

Abstract
Psychiatric disorders are among the most intractable enigmas in medicine. In the past five years, there has been unprecedented progress on the genetics of many of these conditions. In this review, we discuss the genetics of nine cardinal psychiatric disorders (Alzheimer’s disease, attention-deficit hyperactivity disorder, alcohol dependence, anorexia nervosa, autism spectrum disorder, bipolar disorder, major depressive disorder, nicotine dependence, and schizophrenia). Empirical approaches have yielded new hypotheses about etiology, and now provide data on the often debated genetic architectures of these conditions, which have implications for future research strategies. Further study using a balanced portfolio of methods to assess multiple forms of genetic variation is likely to yield many additional new findings.
Keywords: psychiatric disorders, genetics, structural variation, copy number variation, genome-wide association, meta-analysis, sequencing
Introduction
A core set of psychiatric conditions – madness, mania, melancholia – have been perplexing for millennia. Although mortality is increased for many psychiatric disorders, 1 their major impact is on morbidity: psychiatric disorders account for around a third of disability worldwide, 2 and cause enormous personal and societal burden. 3
In the past century, considerable efforts to understand the nature of psychiatric disorders have been undertaken. There have been successes, and a few diseases with prominent psychiatric manifestations that were once prevalent are now rare in many parts of the world (e.g., pellagra 4 and neurosyphilis 5). These few triumphs stand in contrast to decades of frustration and occasional notoriety when highly publicized and plausible findings failed to replicate. Indeed, most psychiatric disorders have been intractable to approaches that were fruitful in other areas of medicine. Thus, psychiatric syndromes are generally referred to as “disorders” (illnesses that disrupt normal function) and only a few as “diseases” (disorders with known pathophysiology or structural pathology). An obvious goal of psychiatric research is to convert idiopathic disorders into pathophysiologically-defined diseases.
Since 2007, numerous robust and replicable genetic findings have been reported for psychiatric disorders. These advances have mostly been via genome-wide association (GWAS) and structural variation (SV) studies, although studies of uncommon or rare exonic variation are likely to play a prominent role in the next few years. These results meet community standards in human genetics for significance and replication. 6 Although these findings often appeared in high-profile journals, sentiments like “genetics has failed in psychiatry” or “there are no genes for psychiatric disorders” are still heard. A review of psychiatric genetics is thus particularly opportune.
Over 300 psychiatric disorders have been described, and nine are covered in this review. The conditions selected are: all psychiatric disorders; have been subjected to intensive genetic study; and have published genome-wide results (usually GWAS and SV but also genome-wide linkage and resequencing). The disorders and their abbreviations are defined in Tables 1 and S1, and the heritabilites and lifetime prevalences are depicted in Figure 1a. Mental retardation could have been included, but its voluminous literature has been reviewed at length. 7–9 Studies of other psychiatric disorders are in progress, but the published data are few (e.g., obsessive-compulsive disorder, Tourette’s syndrome, and panic disorder).
Table 1.
Defining features of nine psychiatric disorders ‡
| Abbrev. | Name | Life prev | Heritability | Essential characteristics | Notable feature |
|---|---|---|---|---|---|
| AD | Alzheimer’s disease | 0.132 | 0.58 | Dementia, defining neuropathology | Of the top 10 causes of death in the US, AD alone has increasing mortality |
| ADHD | Attention-deficit hyperactivity disorder | 0.053 | 0.75 | Persistent inattention, hyperactivity, impulsivity | Costs estimated at ~$US 100×109/year |
| ALC | Alcohol dependence | 0.178 | 0.57 | Persistent ethanol use despite tolerance, withdrawal, dysfunction | Most expensive psychiatric disorder (total costs exceed $US 225×109/year) |
| AN | Anorexia nervosa | 0.006 | 0.56 | Dangerously low weight from self-starvation | Notably high standardized mortality ratio |
| ASD | Autism spectrum disorder | 0.001 | 0.80 | Markedly abnormal social interaction and communication beginning before age 3 | Huge range of function, from people requiring complete daily care to exceptional occupational achievement |
| BIP | Bipolar disorder | 0.007 | 0.75 | Manic-depressive illness, episodes of mania usually with MDD | As a group, nearly as disabling as SCZ |
| MDD | Major depressive disorder | 0.130 | 0.37 | Unipolar depression, marked and persistent dysphoria with physical/cognitive symptoms | Ranks #1 in burden of disease in world |
| NIC | Nicotine dependence | 0.240 | 0.67 | Persistent nicotine use with physical dependence (usually cigarettes) | Major preventable risk factor for many diseases |
| SCZ | Schizophrenia | 0.004 | 0.81 | Long-standing delusions and hallucinations | Life expectancy decreased by 12–15 years |
Figure 1. Results pertaining to genetic architecture.


(a) Plot of heritability by log10 lifetime prevalence for nine psychiatric disorders considered in this review plus three complex diseases for which genetic dissection has been particularly successful (Tables 1 and S1). Each disorder is plotted by as heritability by lifetime prevalence. Color indicates qualitative success in identifying etiological genetic variation (green=notably successful, khaki=some successes, red=minimal or no clear success to date). The bubble sizes are proportional to the numbers of cases studied in GWAS (the smaller circle indicating discovery Ncase and the larger circle the total Ncase for discovery plus replication samples). Abbreviations: ADHD=attention-deficit hyperactivity disorder, ALC=alcohol dependence, AD=Alzheimer’s disease, AN=anorexia nervosa, ASD=autism, BIP=bipolar disorder, BRCA=breast cancer, CD=Crohn’s disease, MDD=major depressive disorder, NIC=nicotine usage (maximum cigarettes per day), SCZ=schizophrenia, and T2DM=type 2 diabetes mellitus.
(b) Allelic spectrum of SCZ. The inset is a conceptual schematic from a 2008 Nature Genetics review. 10 The lower part of the figure depicts empirical results for SCZ. The x-axis is log10(AF) in controls. The y-axis is the point estimate for genotypic relative risk (GRR, log10). For clarity, confidence intervals are not shown. There are no known Mendelian variants for SCZ (AF ≪ 0.0001, GRR ≫ 50). There are no known common variants (AF > 0.05) with GRR > 1.5, and these can be excluded with > 99% statistical power. Nine SVs associated with SCZ are shown as light blue diamonds (Table 2, 1q21.1- is the deletion and 1q21.1+ is the duplication). If AF in controls was 0, AF was set to 0.0001. These SVs do not have a corresponding region in the inset. Seventeen common variants have been associated with SCZ (red circles, Table 3). SNPs contributing to the PGC SCZ risk profile score 58 (21,171 autosomal SNPs with PT < 0.1, Box 3, panel b) are shown in light blue dots with a lowess smoother in dark blue.
The genetic dissection of complex traits has been frequently reviewed. 6,10–12 Box 1 provides an overview of the approaches and study design considerations. Advances in genetics are often yoked to technological advancements. Major approaches that have been informative in psychiatric genetics include assessment of: structural variation (SV) via karyotyping, array-based methods, and high-throughput sequencing (Tables 2 and S2); 13–16 genome-wide association studies (GWAS) using highly multiplexed SNP arrays and, potentially, high-throughput sequencing (Table 3); 12,17–19 and high-throughput sequencing to uncover rare variants of relatively strong effect (perhaps arising de novo). 20,21 Genome-wide linkage and hypothesis-driven candidate gene association studies have also been conducted, and, as in many areas of biomedicine, with uncertain yield. 22–26
Box 1. Study design considerations: simplex and multiplex.
Study design is a crucial component of human genetics research. The major designs are case-control and pedigree-based studies. The most common design is the case-control study in which the frequency of a genetic variant in those with a disorder is contrasted with the appropriate control group. Case-control designs are used in most GWAS 155 and next-generation sequencing studies as they are efficient and conceptually straight-forward. 156 Case-control studies are simpler, and most biases can be surmounted by careful study procedures, but cannot delineate rare inherited from de novo variation. Family-based designs are more complex but can be used for association testing as well as for linkage evaluation of co-segregation of genotypes and phenotypes within pedigrees. They provide protection against a key form of bias (population stratification artifacts), but are less efficient given that the unit of analysis is a set of relatives; however, it is possible to identify mutations that arise de novo.
An additional decision is whether to focus on the presence or absence of other affected family members (“multiplex” and “simplex” pedigrees, respectively). Human genetic studies have classically focused on multiplex pedigrees under the assumption that these pedigrees are enriched for causal genetic variation with higher penetrance. A focus on multiplex pedigrees has led to the identification of specific mutations underlying hundreds of Mendelian disorders (including ASD and AD). Simplex pedigrees have become popular for ASD and SCZ. Simplex-based ascertainment is tailored to evaluate de novo mutations, and predicated on a model in which disorder with dramatically reduced fecundity and a proven role of de novo SV might be explicable as a series of Mendelian disorders attributable to recent high-penetrance mutations in any of a large number of genes.
However, this important choice is not simple, and continues to be moderately controversial. Some investigators believe a focus on simplex pedigrees to be optimal, and other investigators have concerns about the implications of this decision. Some of the issues are listed below. (a) Correct classification as simplex or multiplex requires confident knowledge of family history – many people either do not know their family psychiatric histories, true episodes of illness may have been kept private from other relatives, and some affected individuals can over-call illness in their relatives (e.g., an individual with ALC labeling all relatives who drink as the same). (b) Fecundity is a major confound. If there are greater numbers of relatives, there is a greater chance of multiplex classification. In addition, the presence of a psychiatric disorder can reduce fecundity (e.g., fecundity is reduced in SCZ and having a child with ASD can be a powerful inducement not to reproduce further). If fecundity had not been inhibited due to a psychiatric disorder, some apparently simplex families might have been revealed to be multiplex. (c) Simplex designs often require both parents. This complicates recruitment, increases genetic assay costs, and becomes increasing less practical for disorders with later ages of onset. (d) Both designs have a hidden weakness in the possibility of enriching for environmental causes of illness. Many psychiatric disorders have multiple different but rare environmental risk factors sufficient to cause a disorder. These potent exposures are sometimes very difficult to detect or not routinely evaluated. Examples include mercury poisoning and ASD or viral meningitis and SCZ. Contrary to its intent, simplex cases may be enriched for difficult to detect, individual-specific environmental causes. Multiplex ascertainment could enrich for shared environmental causes.
Some recent data pertain to this choice. Unexpectedly, simplex and multiplex ASD pedigrees show relatively similar de novo mutation rates for SV 80,81 and exonic variation. 83–85 It is possible that larger studies will find simplex/multiplex differences in de novo mutation rates but the magnitude is likely to be smaller than anticipated. For SCZ, the available data are insufficient to resolve this issue. 45,50 It has also been pointed out that de novo events must confer risk in multiplex families, as such mutations increase the chance that an individual is affected and increase risk in that person’s offspring. Intriguingly, there are three instances of ASD cases with de novo deletions of 16p11.2 who also had an affected sibling without this deletion 157–159 along with similar observations for SV in 1q21.1 and 17p12. 158
Table 2.
Structural variation associated with psychiatric disorders.
| SV | Location (mb) | Genes | Type | Disorder | Freqcase | Freqcontrol | OR | P | Citation | Other associations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1q21.1 | chr1:145.0–148.0 | 34 | Deletion | SCZ | 0.0018 | 0.0002 | 9.5 | 8×10−6 | 184 | DD, MR, micro-/macrocephaly, dysmorphia, epilepsy, cataract, cardiac defects, possibly ASD, 185 Thrombocytopenia-Absent Radius syn 48,158,184–188 |
| Duplication | SCZ | 0.0013 | 0.0004 | 4.5 | 0.02 | 184 | ||||
|
| ||||||||||
| 2p16.3 | chr2:50.1–51.2 | NRXN1 | Deletion | ASD | 0.004 | 80 | DD, MR, epilepsy, Pitt-Hopkins-like syndrome-2 | |||
| exons | Deletion | SCZ | 0.0018 | 0.0002 | 7.5 | 1×10−6 | 184 | |||
|
| ||||||||||
| 3q29 | chr3:195.7–197.3 | 19 | Deletion | SCZ | 0.0010 | 0.0 | 3.8 | 4×10−4 | 184 | DD, MR, possibly ASD |
|
| ||||||||||
| 7q11.23 | chr7:72.7–74.1 | 25 | Duplication | ASD | 0.0011 | 0.003 | 80 | DD, MR. Deletion: Williams–Beuren syndrome | ||
|
| ||||||||||
| 7q36.3 | chr7:158.8–158.9 | VIPR2 | Duplication | SCZ | 0.0024 | 0.0001 | 16.4 | 4×10−5 | 44,184 | - |
|
| ||||||||||
| 15q11.2 | chr15:23.6–28.4 | 70 | Duplication | ASD | 0.0018 | 4×10−9 | 80 | DD, MR, Prader-Wili/Angelman syndromes 188 | ||
|
| ||||||||||
| 15q13.3 | chr15:30.9–33.5 | 12 | Duplication | ADHD | 0.0125 | 0.0061 | 2.1 | 2×10−4 | 119 | |
| Duplication | ASD | 0.0013 | 2×10−5 | 80 | ||||||
| Deletion | SCZ | 0.0019 | 0.0002 | 12.1 | 7×10−7 | 184 | DD, MR, epilepsy 188,189 | |||
|
| ||||||||||
| 16p13.11 | chr16:15.4–16.3 | 8 | Duplication | ADHD | 0.0164 | 0.0009 | 13.9 | 8×10−4 | 118 | Deletion: DD, epilepsy 188,189 |
|
| ||||||||||
| 16p11.2 | chr16:29.5–30.2 | 29 | Deletion | ASD | 0.0037 | 5×10−29 | 80 | DD, MR, epilepsy, macrocephaly, obesity 190,191 | ||
| Duplication | ASD | 0.0013 | 2×10−5 | 80 | DD, MR, epilepsy, microcephaly, low BMI 190,191 | |||||
| Duplication | SCZ | 0.0031 | 0.0003 | 9.5 | 3×10−8 | 184 | ||||
|
| ||||||||||
| 17q12 | chr17:34.8–36.2 | 18 | Deletion | ASD | 0.0017 | 0.0 | 6.12 | 9×10−4 | 192 | |
| Deletion | SCZ | 0.0006 | 0.0 | 4.49 | 3×10−4 | |||||
|
| ||||||||||
| 22q11.21 | chr22:18.7–21.8 | 53 | Del or dup | ASD | 0.0013 | 0.002 | 80 | |||
| Deletion | SCZ | 0.0031 | 0.0 | 20.3 | 7×10−13 | 184 | DD, MR, velocardiofacial-DiGeorge syndrome | |||
Locations are NCBI Build 37/UCSC hg19. The positions of these SV are denoted in Figure S1 with yellow circles. For succinctness, the citations refer to the most comprehensive study rather than an initial report. Genes refers to UCSC knownGenes. OR=odds ratio. DD=developmental delay, MR=mental retardation, BMI=body mass index.
Table 3.
GWAS findings for psychiatric disorders.
| Phenotype | SNP | Location | Discovery GWAS | Largest meta-analysis | P value | OR | Nearest gene |
|---|---|---|---|---|---|---|---|
| AD | rs3818361 | chr1:207784968 | 2018/532434 | <19870/3984635 | 3.7×10−14 | 1.18 | CR1 |
| AD | rs744373 | chr2:127894615 | 3006/14642193 | <19870/3984635 | 2.6×10−14 | 1.17 | BIN1 |
| AD | rs9349407 | chr6:47453378 | 8309/736636 | 18762/2982736 | 8.6×10−9 | 1.11 | CD2AP |
| AD | rs11767557 | chr7:143109139 | 8309/736636 | 18762/3559736 | 6.0×10−10 | 1.11 | EPHA1 |
| AD | rs11136000 | chr8:27464519 | 3941/784833 | 8371/26965193 | 1.6×10−16 | 1.18 | CLU |
| AD | rs610932 | chr11:59939307 | 6688/1325135 | >19000/3800035 | 1.2×10−16 | 1.10 | MS4A cluster |
| AD | rs3851179 | chr11:85868640 | 3941/784933 | 8371/26966193 | 3.2×10−12 | 1.15 | PICALM |
| AD | rs3764650 | chr19:1046520 | 5509/1153135 | >17000/3400035 | 5.0×10−21 | 1.23 | ABCA7 |
| AD | rs2075650 | chr19:45395619 | - | 8371/26966193 | 1×10−295 | 2.53 | APOE, TOMM40 |
| AD | rs3865444 | chr19:51727962 | 8309/736636 | 18762/2982736 | 1.6×10−9 | 1.10 | CD33 |
| ALCcon | rs1229984 | chr4:100239319 | -101 | - | 1.3×10−11 | - | ADH1B |
| ALCcon | rs6943555 | chr7:69806023 | -100 | - | 4.1×10−9 | - | AUTS2 |
| ALCcon | rs671 | chr12:112241766 | -99 | - | 3×10−211 | - | ALDH2 |
| BIP | rs12576775 | chr11:79077193 | 7481/925159 | 11974/5179359 | 4.4×10−8 | 1.14 | ODZ4 |
| BIP | rs4765913 | chr12:2419896 | 7481/925059 | 11974/5179259 | 1.5×10−8 | 1.14 | CACNA1C |
| BIP | rs1064395 | chr19:19361735 | 682/1300194 | 8441/35362194 | 2.1×10−9 | 1.17 | NCAN |
| NDsc | rs3025343 | chr9:136478355 | 41,27892 | 64,92492 | 3.6×10−8 | 1.13 | DBH |
| NDcon | rs1329650 | chr10:93348120 | 38,18192 | 73,85392 | 5.7×10−10 | - | LOC100188947 |
| NDint | rs6265 | chr11:27679916 | 74,03592 | 143,02392 | 1.8×10−8 | 0.94 | BDNF |
| NDcon | rs1051730 | chr15:78894339 | 38,18192 | 73,85392 | 2.8×10−73 | - | CHRNA3 |
| NDcon | rs3733829 | chr19:41310571 | 38,18192 | 73,85392 | 1.0×10−8 | - | EGLN2, CYP2A6 |
| SCZ | rs1625579 | chr1:98502934 | 9394/1246258 | 17839/3385958 | 1.6×10−11 | 1.12 | MIR137 |
| SCZ | rs2312147 | chr2:58222928 | - | 18206/42536195 | 1.9×10−9 | 1.09 | VRK2 |
| SCZ | rs1344706 | chr2:185778428 | 479/2937174 | 18945/38675196 | 2.5×10−11 | 1.10 | ZNF804A |
| SCZ | rs17662626 | chr2:193984621 | 9394/1246358 | 17839/3386058 | 4.6×10−8 | 1.20 | PCGEM1 |
| SCZ | rs13211507 | chr6:28257377 | 3322/358769 | 18206/42536195 | 1.4×10−13 | 1.22 | MHC |
| SCZ | rs7004635 | chr8:3360967 | 9394/1246558 | 17839/3386258 | 2.7×10−8 | 1.10 | MMP16 |
| SCZ | rs10503253 | chr8:4180844 | 9394/1246458 | 17839/3386158 | 4.1×10−8 | 1.11 | CSMD1 |
| SCZ | rs16887244 | chr8:38031345 | 3750/646867 | 8133/1100767 | 1.3×10−10 | 1.19 | LSM1 |
| SCZ | rs7914558 | chr10:104775908 | 9394/1246658 | 17839/3386358 | 1.8×10−9 | 1.10 | CNNM2 |
| SCZ | rs11191580 | chr10:104906211 | 9394/1246758 | 17839/3386458 | 1.1×10−8 | 1.15 | NT5C2 |
| SCZ | rs11819869 | chr11:46560680 | 1169/3714197 | 3738/7802197 | 3.9×10−9 | 1.25 | AMBRA1 |
| SCZ | rs12807809 | chr11:124606285 | - | 18206/42536195 | 2.8×10−9 | 1.12 | NRGN |
| SCZ | rs12966547 | chr18:52752017 | 9394/1246858 | 17839/3386558 | 2.6×10−10 | 1.09 | CCDC68 |
| SCZ | rs9960767 | chr18:53155002 | - | 18206/42537195 | 4.2×10−9 | 1.20 | TCF4 |
| SCZ+BIP | rs1344706 | chr2:185778428 | 479/2937174 | 21274/38675196 | 4.1×10−13 | 1.11 | ZNF804A |
| SCZ+BIP | rs2239547 | chr3:52855229 | 9394/1247158 | 16374/1404658 | 7.8×10−9 | 1.12 | ITIH3-ITIH4 |
| SCZ+BIP | rs10994359 | chr10:62222107 | 9394/1247058 | 16374/1404558 | 2.4×10−8 | 1.22 | ANK3 |
| SCZ+BIP | rs4765905 | chr12:2349584 | 9394/1246958 | 16374/14044 | 7.0×10−9 | 1.11 | CACNA1C |
Table 3 focuses on results achieving genome-wide significance in large samples. We use a significance threshold of 5×10−8 (reference 198). Most associations achieving this level of significance are secure but some may ultimately prove not to be. Included are SNPs with p < 5×10−8 that were evaluated in samples of a minimum of around 10,000 cases and 10,000 controls. Discovery sample sizes reflect the primary samples for which full GWAS were conducted. In most cases, discovery P values were > 5×10−8 but met a threshold (typically 1×10−5) for inclusion in replication efforts. In some instances, simultaneous publications based upon overlapping samples were considered “discovery”. Where this occurred, providing samples of roughly equivalent sizes, the most significant primary GWAS findings are given; otherwise, the largest discovery samples are favored. For many studies, it was not possible to extract the exact sample size used for each locus so the sample sizes above are approximate. P-values and ORs are from the meta-analysis with the largest sample sizes. If two meta-analyses based on overlapping samples reported similar results, the “discovery” study is cited. The genes nearest each locus are provided but etiological variants are generally unknown, and it remains likely that some of the associations do not alter the function of the designated genes (e.g., ITIH3-ITIH4 where multiple correlated SNPs span many genes, and TCF4-CCDC68 where statistically independent associations occur in TCF4 and closer to CCDC68). In the TCF4-CCDC68 example, it may be that both associations point to the same functional element but, it is also possible that independent etiological variants occur in adjacent genes.
For the SCZ loci attributed to Steinberg et al., 195 no discovery sample size is listed because the initial P values were modest and, as the authors conducted multiple follow-up analyses, there is no obvious discovery sample. At the MHC, the International Schizophrenia Consortium 69 is designated discovery as it was the only primary GWAS for which genome-wide significance at the MHC was obtained. Steinberg et al. is cited for the meta-analysis at the MHC as it reported the most significant MHC association. 195 The most significant SNP at the MHC across the two studies is not identical, and the one listed is that from Steinberg et al. Multiple statistically independent SNPs have been reported at the MHC. 58,186 We note that genome-wide significance had been reported in BIP for ANK3199 but not in a larger mega-analysis including the same samples. 59 Others have reported genome-wide significance for composite phenotype studies of ITIH3-ITIH4200 (but see reference 201) and CACNA1C 202 but in samples smaller than required for inclusion in the above table.
ALCcon means alcohol consumption. The rs671 association was in East Asian samples. The ADH1B locus was also associated with ALC.
NDcon means nicotine consumption (as maximal cigarettes per day, continuous). NDinit means smoking initiation (ever versus never began smoking). NDsc means smoking cessation (whether regular smokers had quite at time of interview).
In this Review, we summarize the literature for the nine disorders in Table 1 with particular emphasis on the findings that appear to meet community standards for replication in human genetics (i.e., robustly significant with consistent effects across samples). 6 We highlight new hypotheses that have emerged across the allelic spectrum including de novo and rare exonic mutations, rare SV, and common variation from GWAS. Critically, these results provide empirical insights into the genetic architectures of these disorders, data that are essential to guiding future work in this area.
Alzheimer’s Disease (AD)
Rare variation
Prior to 2007, rare autosomal dominant mutations in APP, PSEN1, and PSEN2 were known to cause early-onset familial AD. 27 These loci have atypically large effect sizes, which facilitated identification using “past generation” technologies like candidate gene association and genome-wide linkage studies (Table S2). Treatments for AD based on these findings have been developed and are undergoing testing. Rare SV duplications containing APP have been associated with AD. 28,29 Small exome sequencing studies of AD have been published, 30,31 and larger studies are in progress and should provide a more nuanced understanding of the role of rare exonic mutations in the pathogenesis of AD.
Common variation
In the early 1990’s, APOE was identified by candidate gene association as a susceptibility gene for late-onset AD in no small part due to its unusually large effect size (Table 3). 27,32 In 2009, GWAS from two large consortia 33,34 implicated three novel loci and six additional loci were identified in 2011. 35,36 Full meta-analyses are keenly awaited, but the 10 loci identified to date account for ~20% of the total variation in risk or ~33% of the risk attributable to genetic effects, with the major contribution being from APOE. Note that the association of one gene identified by GWAS, CR1, might result from SV. 37
Intriguingly, pathway analyses (Box 2) of AD implicate cholesterol metabolism and the innate immune response. 38 Genes attaining genome-wide significance point toward immune and inflammatory processes (CLU and CR1), lipid processing (APOE, CLU, and ABCA7), and endocytosis (PICALM, BIN1, CD2AP, and CD33). Altered immune function and lipid metabolism had previously been proposed as AD risk factors, but whether these represented causation or reverse causation was unclear. 39 The genetic findings now strongly point to the former.
Box 2. Pathway analysis.
Pathway analysis is based on the assumption that risk variants for a disease will converge on sets of genes whose functions are more closely related to each other than to random sets of genes. For example, dominant forms of AD are caused by mutations in APP, PSEN1, and PSEN2; the latter two genes encode protein components of γ-secretase, a protease that cleaves APP. The availability of GWAS and SV results for many psychiatric disorders, along with increasing amounts of sequence data, have generated interest in using analytic methods for exploiting non-random functional relationships between genes containing risk variants. Many approaches have been developed (e.g., ALIGATOR, 161 INRICH, 162 162 DAPPLE, 163 and GRAIL 164) and reviewed in detail elsewhere. 146,165,166 Although the algorithms differ, the principle behind these methods is to evaluate whether a given set of genomic regions (i.e., a broadly inclusive definition of “pathway”) is enriched for genetic variants showing some relationship with disease compared to a null expectation.
There are important subsidiary considerations. The first is the definition of a “pathway.” Standard pathways consist of sets of genes found in the Gene Ontology, 167 the Kyoto Encyclopedia of Genes and Genomes, 168 or PANTHER databases. 169 Other pathway gene sets are manually curated by experts in a particular area (e.g., genes known to make proteins that function at the synapse). 170,171 In addition, a “pathway” can consist of genomic regions selected for a particular property such as high degree of conservation 172 or eQTL associations. 173 Finally, other pathways consist of genes known to be connected via experimental data (e.g., via protein-protein interaction screens, micro-RNA target sites, or gene expression modules).
It is advantageous that these pathway datasets are defined independently of genetic studies of psychiatric disorders, but they do have limitations and can contain errors of omission and commission. Standard gene sets can have highly overlapping pathways that complicate some analyses, and the pathway content can have variable quality. Expert-curated pathways can be more specific, but can be vulnerable to post-hoc bias (i.e., including genes in a pathway based on results from genetic studies). Pathways based on empirical approaches depend on the quality and completeness of the primary data (e.g., existing protein-protein interaction databases cover the interaction space partially).
A second question concerns what is required before a member of a pathway is accepted as having some relationship with disease. For common variation, the analysis might be restricted to genes within recombination regions containing SNPs that are genome-wide significant, an approach which was used successfully to implicate broad biological pathways relevant to height. 75 However, much of the interest in pathway analysis involves exploiting much weaker associations under the assumption that these associations more reflect true associations in the context of limited power (signal) rather than chance (noise). If so, those weakly associated SNPs may also be non-random with respect to gene-sets (see also Box 3). The threshold at which SNPs or genes are chosen is arbitrary, and the signal-to-noise ratio for a given arbitrary threshold can vary substantially with sample size and genetic architecture. For rare variants in complex diseases, based on recent empirical results for ASD, it is probably reasonable to assume that pathway analysis will by necessity be based on sets of genes whose involvement in disease is unclear (e.g., genes with a single observed de novo exonic deleterious mutation). 83,84
A third consideration concerns the null expectation to which the observed pathways are compared. Early SV pathway analysis did not account fully for important biases such as that large genes are more likely to be intersected by CNVs by chance and that some functional pathways – often related to brain development – are enriched for large genes. 164 Early GWAS pathway analysis sometimes did not fully allow for the variable numbers of SNPs per gene and their degree of linkage disequilibrium both of which impact the probability of a high-ranking association. 161 Thus, one must be cautious about the utility of pathway based approaches. In psychiatric disorders, some results give cause for optimism. 38,45,81
Finally, in pathway analyses the unit of inference is the pathway. Tempting though it may be, it is generally inappropriate to make strong inferences about specific variants or genes based upon their membership of pathways that attain some level of significance. It may be possible to do so if the variants or genes are subsequently evaluated in datasets independent of those from which the significance of the pathways are derived, using a statistical framework that adequately deals with multiple testing.
It is unclear how the above findings relate to accumulation of beta-amyloid (Aβ) in AD pathogenesis, but some relationship seems likely. For example, PICALM (phosphatidylinositol-binding clathrin assembly protein) and other endocytic molecules can modify Aβ toxicity in yeast and other model systems. 40 Although these genetic findings provide support for novel causal relationships that could be targeted by treatments, the association data point to genomic regions, not genes. Moreover, the proximal steps from genotype to phenotype are unclear, and many of the implicated genes are plausibly involved in multiple relevant functions (e.g., CLU is involved in altered immune function and lipid processing).
Psychotic Disorders (BIP and SCZ)
Rare variation
Unfortunately, unlike AD, no Mendelian forms of BIP and SCZ have been identified. 41 However, rare (frequency < 0.5%) but potent (genotypic relative risk, GRR, 5–20) SVs play a role in a small proportion of cases with SCZ (Table 2, Figure S1). None is fully penetrant, and nearly all appear to be non-specific as risk is often increased for SCZ, ASD, developmental delay, mental retardation, epilepsy, somatic dysmorphism, and extremes of body mass and head size. Most of these SV regions are fairly large (hundreds of kb to mb) and generally center on SV hotspots. 42 Two rare SVs affect single genes (NRXN1 and VIPR2) 43,44 offering opportunities for down-stream functional studies. Pathway analyses of genes intersected by rare SV suggest enrichment for neuronal processes of plausible etiological relevance (e.g., post-synaptic signaling). 45–47 The SV regions in Table 2 probably represent “low-hanging fruit” and more discoveries are likely with application of improved technologies for SV detection to larger samples. 15
A complementary approach is to evaluate SV “burden” in cases compared to controls (e.g., number of SV per person). 48,49 This tests an explicitly multigenic model whereby many rare but different genomic disruptions impact disease risk. Increased SV burden in SCZ cases has been reported by multiple groups. 47,48,50 One report found more rare SV in SCZ cases (odds ratio, OR=1.15), particularly for large deletions (OR=3.6). 48 De novo SV are also more common in SCZ cases. 45 For BIP, there are reports of increased 51–53 and similar SV burden in cases versus controls 54,55 De novo SV may be relevant in BIP (OR=4.8), particularly in cases with earlier ages of onset (OR=6.3). 53
Multiple studies are now evaluating the role of de novo, rare, and uncommon exonic variation in BIP and SCZ using resequencing or genotyping approaches. Two small exome sequencing studies 56,57 reported rates of putatively functional mutations that exceeded null expectations in SCZ cases (although the rate of de novo point mutations was not elevated in cases and specific genes were not identified). Larger studies are ongoing and will illuminate this area in 2012–2013.
Common variation
The Psychiatric Genomics Consortium (PGC) recently published mega-analyses for SCZ and BIP. 58,59 In SCZ, 9,394 cases and 12,462 controls were combined in a single analysis and the top 81 statistically independent loci from that analysis were then tested in over 8,000 cases. The mega-analysis identified seven significant loci (Table 3). A sign test for consistency between the mega-analysis and follow-up stage was highly significant, implying that many of the 81 top loci include true risk loci but that power was insufficient. For BIP, the discovery phase consisted of 7,481 cases and 9,250 controls with follow-up of 34 statistically independent loci in around 4,500 cases. Two loci exceeded genome-wide significance (Table 3). Similarly, a sign test between the discovery and follow-up results was highly significant, again suggesting insufficient power. 59
In BIP, the genome-wide significant association at CACNA1C (α subunit of the L-type voltage-gated calcium channel) deserves specific comment given its mechanistic implications. Indeed, multiple voltage gated calcium channel subunits were among the top 34 loci followed up in the BIP GWAS. Calcium channels regulate neuronal excitability (already a treatment target for BIP) and multiple brain functions including long term potentiation and synaptic plasticity. Combined analysis of the PGC BIP and SCZ samples strengthened the association in the CACNA1C region. Further, results from SGENE+ 60 implicate NRGN (neurogranin) which may act as a calcium sensor. 61 Therefore, detailed investigation of brain calcium biology is warranted for both BIP and SCZ.
For SCZ, the strongest association is in the extended MHC region (chr6:27–33 Mb). The evidence for association is compelling but high gene density and exceptionally high linkage disequilibrium complicate the identification of specific sequence variation. Although tempting to propose that the association supports long-standing hypotheses concerning roles in SCZ for intra-uterine infection, autoimmunity, or even synaptic pruning (in which MHC genes play a role), this lack of precision renders such propositions speculative.
A novel association for SCZ is in Ensembl gene RP11-490G2.1 which encodes the primary transcript for miR-137 (MIR137). 62 Supporting the hypothesis that this association implicates MIR137, predicted targets of miR-137 were significantly enriched for smaller GWAS p-values (p<0.01), and four of the genes that achieved genome-wide significance contain verified miR-137 binding sites. 63 miR-137 is a key regulator of neuronal development with roles in neurogenesis and maturation 64,65 and is highly expressed at synapses in the cortex and hippocampus. 66 Future studies of networks regulated by miR-137 offer the possibility of insights into SCZ pathophysiology.
GWAS of BIP and SCZ have been predominantly based on subjects of European ancestry, but there are increasing reports from other world ancestries. 67,68 Although those findings do not yet provide additional pathophysiological insights, it is worth noting that a chr8 locus found in an East Asian sample 67 has support in the PGC dataset, suggesting that planned mega-analyses across world populations will be informative.
Some of the most intriguing findings for SCZ and BIP are from large sets of genetic markers (Box 3). 69 There are now replicated data that vulnerability to SCZ is influenced by common genetic variation in hundreds of different loci, and this vulnerability partially overlaps that for BIP. 69 Indeed, the large-scale impact of large numbers of common variants may be a general feature of human complex traits 70–77
Box 3. Common variant risk profile.
For SCZ and BIP, sign tests comparing the consistency of association tests from discovery GWAS and replication samples for sets of top signals are usually highly significant even if most loci do not meet genome-wide significance. 58,59,174 This implies that sample sizes are insufficient and that additional loci can be discovered in larger samples. Tests of the existence of large numbers of true but weakly associated variants have been conducted for SCZ, BIP, and many other biomedical disorders.
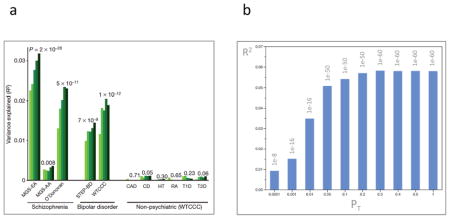
Based on theoretical work by Visscher and colleagues, one study used GWAS results as a discovery set (after removing correlated SNPs), and subjects in 11 independent test GWAS datasets were assigned risk profile scores (i.e., the number of SCZ risk alleles weighted by their effect sizes in the discovery set). The mean risk profile scores for cases were compared to the mean scores for controls in these independent datasets. 69 Panel a of the figure (reproduced from Figure 2 in reference 69) shows risk profile scores for extremely relaxed p-value thresholds (PT < 0.1, 0.2, 0.3, 0.4, and 0.5, light to dark green bars). Risk profile scores were derived (after LD-based SNP pruning) from a discovery SCZ sample, and then applied to three independent SCZ samples, 174,175 two BIP samples, 176,177 and six non-psychiatric diseases (CAD=coronary artery disease, CD=Crohn’s disease, HT=hypertension, RA=rheumatoid arthritis, T1D=type 1 diabetes, and T2D=type 2 diabetes). 177 In three independent GWAS, SCZ cases had significantly higher risk profile scores than controls. Remarkably, the same set of markers also discriminated BIP cases from controls indicating substantial genetic contributions between SCZ and BIP. As an important test of specificity, the SCZ risk profile was not predictive of case status for any of six non-psychiatric diseases. 177 A recent paper evaluated risk profile scores in a trio sample, and could exclude population stratification as an explanation. 178
The proportion of variance explained by the risk profile score increased with relaxation of the significance thresholds. This suggests the discovery sample was insufficiently large to identify many true risk loci at even nominal levels of significance: adding more SNPs contributed more genetic signal than noise. This is partly a feature of sample size. Panel b of the figure (data from Figure S6 in reference 58) shows a similar analyses based upon a larger discovery sample, and the proportion of variance explained is approximately double 58 and, instead of increasing with PT, the proportion of variance reaches a plateau. If sample size were truly adequate, the first PT bin would explain the greatest amount of variance, and relaxing PT would decrease R2.
Finally, estimates from two different methods indicated that the risk profile component for SCZ contributes between a quarter and a third of the overall variance in liability to SCZ, 69,130 a substantial fraction of the 65–81% heritability of SCZ. 179,180 These estimates suggest that “missing heritability” is merely hidden and imperfectly assayed by current genotyping technologies.
Autism Spectrum Disorders (ASD)
Rare variation
For ASD there is a notably strong prima facie case for there being a a cardinal role for rare variation. Karyotyping studies suggested that on the order of 5% of ASD cases have one of a large number of rare but relatively gross chromosomal abnormalities. 14,78 In addition, ASD has been noted as a comorbid feature of >100 single gene, Mendelian medical genetic syndromes, 79 although the penetrance and confidence of the clinical associations are variable. Indeed, ASD mutations with very high penetrance are exceptional (i.e., Rett syndrome mutations in MECP2 and CDKL5), and Mendelian diseases enriched for ASD have far less than complete penetrance (e.g., Fragile X syndrome and tuberous sclerosis). 78
Analysis of SV has been a major focus in ASD research (Table 2, Figure S1). Implicated loci to date are generally rare and potent risk factors but incompletely penetrant and not specific to ASD. As these large events impact the dosages of many genes, biological insight has been slow to emerge; however, pathway analyses of genes within SV do implicate neuronal processes of etiological relevance. 45–47 Large SVs are present in 5–10% of ASD cases, and the number of ASD SVs could total 130–234. 80 There is also consistent evidence for increased SV burden in ASD. 49,80–82 For example, 5.8% of ASD probands had ≥1 rare de novo SV versus 1.7% of their unaffected siblings (OR=3.5), and this difference was more pronounced for SV that intersected genes. 80 The 16p11.2 SV associated with ASD and SCZ has been termed a “mirror image” SV since the deletion and duplication are associated with increased and reduced head and body size. However, it is difficult to understand the clinical features of ASD and SCZ as mirror images and, more importantly, ASD is associated with both 16p11.2 deletions and duplications.
ASD is the first psychiatric disorder for which exome sequencing using substantial numbers of samples has been published. Three recent papers describe the results from exome sequencing of ~600 trios, and identify roles for de novo exonic mutations in SCN2A, KATNAL2, and CHD8 in the pathogenesis of ASD. 83–85 Intriguingly, all three studies noted an increased rate of de novo exonic mutations in older parents (with the mutations generally being of paternal origin), 83–85 and pathway analyses reported in two of the studies found that genes containing de novo exonic variation were more closely connected in reference to protein-protein interaction databases. 83,84 Additional sequencing studies are in progress.
However, a central finding from these papers was that only a minority of cases had a de novo putatively functional variant, suggesting that this class of genetic variation is unlikely fully to explain the clinical entity of ASD. Indeed, estimates from de novo exonic mutations (similar to those from SV data) suggest that ASD is highly polygenic (estimates ranged from 400–1000 genes). 84,85 Importantly, a hypothetical model of ASD caused by rare but fully penetrant mutations in 100 different genes could be confidently rejected. 83
Common variation
Evaluation of rare SV and exonic variation in ASD is particularly advanced. In contrast, evaluation of common variation is far more limited (Figure 1a) and the published GWAS for ASD are small by current standards. 86–89 It is currently not possible to discern or dismiss a role for common genetic variation in risk for ASD. In our opinion, GWAS with larger samples are needed for ASD, given that detailed studies of rare variation currently explain a fraction of risk and that common variation plays a clear role in other psychiatric disorders. Indeed, there were few confident findings for GWAS of SCZ when the sample sizes were similar to those now available for ASD. Additional support for our recommendation for more GWAS is provided by Voineagu et al. who identified a gene expression module that had attenuated expression in post-mortem brain samples of individuals with ASD and which also had enrichment for GWAS signals. 90
Alcohol and Nicotine Dependence (ALC & NIC)
ALC and NIC are complex conditions to study, given the requirement for ingestion of a psychoactive substance and cohort effects due to temporal and geographic variation in the availability of ethanol and nicotine. Many investigators focus on ALC and NIC, which are clinically salient but multi-component syndromes. 91 As part of the TAG consortium, 92 we determined that the components of the Fagerstrom Test for Nicotine Dependence (a measure of NIC) had heritabilities ranging from relatively high to near zero with important common environmental effects. Other investigators evaluated self-reported lifetime maximum use of ethanol (grams/day) or nicotine (cigarettes per day), and such continuous measures of consumption are often available for secondary analysis of samples studied for other diseases.
For ALC, the published GWAS are small and no large-scale meta-analysis has been conducted. 93–97 In our opinion, there are clear needs for a high-quality meta-analysis and to increase the number of samples with GWAS data – particularly given that risk profile analysis (Box 3) suggested that larger samples would yield more associations. 97 For alcohol consumption, GWAS in East Asian samples confirmed the role of ALDH2, 98,99 and AUTS2 was implicated in alcohol consumption in European subjects. 100 Using a candidate gene approach, the association of ADH1B with ALC and alcohol consumption was extended to European ancestry subjects. 101
For NIC, a field-wide meta-analysis is also needed. For smoking behavior, large meta-analyses have been conducted. 92,102,103 The strongest finding is an association of smoking quantity with a cluster of nicotinic receptor genes (CHRNA5-CHRNA3-CHRNB4) with an effect size corresponding to one cigarette per day, and there may be several independent associations. 104 Associations to this region have also bee reported for lung cancer. 105,106 A recent study showed that Chrna5 null mice had higher nicotine intake due to loss of an inhibitory effect on brain reward systems. 107
Major Depressive Disorder (MDD)
The PGC GWAS mega-analysis of 9,240 MDD cases and 9,519 controls (replication in 6,783 MDD cases) revealed no findings of genome-wide significance. 108 These null results are intriguing as almost all other published GWAS with N>11,000 for any disease has found at least one genome-wide significant finding. The most likely reasons for these results are particularly high heterogeneity of MDD and insufficient power arising from its lower heritability. 108 There are few published data on SV although one study found increased SV burden in MDD cases versus controls (OR=1.31). 109
A provocative finding from 2003 was that risk for MDD might be influenced by a gene-environment interaction with genetic variation near the serotonin transporter. 110 Meta-analyses have supported 111,112 and not supported 113,114 this finding. This association did not replicate in an independent but similar study from the same geographic region, casting particular doubt on the reported association. 115
Other disorders (ADHD and AN)
The published GWAS for ADHD 116 and AN 117 are small, but larger samples are in progress (e.g., by the Wellcome Trust Case-Control Consortium for AN). Given low power, no conclusions about common variation can be made. In ADHD, increased SV burden has been reported (OR=2.1), 118,119 an effect higher in ADHD cases with MR (OR=5.7). 118 Pathway analysis in ADHD found association signals enriched in the same GO categories also overrepresented for large SV. 120 The weak signals in ADHD GWAS are not randomly distributed but index the same pathophysiological pathways as rare SV. Thus, it appears that the reason no common variants have yet confidently been implicated in ADHD by GWAS is lack of power, not lack of variants to be found.
What is the Emerging Picture
Knowledge of psychiatric genetics is vastly greater than it was five years ago. Specifically, there are now multiple high-confidence SV (Table 2), rare exonic variants (currently only for ASD, AD, and ALC), and an increasing number of robustly significant and replicated common variants (Table 3). The data support multiple novel biological hypotheses (for example, cholesterol metabolism and the innate immune response in AD, a network involving miR-137 for SCZ, calcium signaling for BIP and SCZ, and chromatin remodeling for ASD) and reinforce previous hypotheses such as synaptic biology for SCZ and ASD).
Genetic architecture
These results also provide insights into genetic architecture that are critical for planning more complete attempts at the genetic dissection of these major public health conditions; now we can make informed predictions about the types of future studies that can increase understanding in order to generate well-grounded biological hypotheses.
For several disorders, there are now data to replace the interminable debate about the fundamental nature of these illnesses. 121 These occasionally vociferous debates 70 have generally been of an “either/or” nature: psychiatric disorders as collections of Mendelian-like, single gene disorders (multiple rare variant models) “versus” psychiatric disorders are caused by many common variants of small effects (common disease/common variant models). 15,122 Although we were initially agnostic 123 we now believe that the data support both positions.
For disorders with sufficient data (AD, BIP, and SCZ), the results are consistent with an allelic spectrum and an etiological role for both rare and common variation. As an example, Figure 1b synthesizes current knowledge of SCZ as an empirical allelic spectrum map compared to a conceptual schematic from a 2008 review in this journal. 10 There are no known Mendelian variants, and power analyses can exclude common variants of modest effect (genotypic relative risk > 1.5 for allele frequencies > 0.1). There are multiple SVs that are rare, strong, but non-specific risk factors (Table 2), and 17 common variant associations of subtle effects (Table 3). There is an important component arising from common variation in hundreds of different loci (Box 3), and larger sample sizes are likely to convert many of these to genome-wide significance. The frequency region between 0.001–0.05 is under investigation by studies evaluating exon variation, and more should be known in 2012–2013. This allelic spectrum map might well be replicated for other psychiatric disorders should larger studies of both rare, uncommon, and common variation be achieved.
Hypothesized genetic architectures consisting entirely of rare variants are inconsistent with the data for AD, ASD, BIP, and SCZ (as well as for multiple other complex biomedical diseases). 70–77 The Procrustean theory that common variant signals inevitably reflect “synthetic associations” 124 to rare, high penetrance mutations is not credible. 77,125–127
Psychiatric disorders are polygenic. The evidence is strong that many genes are involved in the etiology of AD (currently evidence of rare exonic, rare SV, and common variation), ALC (currently evidence of common variation), ASD (currently evidence of de novo exonic variation and SV), BIP (currently evidence of common variation), NIC (currently evidence of common variation), and SCZ (currently evidence of rare SV and common variation). Projections for ASD and SCZ suggest that variation at hundreds of different genes will ultimately be shown to be involved. 58,80,85 There are statistical hints that ADHD and MDD might also be polygenic.
Polygenicity may be a general feature of complex biomedical diseases. 128,129 Common variant SNP effects have been estimated to explain large proportions of the phenotypic heritability for a wide range of diseases: BIP and SCZ; 69,130 T1DM, T2DM, Crohn’s disease, rheumatoid arthritis, celiac disease, and coronary artery disease; 70,77 and continuous traits (height, intelligence, and body mass). 70,131,132 These results are consistent with suggestions that the “missing heritability” 133 is merely hidden. 132
As discussed further below, currently we do not now possess a comprehensive enumeration of loci associated with any psychiatric disorder (i.e., the “parts list”), regardless of where genetic variation might lie in the allelic spectrum-effect size space.
Implications and Future Directions
Why these successes matter
As other commentators have written, 70,128,134,135 and as we argued in early 2009, 123 the proximal purpose of genetic studies is to gain insight into biology. This goal is crucial for psychiatric disorders as so little is known about pathophysiology, and as highly publicized but ultimately false leads have occurred. For this primary goal, there have been unequivocal successes for many psychiatric disorders. This crucial point is sometimes overlooked: the knowledge base in psychiatric genetics is vastly greater than five years ago, and the rate of change is unprecedented in the history of the field.
What about clinical utility? So-called personalized medicine has been touted as the critical yardstick against which to measure the success of genetic studies. We believe clinical utility is the ultimate goal, but an inappropriate proximal goal. Still, there are a number of findings whose clinical significance should be evaluated. For example, SV testing is often part of the clinical evaluation of ASD, and careful evaluation of its utility in psychosis is warranted. As another example, Dr Roy Perlis and colleagues are evaluating the “repurposing” of isradipine (an approved antihypertensive that interacts with the protein product of CACNA1C) for the treatment of BIP. It is possible that risk profile scores, SV burden, or rare variant burden could have clinical utility. If these assess latent liability, they might be useful in selected clinical scenarios (e.g., predicting which patients require aggressive treatment in the psychosis prodrome). 136
The polygenicity of psychiatric disorders poses intriguing difficulties: how can these many genes be coherently tied together? A parsimonious hypothesis is that the polygenic basis of a psychiatric disorder is manifest in the regulation or function of one or more known or novel pathways. Genetic variation at many different loci could introduce numerous slight alterations that result in a pathway that is insufficiently robust in response to an environmental insult or that leads to an inappropriate developmental program. 137 Risk for a complex psychiatric disorder could be conferred by the emergent properties of the pathway itself rather than any single component. For SCZ, this conceptualization is supported by the risk profile findings for SCZ, and by the miR-137 results that hint at an underlying regulatory network. For ASD, typical patterns of cortical gene expression in frontal and temporal cortex have been found to be attenuated in ASD cases compared to controls, and an empirically-derived gene expression module that is under-expressed in ASD was found to be enriched for known ASD susceptibility genes and genetic association signals. 90
Alternative modes of investigation, such as network medicine, are needed to further our understanding of the roles of pathways in complex biological traits. 138 If polygenicity is indeed fundamental to complex psychiatric disorders and if some psychiatric disorders eventually prove to be pathway diseases, 137 then we need to confront this directly and to develop innovative methods. Developing such methods is more constructive and more likely to advance our understanding of these devastating diseases than raging against nature for not delivering common diseases in simpler Mendelian units.
Indeed, if one or more psychiatric disorders eventually prove to be pathway diseases, there could be clinical benefit. We conjecture that it might be considerably easier to coax an existing but dysfunctional biological pathway into the normal range than to replace components broken by Mendelian mutations. Moreover, in an era where many drug companies have moved away from drug development for psychiatric disorders, 139 the ability to measure such a hypothetical pathway in an appropriate cellular system could enable chemical biology screens of existing and novel compounds as well as the evaluation of the rational use of multiple compounds simultaneously.
Implications for strategy
A comprehensive portrait of genetic architecture does not now exist for any psychiatric disorder. Gaining more complete knowledge of the “parts list” for each disorder - the specific loci etiologically involved plus the identity, frequency, and impact of genetic variation at each locus - would be of exceptional importance. Such an enumeration would catalyze an array of specific, targeted and nuanced scientific studies. For example, such studies might lead to elucidation of biological mechanisms between the genotype and psychiatric phenotype, enablement of cell-based chemical biology and pharmacological screening, evaluation of gene action over developmental time, addressing the critical roles of gene-gene and gene-environment interactions, understanding the role played by epigenetic modifications, evaluation of disease prediction models, and so forth.
This is an attainable goal. The genomic search space is large but finite and so, in theory, elucidating the parts list for a psychiatric disorder could be achieved. Based on the evidence to date, thorough and well-powered genomic evaluations across the allelic spectrum are needed. We believe that a balanced portfolio of genomic assessments is required, as there are clear roles for common variation, SV, rare variation, and de novo variation for most disorders. Most discoveries in psychiatric genetics to date are from GWAS and SV evaluation (both often based on the use of GWAS chips), and larger and more comprehensive GWAS and SV studies are highly likely to increase knowledge. 135 It is possible to provide realistic estimates of power and to predict the number of new associations for each increment in sample size 128,129,140 (e.g., predictions have been made for GWAS and SV in 50,000 SCZ cases and 50,000 controls 140 ). Based on the recent ASD studies, sequencing directed at rare and de novo variation will have a role in a balanced portfolio of approaches. 83–85 Indeed, with improvements in accuracy, coverage, and pricing, it is possible that sequencing could evolve into the technology of choice for genotyping all major classes of genetic variation.
Such a series of studies would be costly, so a critical challenge is funding. These costs deserve to be placed in context of the public health implications of these disorders and, historically, psychiatric research has ben underfunded in comparison to public health impact (with the possible exception of AD). 141–144 For example, the lifetime cost per person with SCZ is on the order of $US 1.4 million: 145 if this program of research were eventually able to prevent only several dozen cases, it would likely prove to be cost-effective.
Continued cooperation
The successful study of any type of genetic variation in complex biomedical diseases requires very large sample sizes as a means to cut the Gordian knot posed by genetic architecture, etiological complexity, and phenotypic uncertainty. To achieve this end, there have been multiple meta-analysis 146,147 consortia in psychiatric genetics, of which the Psychiatric Genomics Consortium (http://pgc.unc.edu) is the largest and most encompassing. 123,148,149 Indeed, a GWAS co-authorship network graph demonstrates the high connectedness of researchers in the field (Figure S2).
An initial concern regarding GWAS meta-analysis was that increased signal from combining multiple samples would be negated by “noise” due to inter-site differences. This theoretical concern has not been borne out in practice, as illustrated above with the examples in SCZ, 58 BIP, 59 smoking behavior, 92 alcohol consumption, 100 and AD. 35,36 These meta-analyses are designed to identify risk or protective loci that have relatively similar effects across populations and that are not particularly sensitive to sample-specific factors. For example, T2DM and breast cancer loci identified in European samples tend to replicate in samples of East Asian ancestry. 150,151 It is possible that some genetic variants associated with phenotype risk are only found in certain population groups and are missed in meta-analysis; however, conclusive identification of such loci is likely to be challenging unless the effect sizes are relatively large.
Statistical rigor
In our opinion, a key ingredient of progress in psychiatric genetics has been uncompromising statistical rigor. Genomic technologies routinely posit 105–108 hypotheses, and false positives are a serious concern. For some investigators, suggestive statistical evidence combined with intriguing biology is sufficient. However, we fear that any benefit from relaxing statistical standards will be outweighed by the negative consequences of false positive claims.
This issue is particularly salient for exonic variation. Humans carry a huge pool of phenotypically-neutral background variation that adds noise to genetic analyses (for example, each person has ~100 loss-of-function variants, most of which are rare in a population), 152,153 and the presence of such variation complicates identification of disease-relevant variants. Thus, owing to chance, a researcher would expect to find a functional exonic mutation – possibly in a gene with intriguing biology – in one case sample and none of their control samples. More quantitatively, if 1% of cases are caused by fully penetrant mutations in a single gene with no background confounding variation, then observing 10 deleterious mutations in 1,000 cases and 0 in 1,000 controls would not stand out in test statistics from 20,000 genes. More realistic scenarios (including locus heterogeneity, incomplete penetrance, and background variation) will substantially erode the signal. The published results for ASD 83–85 and unpublished data on SCZ suggest these issues will be important and underscore the need for sequencing studies to have the same emphasis on statistical rigor and large sample sizes that has enabled GWAS to realize success for multiple psychiatric disorders. 154
Psychiatric genetics is not “post-genomic”
In psychiatric genetics, we are at the end of the beginning, not the beginning of the end. Remarkably, in a field characterized by a checkered history and few confident etiological clues, the genetics knowledge base has advanced considerably during the past five years, and results to date contain clear indications that further study will yield greater insight. Elucidation of the genetic architectures of psychiatric disorders is an attainable goal with existing technologies (albeit both costly and cost-effective). Few predictions are perfectly safe, but we would argue that genetics is a particularly good bet for psychiatry.
Supplementary Material
Acknowledgments
We remain indebted to the tens of thousand of individuals with psychiatric disorders for their participation in genetic studies of these disorders. We thank many colleagues for helpful critiques, plus a wealth of conversations with colleagues in the Psychiatric Genomics Consortium. We thank the NIH for funding (MH077139 and MH085520), Dr Thomas Lehner of the NIMH for his continued support, Drs Ben Voight and Joel Hirschhorn for data relevant to power estimation, Dr Stephan Ripke for help with figures, Dr Jin Szatkiewicz for assistance in reviewing the SV literature, and Drs Cynthia Bulik and James Crowley for helpful comments.
This study makes use of data generated by DECIPHER. A full list of centers that contributed to the generation of the data is available from http://decipher.sanger.ac.uk. Funding for DECIPHER was provided by the Wellcome Trust.
Glossary
Note. This is a placeholder, will be completed prior to publication
- Genome-wide association
an unbiased genome screen unrelated cases and appropriately matched controls or parent-affected child trios. The dominant technology has been individual genotyping using highly multiplexed SNP arrays
- Genome-wide linkage
a type of unbiased genome screen based on multiplex pedigrees. Genotyping approaches have included restriction fragment length polymorphisms, microsatellites, and SNP arrays. After adjustment for multiple comparisons, the signal is the co-segregation of a genotype with a disease phenotype within the pedigrees
- Multiplex pedigree
a family constellation containing more than one affected individual
- Polygenic
“many genes”, with no implications about the frequencies, modes of action, or effect sizes of any relevant genetic variation
- Risk profile
defined in Box 3
- Simplex pedigree
a family constellation containing one affected individual
- Karyotyping
- Structural variation
Footnotes
Conflicts of Interest
The authors report no conflicts.
Author Contributions
The authors jointly conceived and wrote this paper, and take responsibility for its content.
References
- 1.Eaton WW, et al. The burden of mental disorders. Epidemiologic reviews. 2008;30:1–14. doi: 10.1093/epirev/mxn011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. The Global Burden of Disease: 2004 Update. WHO Press; Geneva: 2008. [Google Scholar]
- 3.Collins PY, et al. Grand challenges in global mental health. Nature. 2011;475:27–30. doi: 10.1038/475027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park YK, Sempos CT, Barton CN, Vanderveen JE, Yetley EA. Effectiveness of food fortification in the United States: the case of pellagra. American journal of public health. 2000;90:727–38. doi: 10.2105/ajph.90.5.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2009. U.S. Department of Health and Human Services; Atlanta, GA: 2010. [Google Scholar]
- 6.Chanock SJ, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 7.McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. European journal of human genetics : EJHG. 2008;16:422–34. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- 9.Inlow JK, Restifo LL. Molecular and comparative genetics of mental retardation. Genetics. 2004;166:835–81. doi: 10.1093/genetics/166.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy MI, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Reviews Genetics. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 11.Altshuler D, Daly M. Guilt beyond a reasonable doubt. Nat Genet. 2007;39:813–5. doi: 10.1038/ng0707-813. [DOI] [PubMed] [Google Scholar]
- 12.Corvin A, Craddock N, Sullivan PF. Genome-wide association studies: a primer. Psychologal Medicine. 2010;40:1063–77. doi: 10.1017/S0033291709991723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bassett AS, Chow EW, Weksberg R. Chromosomal abnormalities and schizophrenia. Am J Med Genet. 2000;97:45–51. doi: 10.1002/(sici)1096-8628(200021)97:1<45::aid-ajmg6>3.0.co;2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vorstman JA, et al. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11:18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 15.Malhotra D, Sebat J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell. 2012;148:1223–41. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nature Reviews Genetics. 2011;12:363–76. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Attia J, et al. How to use an article about genetic association: A: Background concepts. Jama. 2009;301:74–81. doi: 10.1001/jama.2008.901. [DOI] [PubMed] [Google Scholar]
- 18.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasaniuc B, et al. Extremely low-coverage sequencing enables cost effective GWAS. Nature Genetics. (In press) [Google Scholar]
- 20.Ng SB, Nickerson DA, Bamshad MJ, Shendure J. Massively parallel sequencing and rare disease. Hum Mol Genet. 2010;19:R119–24. doi: 10.1093/hmg/ddq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature reviews. Genetics. 2010;11:415–25. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 22.Collins AL, et al. Hypothesis-driven candidate genes for schizophrenia compared to genome-wide association results. Psychological Medicine. 2012;42:607–16. doi: 10.1017/S0033291711001607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng MY, et al. Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol Psychiatry. 2009;14:774–85. doi: 10.1038/mp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McQueen MB, et al. Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet. 2005;77:582–95. doi: 10.1086/491603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trikalinos TA, et al. A heterogeneity-based genome search meta-analysis for autism-spectrum disorders. Mol Psychiatry. 2006;11:29–36. doi: 10.1038/sj.mp.4001750. [DOI] [PubMed] [Google Scholar]
- 26.Zhou K, et al. Meta-analysis of genome-wide linkage scans of attention deficit hyperactivity disorder. American journal of medical genetics. Part B. 2008;147B:1392–8. doi: 10.1002/ajmg.b.30878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nature Reviews Neuroscience. 2008;9:768–78. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- 28.McNaughton D, et al. Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiology of aging. 2012;33:426, e13–21. doi: 10.1016/j.neurobiolaging.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rovelet-Lecrux A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature genetics. 2006;38:24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 30.Guerreiro RJ, et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiology of aging. 2012;33:1008, e17–23. doi: 10.1016/j.neurobiolaging.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pottier C, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Molecular psychiatry. 2012 doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 32.Strittmatter WJ, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harold D, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature genetics. 2009;41:1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature genetics. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 35.Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nature genetics. 2011;43:429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature genetics. 2011;43:436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brouwers N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Molecular psychiatry. 2012;17:223–33. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones L, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PloS one. 2010;5:e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerreiro RJ, Hardy J. Alzheimer’s disease genetics: lessons to improve disease modelling. Biochemical Society transactions. 2011;39:910–6. doi: 10.1042/BST0390910. [DOI] [PubMed] [Google Scholar]
- 40.Treusch S, et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science. 2011;334:1241–5. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owen MJ, Craddock N, O’Donovan MC. Suggestion of roles for both common and rare risk variants in genome-wide studies of schizophrenia. Archives of general psychiatry. 2010;67:667–73. doi: 10.1001/archgenpsychiatry.2010.69. [DOI] [PubMed] [Google Scholar]
- 42.Itsara A, et al. Population analysis of large copy number variants and hotspots of human genetic disease. American journal of human genetics. 2009;84:148–61. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009;18:988–96. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vacic V, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raychaudhuri S, et al. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 48.International Schizophrenia Consortium. Greater burden of rare copy number variants in schizophrenia. Nature. 2008;455:237–41. [Google Scholar]
- 49.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007 doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008 doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 51.Zhang D, et al. Singleton deletions throughout the genome increase risk of bipolar disorder. Mol Psychiatry. 2009;14:376–80. doi: 10.1038/mp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Priebe L, et al. Genome-wide survey implicates the influence of copy number variants (CNVs) in the development of early-onset bipolar disorder. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.8. [DOI] [PubMed] [Google Scholar]
- 53.Malhotra D, et al. High Frequencies of De Novo CNVs in Bipolar Disorder and Schizophrenia. Neuron. 2011;72:951–63. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grozeva D, et al. Rare copy number variants: a point of rarity in genetic risk for bipolar disorder and schizophrenia. Archives of general psychiatry. 2010;67:318–27. doi: 10.1001/archgenpsychiatry.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McQuillin A, et al. Analysis of genetic deletions and duplications in the University College London bipolar disorder case control sample. European journal of human genetics : EJHG. 2011;19:588–92. doi: 10.1038/ejhg.2010.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Girard SL, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nature genetics. 2011;43:860–3. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 57.Xu B, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nature genetics. 2011;43:864–8. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schizophrenia Psychiatric Genome-Wide Association Study Consortium. Genome-wide association study of schizophrenia identifies five novel loci. Nature Genetics. 2011;43:969–76. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature genetics. 2011;43:977–83. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. The EMBO journal. 2009;28:3027–39. doi: 10.1038/emboj.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bemis LT, et al. MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer research. 2008;68:1362–8. doi: 10.1158/0008-5472.CAN-07-2912. [DOI] [PubMed] [Google Scholar]
- 63.Kwon E, Wang W, Tsai LH. Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.170. [DOI] [PubMed] [Google Scholar]
- 64.Szulwach KE, et al. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–41. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smrt RD, et al. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells. 2010;28:1060–70. doi: 10.1002/stem.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willemsen MH, et al. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. Journal of medical genetics. 2011;48:810–8. doi: 10.1136/jmedgenet-2011-100294. [DOI] [PubMed] [Google Scholar]
- 67.Shi Y, et al. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nature genetics. 2011;43:1224–7. doi: 10.1038/ng.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yue WH, et al. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11.2. Nature genetics. 2011;43:1228–31. doi: 10.1038/ng.979. [DOI] [PubMed] [Google Scholar]
- 69.International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Visscher PM, Brown MA, McCarthy MI, Yang J. Five Years of GWAS Discovery. American journal of human genetics. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Houlston RS, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nature genetics. 2010;42:973–7. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turnbull C, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nature genetics. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stahl EA, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nature genetics. 2012 doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2:943–55. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 79.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 80.Sanders SJ, et al. Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron. 2011;70:863–85. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012 doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012 doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012 doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang K, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–33. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–8. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anney R, et al. A genome-wide scan for common alleles affecting risk for autism. Human molecular genetics. 2010;19:4072–82. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma D, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Annals of human genetics. 2009;73:263–73. doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- 92.Tobacco and Genetics Consortium. Meta-analyses of genome-wide association studies implicate multiple loci for smoking behavior. Nature Genetics. 2010;42:441–7. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Edenberg HJ, et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcoholism, clinical and experimental research. 2010;34:840–52. doi: 10.1111/j.1530-0277.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bierut LJ, et al. A genome-wide association study of alcohol dependence. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5082–7. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kendler KS, et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcoholism, clinical and experimental research. 2011;35:963–75. doi: 10.1111/j.1530-0277.2010.01427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heath AC, et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biological psychiatry. 2011;70:513–8. doi: 10.1016/j.biopsych.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frank J, et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addiction biology. 2012;17:171–180. doi: 10.1111/j.1369-1600.2011.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baik I, Cho NH, Kim SH, Han BG, Shin C. Genome-wide association studies identify genetic loci related to alcohol consumption in Korean men. The American journal of clinical nutrition. 2011;93:809–16. doi: 10.3945/ajcn.110.001776. [DOI] [PubMed] [Google Scholar]
- 99.Takeuchi F, et al. Confirmation of ALDH2 as a Major locus of drinking behavior and of its variants regulating multiple metabolic phenotypes in a Japanese population. Circulation journal : official journal of the Japanese Circulation Society. 2011;75:911–8. doi: 10.1253/circj.cj-10-0774. [DOI] [PubMed] [Google Scholar]
- 100.Schumann G, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7119–24. doi: 10.1073/pnas.1017288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bierut LJ, et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thorgeirsson TE, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nature genetics. 2010;42:448–53. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu JZ, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nature genetics. 2010;42:436–40. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saccone NL, et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thorgeirsson TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Truong T, et al. Replication of lung cancer susceptibility loci at chromosomes 15q25, 5p15, and 6p21: a pooled analysis from the International Lung Cancer Consortium. J Natl Cancer Inst. 2010;102:959–71. doi: 10.1093/jnci/djq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Major Depressive Disorder Working Group of the PGC. A mega-analysis of genome-wide association studies for major depressive disorder. Molecular Psychiatry. doi: 10.1038/mp.2012.21. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rucker JJ, et al. Genome-wide association analysis of copy number variation in recurrent depressive disorder. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Caspi A, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–9. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 111.Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Archives of general psychiatry. 2011;68:444–54. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Uher R, McGuffin P. The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Molecular psychiatry. 2010;15:18–22. doi: 10.1038/mp.2009.123. [DOI] [PubMed] [Google Scholar]
- 113.Munafo MR, Durrant C, Lewis G, Flint J. Gene X environment interactions at the serotonin transporter locus. Biological psychiatry. 2009;65:211–9. doi: 10.1016/j.biopsych.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 114.Risch N, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. Jama. 2009;301:2462–71. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fergusson DM, Horwood LJ, Miller AL, Kennedy MA. Life stress, 5-HTTLPR and mental disorder: findings from a 30-year longitudinal study. The British journal of psychiatry : the journal of mental science. 2011;198:129–35. doi: 10.1192/bjp.bp.110.085993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Neale BM, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. Journal of the American Academy of Child and Adolescent Psychiatry. 2010;49:884–97. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang K, et al. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Molecular psychiatry. 2011;16:949–59. doi: 10.1038/mp.2010.107. [DOI] [PubMed] [Google Scholar]
- 118.Williams NM, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376:1401–8. doi: 10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Williams NM, et al. Genome-Wide Analysis of Copy Number Variants in Attention Deficit Hyperactivity Disorder: The Role of Rare Variants and Duplications at 15q13.3. The American journal of psychiatry. 2011 doi: 10.1176/appi.ajp.2011.11060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stergiakouli E, et al. Investigating the Contribution of Common Genetic Variants to the Risk and Pathogenesis of ADHD. The American journal of psychiatry. 2011 doi: 10.1176/appi.ajp.2011.11040551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wray NR, Visscher PM. Narrowing the boundaries of the genetic architecture of schizophrenia. Schizophr Bull. 2010;36:14–23. doi: 10.1093/schbul/sbp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McClellan J, King MC. Genomic analysis of mental illness: a changing landscape. JAMA. 2010;303:2523–4. doi: 10.1001/jama.2010.869. [DOI] [PubMed] [Google Scholar]
- 123.Psychiatric GWAS Consortium. A framework for interpreting genomewide association studies of psychiatric disorders. Molecular Psychiatry. 2009;14:10–7. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- 124.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wray NR, Purcell SM, Visscher PM. Synthetic associations created by rare variants do not explain most GWAS results. PLoS Biol. 2011;9:e1000579. doi: 10.1371/journal.pbio.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011;9:e1000580. doi: 10.1371/journal.pbio.1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Orozco G, Barrett JC, Zeggini E. Synthetic associations in the context of genome-wide association scan signals. Hum Mol Genet. 2010;19:R137–44. doi: 10.1093/hmg/ddq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–97. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 129.Kim Y, Zerwas S, Trace SE, Sullivan PF. Schizophrenia genetics: where next? Schizophrenia Bulletin. 2011;37:456–63. doi: 10.1093/schbul/sbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee S, et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nature Genetics. 2012 doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Davies G, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Molecular psychiatry. 2011;16:996–1005. doi: 10.1038/mp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Eichler EE, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nature reviews. Genetics. 2010;11:446–50. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 135.Sullivan PF. Don’t give up on GWAS. Molecular Psychiatry. 2011 [Google Scholar]
- 136.Addington J, Heinssen R. Prediction and prevention of psychosis in youth at clinical high risk. Annual review of clinical psychology. 2012;8:269–89. doi: 10.1146/annurev-clinpsy-032511-143146. [DOI] [PubMed] [Google Scholar]
- 137.Sullivan PF. Schizophrenia as a pathway disease. Nature Medicine. 2012;18:210–11. doi: 10.1038/nm.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature reviews. Genetics. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Miller G. Is pharma running out of brainy ideas? Science. 2010;329:502–4. doi: 10.1126/science.329.5991.502. [DOI] [PubMed] [Google Scholar]
- 140.Yang J, Wray NR, Visscher PM. Comparing apples and oranges: equating the power of case-control and quantitative trait association studies. Genet Epidemiol. 2010;34:254–7. doi: 10.1002/gepi.20456. [DOI] [PubMed] [Google Scholar]
- 141.Gross CP, Anderson GF, Powe NR. The relation between funding by the National Institutes of Health and the burden of disease. The New England journal of medicine. 1999;340:1881–7. doi: 10.1056/NEJM199906173402406. [DOI] [PubMed] [Google Scholar]
- 142.Aoun S, Pennebaker D, Pascal R. To what extent is health and medical research funding associated with the burden of disease in Australia? Australian and New Zealand journal of public health. 2004;28:80–6. doi: 10.1111/j.1467-842x.2004.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 143.Gillum LA, et al. NIH disease funding levels and burden of disease. PloS one. 2011;6:e16837. doi: 10.1371/journal.pone.0016837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lamarre-Cliche M, Castilloux AM, LeLorier J. Association between the burden of disease and research funding by the Medical Research Council of Canada and the National Institutes of Health. A cross-sectional study. Clinical and investigative medicine. Medecine clinique et experimentale. 2001;24:83–9. [PubMed] [Google Scholar]
- 145.Wu EQ, et al. The economic burden of schizophrenia in the United States in 2002. The Journal of clinical psychiatry. 2005;66:1122–9. doi: 10.4088/jcp.v66n0906. [DOI] [PubMed] [Google Scholar]
- 146.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. American journal of human genetics. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.de Bakker PI, Neale BM, Daly MJ. Meta-analysis of genome-wide association studies. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.top81. pdb top81. [DOI] [PubMed] [Google Scholar]
- 148.Psychiatric GWAS Consortium. Genome-wide association studies: history rationale and prospects for psychiatric disorders. American Journal of Psychiatry. 2009;166:540–6. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sullivan PF. The Psychiatric GWAS Consortium: big science comes to psychiatry. Neuron. 2010;68:182–6. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Waters KM, et al. Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS genetics. 2010;6 doi: 10.1371/journal.pgen.1001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Easton DF, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–8. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kiezun A, et al. Exome sequencing and the genetic basis of complex traits. doi: 10.1038/ng.2303. (Submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–8. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ott J. Analysis of Human Genetic Linkage. The Johns Hopkins University Press; Baltimore, MD: 1999. [Google Scholar]
- 157.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. New England Journal of Medicine. 2008;358:667–75. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 158.Szatmari P, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–28. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kumar RA, et al. Recurrent 16p11.2 microdeletions in autism. Human molecular genetics. 2008;17:628–38. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 160.Vassos E, et al. Penetrance for copy number variants associated with schizophrenia. Human molecular genetics. 2010;19:3477–81. doi: 10.1093/hmg/ddq259. [DOI] [PubMed] [Google Scholar]
- 161.Holmans P, et al. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee P, O’Dushlaine C, Thomas B, Purcell S. InRich: Interval-based enrichment analysis for genome-wide association studies. Bioinformatics. doi: 10.1093/bioinformatics/bts191. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rossin EJ, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS genetics. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Raychaudhuri S, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS computational biology. 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yaspan BL, Veatch OJ. Strategies for pathway analysis from GWAS data. Current protocols in human genetics/editorial board, Jonathan L. Haines … [et al.] 2011;Chapter 1(Unit 1):20. doi: 10.1002/0471142905.hg0120s71. [DOI] [PubMed] [Google Scholar]
- 167.GO Project. The Gene Ontology: enhancements for 2011. Nucleic acids research. 2012;40:D559–64. doi: 10.1093/nar/gkr1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mi H, et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic acids research. 2010;38:D204–10. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Croning MD, Marshall MC, McLaren P, Armstrong JD, Grant SG. G2Cdb: the Genes to Cognition database. Nucleic acids research. 2009;37:D846–51. doi: 10.1093/nar/gkn700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ruano D, et al. Functional gene group analysis reveals a role of synaptic heterotrimeric G proteins in cognitive ability. American journal of human genetics. 2010;86:113–25. doi: 10.1016/j.ajhg.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lindblad-Toh K, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–82. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS genetics. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.O’Donovan M, et al. Identification of novel schizophrenia loci by genome-wide association and follow-up. Nature Genetics. 2008;40:1053–5. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 175.Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–7. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sklar P, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008 doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.WTCCC. Genome-wide association study of 14, 000 cases of seven common diseases 3, 000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ruderfer DM, et al. A family-based study of common polygenic variation and risk of schizophrenia. Molecular psychiatry. 2011;16:887–8. doi: 10.1038/mp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Archives of General Psychiatry. 2003;60:1187–92. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 180.Lichtenstein P, et al. Common genetic influences for schizophrenia and bipolar disorder: A population-based study of 2 million nuclear families. Lancet. 2009;373:234–9. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ropers HH. Genetics of early onset cognitive impairment. Annual review of genomics and human genetics. 2010;11:161–87. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 182.Arcelus J, Mitchell AJ, Wales J, Nielsen S. Mortality rates in patients with anorexia nervosa and other eating disorders. A meta-analysis of 36 studies. Archives of general psychiatry. 2011;68:724–31. doi: 10.1001/archgenpsychiatry.2011.74. [DOI] [PubMed] [Google Scholar]
- 183.Biederman J, Faraone SV. The effects of attention-deficit/hyperactivity disorder on employment and household income. MedGenMed : Medscape general medicine. 2006;8:12. [PMC free article] [PubMed] [Google Scholar]
- 184.Levinson DF, et al. Copy number variants in schizophrenia: Confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–16. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. The New England journal of medicine. 2008;359:1685–99. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–6. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Brunetti-Pierri N, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nature genetics. 2008;40:1466–71. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nature genetics. 2011;43:838–46. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mefford HC, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS genetics. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jacquemont S, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shinawi M, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. Journal of medical genetics. 2010;47:332–41. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Moreno-De-Luca D, et al. Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. American journal of human genetics. 2010;87:618–30. doi: 10.1016/j.ajhg.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Seshadri S, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cichon S, et al. Genome-wide Association Study Identifies Genetic Variation in Neurocan as a Susceptibility Factor for Bipolar Disorder. Am J Hum Genet. 2011;88:372–81. doi: 10.1016/j.ajhg.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Steinberg S, et al. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Human molecular genetics. 2011;20:4076–81. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Williams HJ, et al. Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rietschel M, et al. Association between genetic variation in a region on chromosome 11 and schizophrenia in large samples from Europe. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.80. [DOI] [PubMed] [Google Scholar]
- 198.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 199.Ferreira M, et al. Collaborative genome-wide association analysis of 10,596 individuals supports a role for Ankyrin-G (ANK3) and the alpha-1C subunit of the L-type voltage-gated calcium channel (CACNA1C) in bipolar disorder. Nature Genetics. 2008;40:1056–8. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.McMahon FJ, et al. Meta-analysis of genome-wide association data identifies a risk locus for major mood disorders on 3p21.1. Nat Genet. 2010;42:128–31. doi: 10.1038/ng.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Breen G, et al. Replication of association of 3p21.1 with susceptibility to bipolar disorder but not major depression. Nature Genetics. 2011;43:3–5. doi: 10.1038/ng0111-3. [DOI] [PubMed] [Google Scholar]
- 202.Green EK, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Molecular psychiatry. 2010;15:1016–22. doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.