 hr at room temperature and incubated with primary antibody against p-synapsin I Ser9 (1:2000) (Cell Signaling, Beverly, MA), p-synapsin I Ser62/67 (1:1000), p-synapsin I Ser603 (1:3000), p-GluA1 Ser845 (1:1000) or p-CREB Ser133 (1:1000) (Millipore, Bedford, MA) overnight at 4°C. After three washes with TBST, membranes were incubated with their appropriate secondary antibodies for 1 hr at room temperature followed by three more washes with TBST. Antibody binding was detected by using an enhanced chemiluminescence kit (ECL Plus; GE Healthcare Bio-Sciences, Piscataway, NJ). Membranes were then stripped and re-probed with total protein by antisera against synapsin I (1:2000; Cell Signaling), GluA1 (1:5000; Abcam, Cambridge, MA) or CREB (1:500; Millipore) for quantification. Equal loading proteins were further confirmed by probing with anti-calnexin antiserum (1:10000; Enzo Life Sciences, Farmingdale, NY). All Western blot analyses were performed a minimum of two times. The integrated density of each phosphoprotein and total protein band was measured using Image J software (National Institute of Health, Bethesda, MD).
Experimental design
In experiment 1, following 10 days of cocaine or yoked-saline self-administration and 7 days of abstinence, rats were euthanized immediately with or without a 30 min cue test that induced cocaine-seeking under extinction conditions. The group that underwent the cue test is referred to as the “relapse” group instead of the “reinstatement” group because the rats’ lever pressing behavior was not extinguished. The experimental design is based on our previous study (Whitfield et al., 2011) in which a significant p-CREB induction in the dmPFC occurred after 7 days of abstinence. The dmPFC and NAc were processed for western blotting to evaluate the effects of abstinence and cue-induced cocaine-seeking upon phosphoproteins. In experiment 2, the PKA inhibitor, 8-bromo-Rp-cyclic adenosine 3′,5′-monophosphorothioate, sodium salt (Rp-cAMPs, 40 nmol/0.5 μl/side; EMD Millipore, Billerica, MA) or its vehicle, aCSF (pH=7.4), was infused into the dmPFC on the 7th day of abstinence. Each bilateral infusion was administered through an injector (33 gauge; Plastics One) inserted into the guide cannula so that 1 mm of the injector length extended beyond the guide cannula. After a 2 min infusion (0.25 μl/min), the injectors remained in the cannulae for 1 min to allow complete diffusion. This concentration of Rp-cAMPs has previously been shown to alter cocaine seeking and inhibit amphetamine-induced PKA-related phosphoproteins in the NAc of anesthetized rats (Self et al., 1998). Thirty minutes after infusion, rats underwent a cue-induced cocaine-seeking test under extinction conditions for 30 min to examine the effect of PKA inhibition on cocaine seeking. Upon the completion of the test, rats were subjected to a locomotor activity test for 1 hr. An additional cohort of animals received infusions during the last day of abstinence to examine whether PKA inhibition would abrogate the abstinence-induced phosphoproteins 30 min later without a behavioral test.
Cannula placement
At the end of the locomotor activity test in experiment 2, rats were rapidly decapitated and brains were immediately removed and flash frozen in 2-methylbutane (−40°C) and stored at −80°C. Forty μm coronal sections throughout the PFC of each brain were cut in a cryostat and placed onto slides for cannulae placement verification after Nissl-staining. For rats used for western blot analysis, rats were rapidly decapitated and cannula placements at the site of injection (dmPFC) were immediately verified by observation when the dmPFC was dissected.
Data analysis
Two-way ANOVA or independent t-tests were used for cocaine self-administration and western blot data. Student–Newman–Keuls (SNK) multiple-comparison tests were performed when a significant interaction or main effect was found after ANOVA. The locomotor data (experiment 2) were analyzed by calculating the area under the curve (AUC) for the activity counts plotted against time followed by an independent t-test. Determination of statistically significant differences was considered at a 0.05 level (P<0.05).
RESULTS
Histology
Cannula placements from experiment 2 are illustrated in Fig 1. All cannula tips were located on the anterior cingulate/prelimbic cortex border or within the prelimbic cortex.
Figure 1.

Representative histological cannula placements in the dmPFC from experiment 2. Bilateral cannula placements were at the anterior cingulate/prelimbic border and within the prelimbic cortex. Rats with track placement that were not in the dmPFC were excluded from data analysis (n=1).
Experiment 1: Effects of abstinence on cue-induced relapse and phosphoproteins in the dmPFC and NAc
During the last three SA sessions, cocaine self-administering rats exhibited higher active lever pressing and lower inactive lever pressing than yoked-saline controls, indicating the acquisition of cocaine-associated reinforcing effects (active lever: t (34) = 11.76, p<0.001; inactive lever t (34) = 6.86, p<0.001; Fig. 2A-left). After 7 days of abstinence, re-exposure to the conditioned light and tone cues resulted in significantly greater active lever pressing by rats with a cocaine history than by saline controls (t (14) = 4.76, p<0.001; Fig 2A-right). Active lever pressing during the cue test was significantly greater than during SA for the cocaine group (t (30) = 4.88, p<0.0001). With regard to protein expression, there were significant main effects of cocaine and relapse for p-CREB (cocaine: F (1,34) = 8.52, p<0.01; relapse: F (1,34) = 10.68, p<0.01) with a marginal cocaine by relapse interaction (F (1,34) = 3.57, p=0.067). A SNK test revealed that p-CREB was only increased in cocaine animals after abstinence (p<0.01; Fig. 2B). Similarly, for p-GluA1, there were significant cocaine and relapse main effects as well as a cocaine by relapse interaction (cocaine: F (1,34) = 5.81, p<0.05; relapse: F (1,34) = 15.10, p<0.001; cocaine×relapse: F (1,34) = 4.38, p<0.05). The cocaine group after abstinence had significantly higher p-GluA1 in the dmPFC than other groups (p<0.05; Fig. 2C). There was no effect of cocaine on p-ERK expression in the dmPFC after abstinence (Supplementary Fig. S1A). The effects of abstinence and relapse on PKA/CaMKI-mediated p-synapsin I Ser9 in the NAc are shown in Fig. 2D. There were significant cocaine and relapse main effects as well as a cocaine by relapse interaction (cocaine: F (1,29) = 5.47, p<0.05; relapse: F (1,29) = 6.99, p<0.05; cocaine×relapse: F (1,29) = 4.33, p<0.05). Abstinence, but not relapse, resulted in a higher p-synapsin I Ser9 level in the cocaine group (p<0.05). Abstinence from cocaine did not have an effect on p-synapsin I Ser62/67 or Ser603 in the NAc (Supplementary Fig. S1B and C, respectively). In contrast, the abstinence-induced elevations in phosphoprotein levels in both brain regions were attenuated after cue-induced relapse to cocaine seeking (p>0.05 when compared to saline controls; Fig. 2B–D).
Figure 2.
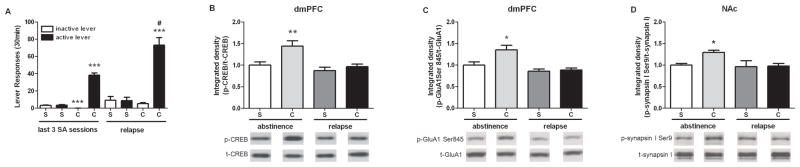
Effects of abstinence and cue-induced relapse on cocaine-seeking and PKA phosphoprotein targets. (A) The average number of active and inactive lever responses during the last 3 sessions of yoked-saline or cocaine SA (left) and lever responses during a 30 min cue-induced relapse after 7 days of abstinence (right) (n = 6–21/group; ***p<0.001, cocaine SA versus yoked-saline; #p<0.0001 active lever responses during cue test versus during cocaine SA, t-test). Protein levels of p-CREB (B), and p-GluA1 (C) in the dmPFC after abstinence or relapse (n = 6–12/group; *p<0.05 and **p<0.01 compared to saline controls, two-way ANOVA followed by SNK test). (D) Protein level of p-synapsin I Ser9 in the NAc after abstinence or relapse (n = 5–11/group; *p<0.05 compared with saline controls, two-way ANOVA followed by SNK test). The bar graphs represent the mean ± SEM. S=saline; C=cocaine.
Experiment 2: Effects of intra-dmPFC Rp-cAMPs infusion on cue-induced relapse and abstinence-induced increase in phosphoprotein levels
PKA activation is necessary for the phosphorylation of p-CREB, GluA1 Ser845, and, in part, for synapsin I Ser9. Thus, the effect of PKA inhibition on cue-induced responding and abstinence-induced phosphoprotein signaling was evaluated in experiment 2. Before infusion, all rats in the cocaine groups showed similar amounts of active lever pressing during the last three sessions of cocaine SA (t (18) = 0.43, p>0.05; Fig. 3A-left). During the cue-induced relapse test, vehicle-infused rats exhibited significantly higher active lever pressing than during cocaine SA (t (17) = 03.66, p <0.001). In contrast, intra-dmPFC Rp-cAMPs infusion 30 min before the cue test attenuated cue-induced cocaine-seeking (t (16) = 2.63, p <0.05; Fig. 3A-right). Locomotor activity was not different between the Rp-cAMPs and vehicle-infused groups, indicating a lack of nonspecific sedation (Fig. 3B–C).
Figure 3.
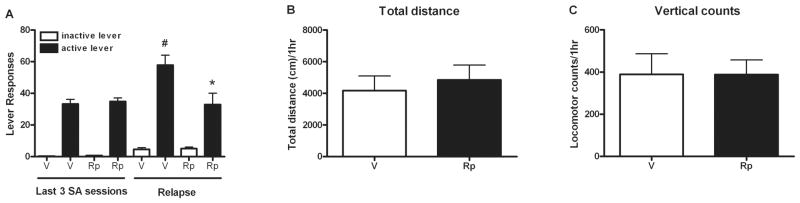
Suppressive effects of intra-dmPFC Rp-cAMPs infusion on cue-induced relapse to cocaine-seeking but not on locomotor activity. (A) The average lever responses during the last 3 sessions of cocaine SA (left) and the lever responses during a cue-induced relapse test 30 min after intra-dmPFC infusion (right). (n = 9–10/group; *p<0.05 compared with vehicle-infused group; #p<0.01 when compared with active lever pressing during cocaine SA, t-test). (B–C) One hr after intra-dmPFC Rp-cAMPs infusion, the locomotor activity, including total distance traveled (B) and vertical activity (C), was not changed The bar graphs represent the mean ± SEM. V=vehicle infusion; Rp=Rp-cAMPs infusion.
There were significant main effects of cocaine and Rp-cAMPs infusion as well as a cocaine by infusion interaction for p-CREB (cocaine: F (1,20) = 21.12, p<0.001; infusion: F (1,20) = 4.76, p<0.05; cocaine×infusion: F (1,20) = 4.66, p<0.05). A SNK test indicated that abstinence-induced p-CREB was decreased by intra-dmPFC Rp-cAMPs infusion (p<0.01; Fig 4A). In addition, there was a significant main effect of Rp-cAMPs infusion with a marginally significant cocaine by infusion interaction for the p-GluA1 in the dmPFC (infusion: F (1,20) = 22.77, p<0.001; cocaine×infusion: F (1,20) = 4.16, p<0.06). A SNK test revealed that Rp-cAMPs antagonized abstinence-induced p-GluA1 in the cocaine SA group (p<0.001; Fig. 4B). There was a significant cocaine by Rp-cAMPS infusion interaction for p-synapsin I Ser9 in the NAc (F (1,20) = 6.28, p<0.05) and intra-dmPFC Rp-cAMPs infusion suppressed abstinence-induced p-synapsin I Ser9 in the cocaine SA group (p<0.05; Fig. 4C). After Rp-cAMPs infusion, phosphoprotein levels in both brain regions were not different than in vehicle-infused rats with a yoked-saline history.
Figure 4.
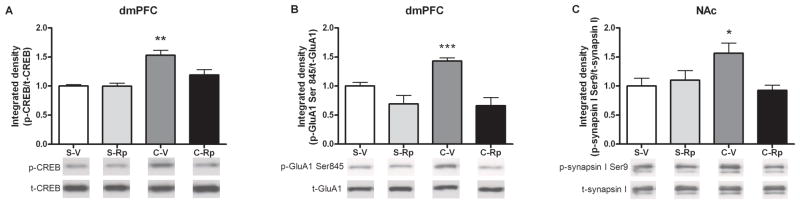
Protein levels of p-CREB and p-GluA1 Ser845 in the dmPFC and p-synapsin I Ser9 in the NAc on the 7th day of abstinence after intra-dmPFC Rp-cAMPs infusion. (A) p-CREB, (B) p-GluA1 Ser845 in the dmPFC and (C) p-synapsin I Ser9 in the NAc 30 min after infusion without cue-induced relapse test. (n = 5–7/group; *p<0.05, **p<0.001, and ***p<0.001 compared to saline-vehicle controls, two-way ANOVA followed by SNK test). The bar graphs represent the mean ± SEM. S-V=Saline with vehicle infusion; S-Rp=Saline with Rp-cAMPs infusion; C-V=Cocaine with vehicle infusion; C-Rp=Cocaine with Rp-cAMPs infusion.
DISCUSSION
In the present study, we demonstrated that abstinence-induced phosphoproteins, p-CREB and p-GluA1 Ser845 in the dmPFC, and p-synapsin I Ser9 in the NAc, are dependent on PKA activation in the dmPFC after cocaine SA. Interestingly, re-exposure to drug-associated cues after 7 days of abstinence normalized these phosphoproteins, suggesting that relapse alleviates abstinence-induced PKA activity in the PFC. Moreover, Rp-cAMPs suppressed cue-induced cocaine-seeking, suggesting that dmPFC PKA activation may represent an essential contributor to the expression of cue-elicited cocaine-seeking after abstinence. These experiments have specifically identified the role of PKA and its downstream targets in the dmPFC-NAc pathway in the emergence of relapse to cocaine-seeking.
The finding that PKA, but not ERK MAP kinase, activation in the dmPFC is necessary for abstinence-induced augmentation of p-CREB and p-GluA1 Ser845 in the dmPFC, and PKA/CaMKI-mediated p-synapsin I Ser9 in the NAc, suggests that there is a biphasic alteration in PFC signaling that shifts from ERK dependence during early withdrawal (Whitfield et al., 2011) to PKA dependence during more prolonged abstinence. This idea is consistent with the ability of intra-PFC infusion of Rp-cAMPs to attenuate abstinence-induced neuroadaptations in both PFC and NAc and suggests that these PKA protein targets are expressed in NAc-projecting pyramidal neurons originating in the PFC. The present findings, along with other studies (Boudreau et al., 2009; Hope et al., 2005; Lu et al., 2003; Whitfield et al., 2011) indicate increased PKA-mediated signaling in both dmPFC and NAc after abstinence from repeated cocaine. PKA phosphorylates many targets that facilitate excitatory neurotransmission, including GluA1, NMDA receptor subunits, and L-type Ca2+ channels (Ford et al., 2009), pointing to augmented excitability in the forebrain during abstinence. Indeed, PKA activity and L-type Ca2+ channel surface expression in the PFC are elevated in cocaine sensitized animals after 3 and 21 days of withdrawal, which is consistent with higher basal activity of pyramidal neurons (Ford et al., 2009; Nasif et al., 2005). Further, 8 days after repeated cocaine exposure, there is a delayed increase of BDNF-regulated long-term potentiation (LTP) coupled with suppressed GABAergic inhibition in the mPFC (Lu et al., 2010). PFC hyperexcitability has been detected during repeated cocaine exposure and may be augmented by basal hypoactivity between cocaine infusions (Sun and Rebec, 2006). In fact, a rapid dephosphorylation of CREB and ERK, as well as a reduction of Bdnf mRNA and other immediate early genes were found in the mPFC 2 and 22 hr after the end of cocaine SA (McGinty et al.; Whitfield et al., 2011), suggesting basal hypoactivity in the PFC during early withdrawal. In contrast, the increase of p-GluA1 Ser845 after 7 days of abstinence is consistent with enhanced AMPA receptor-mediated excitatory activity mediated by GluA1 plasma membrane insertion (Banke et al., 2000; Oh et al., 2006; Roche et al., 1996). Therefore, it is plausible that cocaine SA results in an initial decline in dmPFC activity during early withdrawal followed by a rebound and/or potentiated activity mediated by PKA that may increase relapse vulnerability after prolonged abstinence.
Numerous studies have shown that cue-elicited drug seeking after cocaine SA increases immediate early gene expression in the dmPFC (Ciccocioppo et al., 2001; Hearing et al., 2008; Kufahl et al., 2009; Thomas et al., 2003; Zavala et al., 2008; Ziolkowska et al., 2011) among other regions, suggesting neuronal activation induced by drug-associated cues. In contrast, this study demonstrated a normalizing effect of relapse on abstinence-induced p-CREB, p-GluA1 Ser845, and p-synapsin I Ser9 in the dmPFC and NA, respectively. Other studies have reported similar events that suggest that relapse to drug-seeking can reverse an abstinence-induced suppression of GABAergic tone or AMPA/NMDA ratio in the PFC or NAc. For example, cue-induced reinstatement of heroin seeking reversed suppressed extracellular matrix proteins in perineuronal nets surrounding GABAergic neurons in the mPFC that correlated with enhancement of GABAergic inhibition of mPFC neurons (Van den Oever et al., 2010). Additionally, in a conditioned place preference model, cocaine-associated cue re-exposure induced robust Fos-immunoreactivity in GABAergic interneurons, not pyramidal neurons, in the prelimbic cortex (Miller and Marshall, 2004). Similarly, a cocaine challenge or re-exposure to cocaine-related cues attenuated withdrawal-induced surface expression of GluA1/2-containing AMPA receptors in the NAc of cocaine-sensitized animals (Boudreau et al., 2007) and a reduction in withdrawal-induced potentiation of the AMPA/NMDA ratio termed “re-exposure LTD” (Kourrich et al., 2007). Taken together, it is plausible that cue-induced relapse normalizes PKA signaling in the PFC-NAc pathway, possibly by re-activating suppressed GABAergic interneuronal tone (Lu et al., 2010) leading to a reduction in glutamatergic neurotransmission in the PFC-NAc pathway.
The suppressive effect of dmPFC PKA inhibition on relapse to cocaine-seeking is consistent with previous studies indicating that inactivation of the dmPFC with a GABAA/B agonist cocktail or TTX prevents reinstatement induced by stress, cue, and a cocaine prime (Capriles et al., 2003; McFarland and Kalivas, 2001; McLaughlin and See, 2003) and that microinfusion of cocaine or dopamine into dmPFC reinstates cocaine-seeking behavior (McFarland and Kalivas, 2001; Park et al., 2002), presumably by augmenting PKA activity. Increased PKA activation in the dmPFC results in abnormal working memory performance that can be rescued by Rp-cAMPs infusion (Taylor et al., 1999). The ability of Rp-cAMPs infusion to suppress cue-induced relapse (Fig. 3A) and abstinence-induced p-GluA1 (Fig. 4C) supports the notion that dmPFC activation is necessary for the expression of cocaine-seeking and that potentiated PKA-mediated signaling during abstinence is indicative of abnormal PFC functioning. PKA phosphorylates AMPA receptors, NMDA receptors, and L-type Ca2+ channels (Ford et al., 2009). Therefore, Rp-cAMPs’ inhibitory effect on cue-induced drug-seeking may result from its interaction with these receptors to restore normal PFC activity during abstinence. Similarly, intra-NAc Rp-cAMPs infusion attenuates cocaine self-administration and the motivation to seek cocaine when rats are on a progressive ratio schedule (Lynch and Taylor, 2005; Self et al., 1998). Self et al. (1998) demonstrated that intra-NAc Rp-cAMPs (40 or 80 nmol/side) infusion reinstated cocaine seeking. There are several possible explanations for this apparent discrepancy. First, in their experiment, a within-subjects design was used with multiple drug infusions during extinction and cocaine SA infusions occurred prior to each reinstatement test. Second, Rp-cAMPs infusion into the NAc would have a major inhibitory effect on enhanced PKA activity in postsynaptic medium spiny neurons (Lu et al., 2003) that may lead to different consequences than that observed after Rp-cAMPs infusion into dmPFC in abstinent rats.
We demonstrated that 1 hr after intra-dmPFC Rp-cAMPs infusion, basal locomotor activity was not affected, suggesting a lack of a nonspecific sedative effect on behavior. In the NAc of anesthetized rats, a previous study indicated that intra-NAcRp-cAMPs (80 nmol) infusion attenuated basal or acute amphetamine-induced p-DARPP32 Thr34 and p-CREB up to 1–2 hr after infusion (Self et al., 1998). Additionally, a single Rp-cAMPs (20 nmol) infusion into the NAc resulted in a long-lasting inhibitory effect on the motivation to seek cocaine by repeated progressive ratio schedule testing for 4 days (Lynch and Taylor 2005). Together, these studies indicate a long half-live of Rp-cAMPs after intracranial infusion. Thus, it seems implausible that a lack of an effect of Rp-cAMPs on locomotor activity in this study was due to its short half-life. During the cue-induced cocainae-seeking test, both vehicle- and Rp-cAMPs-infused rats showed similar inactive lever pressing (Fig. 3A), further supporting the specific effect of Rp-cAMPs on active lever pressing.
In the present study, the increase in p-synapsin I Ser9 in the NAc after 7 days of abstinence from cocaine SA was mediated by PKA activation in the dmPFC, consistent with an increase in PKA-mediated signaling in the PFC-NAc projection after abstinence. A similar p-synapsin I Ser9 enhancement was found in the striatum following 7 days of abstinence after repeated, non-contingent amphetamine administration (Iwata et al., 1996). In contrast, in a study of cocaine SA on PKA targets, Edwards et al. (2007) reported that an increase of p-GluA1 Ser845 in the PFC and p-synapsin I Ser9 in the NAc shell, but not in the core, occurred immediately after the last cocaine SA session. In their study, a different cocaine SA procedure was used (4 hr daily sessions×18 days) and acute cocaine or yoked-cocaine regimens also increased both phosphoproteins in the PFC and NAc shell. However, later timepoints and the functional relevance of the phosphoprotein changes were not evaluated.
The Ser9 site of synapsin I is phosphorylated by PKA and CaMKI (Jovanovic et al., 2001) and a role for CaMKI in this study cannot be excluded. However, activation of PKA and CaMKI cascades may have different effects on p-synapsin I Ser9 (Matsushita and Nairn, 1999). Thus, unlike pGluA1 Ser845, changes in phosphorylation of synapsin I Ser9 after abstinence and relapse may depend on fine tuning between PKA and CAMKI. Synapsin I phosphorylation at Ser9 promotes vesicle mobilization whereas activity-dependent phosphorylation of CaMKII sites (Ser566 and 603) has a stronger effect on promoting vesicle dispersion (Chi et al., 2001, 2003), suggesting a synergistic action on neurotransmitter release through these two different sites. However, cocaine SA caused an increase in p-synapsin I Ser9 after abstinence but there was no effect on p-synapsin I Ser603 or Ser62/67. It is possible that phosphorylation of synapsin I Ser9 alone facilitates preterminal axonal dispersion of vesicles but is not sufficient to mobilize them into the terminal in the absence of action potential-dependent CAMKII activity. This hypothesis is supported by findings that synaptogenesis mediated by PKA-mediated p-synapsin I Ser9 is independent of activity-dependent synaptogenesis (Perlini et al., 2011). The latter study also suggested that PKA-dependent phosphorylation of synapsin I Ser9 may compensate for impairments in activity-dependent synaptogenesis (Perlini et al., 2011). Thus, it is possible that abstinence-induced elevation in p-synapsin I Ser9 in PFC-NAc neurons is an attempt to compensate for cocaine SA-induced impairments in glutamatergic neurotransmission.
In conclusion, we demonstrated that an increase in abstinence-induced PKA-mediated signaling in the PFC-NAc pathway is necessary for the expression of cue-induced cocaine seeking. In contrast, PKA-mediated molecular targets are normalized after cue-induced relapse which may represent a shift in neuronal activity in response to drug-associated cues during abstinence. Understanding the dynamic change in PKA-regulated phosphoproteins in the PFC-NAc pathway after abstinence from cocaine SA may provide novel molecular targets for the development of therapeutic approaches to cocaine addiction.
Supplementary Material
Figure 1 SuppInfo. No effects of abstinence on p-ERK in the dmPFC and p-synapsin I Ser62/67 and Ser603 in the NAc after cocaine self-administration. (A) p-ERK, (B) p-synapsin I Ser62/67, and (C) p-synapsin I Ser603. The bar graphs represent the mean ± SEM. S=Saline; C=Cocaine.
Acknowledgments
We thank Phong Do, Andrew Nowak, and John Yang for excellent technical assistance. This research was supported by P50 DA015369 and the MUSC Neuroscience Institute.
Footnotes
Author contributions: WLS and JFM were responsible for the study concept and design. WLS, NTC, AZM, TWW, and SMB conducted the behavioral and/or molecular experiments. WLS, TWW, and JFM wrote the manuscript. All authors critically reviewed content and approved the final version for publication.
The authors report no biomedical financial interests or potential conflicts of interest.
References
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluA1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Ferrario CR, Glucksman MJ, Wolf ME. Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J Neurochem. 2009;110:363–377. doi: 10.1111/j.1471-4159.2009.06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriles N, Rodaros D, Sorge RE, Stewart J. A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2003;168:66–74. doi: 10.1007/s00213-002-1283-z. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Sanna PP, Weiss F. Cocaine-predictive stimulus induces drug-seeking behavior and neural activation in limbic brain regions after multiple months of abstinence: reversal by D(1) antagonists. Proc Natl Acad Sci U S A. 2001;98:1976–1981. doi: 10.1073/pnas.98.4.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng L-J, Shaham S, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–124. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Graham DL, Bachtell RK, Self DW. Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci. 2007;25:2201–2213. doi: 10.1111/j.1460-9568.2007.05473.x. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi S-H, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nature Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Ford KA, Wolf ME, Hu XT. Plasticity of L-type Ca2+ channels after cocaine withdrawal. Synapse. 2009;63:690–697. doi: 10.1002/syn.20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Hearing MC, Miller SW, See RE, McGinty JF. Relapse to cocaine seeking increases activity-regulated gene expression differentially in the prefrontal cortex of abstinent rats. Psychopharmacology (Berl) 2008;198:77–91. doi: 10.1007/s00213-008-1090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope BT, Crombag HS, Jedynak JP, Wise RA. Neuroadaptations of total levels of adenylate cyclase, protein kinase A, tyrosine hydroxylase, cdk5 and neurofilaments in the nucleus accumbens and ventral tegmental area do not correlate with expression of sensitized or tolerant locomotor responses to cocaine. J Neurochem. 2005;92:536–545. doi: 10.1111/j.1471-4159.2004.02891.x. [DOI] [PubMed] [Google Scholar]
- Iwata S, Hewlett GH, Ferrell ST, Czernik AJ, Meiri KF, Gnegy ME. Increased in vivo phosphorylation state of neuromodulin and synapsin I in striatum from rats treated with repeated amphetamine. J Pharmacol Exp Ther. 1996;278:1428–1434. [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Caine SB, Parsons L, Markou A, Weiss F. Opponent process model and psychostimulant addiction. Review Pharmacology, Biochemistry & Behavior. 1997;57:513–521. doi: 10.1016/s0091-3057(96)00438-8. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufahl PR, Zavala AR, Singh A, Thiel KJ, Dickey ED, Joyce JN, Neisewander JL. c-Fos expression associated with reinstatement of cocaine-seeking behavior by response-contingent conditioned cues. Synapse. 2009;63:823–835. doi: 10.1002/syn.20666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Shaham Y, Hope BT. Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem. 2003;85:1604–1613. doi: 10.1046/j.1471-4159.2003.01824.x. [DOI] [PubMed] [Google Scholar]
- Lynch WJ, Taylor JR. Persistent changes in motivation to self-administer cocaine following modulation of cyclic AMP-dependent protein kinase A (PKA) activity in the nucleus accumbens. Eur J Neurosci. 2005;22:1214–1220. doi: 10.1111/j.1460-9568.2005.04305.x. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Nairn AC. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J Biol Chem. 1999;274:10086–10093. doi: 10.1074/jbc.274.15.10086. [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- Mendelson JH, Mello NK. Management of cocaine abuse and dependence. N Engl J Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF. Altered prelimbic cortex output during cue-elicited drug seeking. J Neurosci. 2004;24:6889–6897. doi: 10.1523/JNEUROSCI.1685-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 2005;312:1305–1313. doi: 10.1124/jpet.104.075184. [DOI] [PubMed] [Google Scholar]
- O’Brien CP. A range of research-based pharmacotherapies for addiction. Science. 1997;278:66–70. doi: 10.1126/science.278.5335.66. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluA1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci. 2002;22:2916–2925. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Waston C. The rat brain in stereotaxic coordinates. 5. Academic; London: 2005. [DOI] [PubMed] [Google Scholar]
- Perlini LE, Botti F, Fornasiero EF, Giannandrea M, Bonanomi D, Amendola M, Naldini L, Benfenati F, Valtorta F. Effects of phosphorylation and neuronal activity on the control of synapse formation by synapsin I. J Cell Sci. 2011;124:3643–3653. doi: 10.1242/jcs.086223. [DOI] [PubMed] [Google Scholar]
- Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluA1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Rebec GV. Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci. 2006;26:8004–8008. doi: 10.1523/JNEUROSCI.1413-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zhou L, Hazim R, Quinones-Jenab V, Jenab S. Effects of acute cocaine on ERK and DARPP-32 phosphorylation pathways in the caudate-putamen of Fischer rats. Brain Res. 2007;1178:12–19. doi: 10.1016/j.brainres.2007.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Birnbaum S, Ubriani R, Arnsten AF. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci. 1999;19:RC23. doi: 10.1523/JNEUROSCI.19-18-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KL, Arroyo M, Everitt BJ. Induction of the learning and plasticity-associated gene Zif268 following exposure to a discrete cocaine-associated stimulus. Eur J Neurosci. 2003;17:1964–1972. doi: 10.1046/j.1460-9568.2003.02617.x. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Lubbers BR, Goriounova NA, Li KW, Van der Schors RC, Loos M, Riga D, Wiskerke J, Binnekade R, Stegeman M, Schoffelmeer AN, Mansvelder HD, Smit AB, De Vries TJ, Spijker S. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology. 2010;35:2120–2133. doi: 10.1038/npp.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton BJ, Seipel AT, Phillips PE, Wetsel WC, Gitler D, Greengard P, Augustine GJ, Wightman RM. Cocaine increases dopamine release by mobilization of a synapsin-dependent reserve pool. J Neurosci. 2006;26:3206–3209. doi: 10.1523/JNEUROSCI.4901-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, McGinty JF. Glutamate-dopamine interactions mediate the effects of psychostimulant drugs. Addiction Biology. 1999;4:141–150. doi: 10.1080/13556219971641. [DOI] [PubMed] [Google Scholar]
- White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- Whitfield TW, Jr, Shi X, Sun WL, McGinty JF. The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci. 2011;31:834–842. doi: 10.1523/JNEUROSCI.4986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavala AR, Osredkar T, Joyce JN, Neisewander JL. Upregulation of Arc mRNA expression in the prefrontal cortex following cue-induced reinstatement of extinguished cocaine-seeking behavior. Synapse. 2008;62:421–431. doi: 10.1002/syn.20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowska B, Kielbinski M, Gieryk A, Soria G, Maldonado R, Przewlocki R. Regulation of the immediate-early genes arc and zif268 in a mouse operant model of cocaine seeking reinstatement. J Neural Transm. 2011;118:877–887. doi: 10.1007/s00702-011-0583-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1 SuppInfo. No effects of abstinence on p-ERK in the dmPFC and p-synapsin I Ser62/67 and Ser603 in the NAc after cocaine self-administration. (A) p-ERK, (B) p-synapsin I Ser62/67, and (C) p-synapsin I Ser603. The bar graphs represent the mean ± SEM. S=Saline; C=Cocaine.