

Abstract
As a major component of the crucial nonlysosomal protein degradation pathway in the cells, the proteasome has been implicated in many diseases such as Alzheimer’s disease, Huntington’s disease, inflammatory bowel diseases, autoimmune diseases, multiple myeloma (MM) and other cancers. There are two main proteasome subtypes: the constitutive proteasome which is expressed in all eukaryotic cells and the immunoproteasome which is expressed in immune cells and can be induced in other cell types. Majority of currently available proteasome inhibitors are peptide backbone-based, having short half-lives in the body. It is highly desirable to identify novel, immunoproteasome-selective inhibitors with non-peptide scaffolds for development of novel therapeutics. Through combined virtual screening and experimental studies targeting the immunoproteasome, we have identified a set of novel immunoproteasome inhibitors with diverse non-peptide scaffolds. Some of the identified inhibitors have significant selectivity for the immunoproteasome over the constitutive proteasome. Unlike most of the currently available proteasome inhibitors, these new inhibitors lacking electrophilic pharmacophores are not expected to form a covalent bond with proteasome after the binding. These non-peptide scaffolds may provide a new platform for future rational drug design and discovery targeting the immunoproteasome.
Keywords: Immunoproteasome, selective inhibitor, non-peptide scaffold, inhibitor identification
Proteasome plays a central role in maintaining cellular homeostasis, controlling the cell cycle, removing misfolded proteins that can be toxic, and regulating the immune system.1 This crucial protein degradation machinery has been implicated in multiple diseases such as Alzheimer’s disease, Huntington’s disease (HD), inflammatory bowel diseases (IBD), autoimmune diseases, multiple myeloma (MM) and other cancers.2 In particular, proteasome has been recognized as a promising cancer target, and the Food and Drug Administration (FDA) has approved proteasome inhibitors bortezomib (Velcade®) in 2003 and carfilzomib (Kyprolis®) in 2012 for the MM treatment. The FDA approvals of these proteasome inhibitors as chemotherapeutic agents have dramatically improved the therapeutic landscape for patients with MM.3 Despite the remarkable successes of these proteasome inhibitors in the clinic, intrinsic and acquired drug resistance remains a major clinical challenge. In addition, these drugs have failed to provide clinical benefit to patients with solid cancers,4-6 further highlighting the need for next-generation of proteasome inhibitors.
There are two main proteasome subtypes: the constitutive proteasome (CP) which is expressed in all eukaryotic cells and the immunoproteasome (IP) which is expressed in immune cells and can be induced in other cell types. Constitutive proteasome contains three catalytic subunits denoted as β1, β2, and β5, whereas the three corresponding catalytic subunits of immunoproteasome are denoted as β1i, β2i, and β5i. The catalytic subunits responsible for the chymotrypsin-like (CT-L) activity (β5 and β5i) are thought to be most physiologically important and have been recognized as the key targets of bortezomib and carfilzomib.7, 8 However, these drugs have failed to achieve efficacy in patients with solid cancers despite strong indications of activity in preclinical animal models. The failure of these drugs has been attributed to their poor metabolic stability.9 Further, recent reports revealed the importance of targeting the immunoproteasome catalytic subunit β5i in killing cancer cells.8 Therefore, it is highly desirable to identify novel compounds with non-peptide scaffolds that can selectively inhibit the immunoproteasome β5i as the next-generation of therapeutic agents.
Here, we report the identification of a set of novel, selective inhibitors of the immunoproteasome with diverse non-peptide scaffolds through combined virtual screening and experimental studies targeting the catalytic subunit β5i (which is also known as LMP7).
Our virtual screening was based on our previously modeled three-dimensional (3D) structure of the immunoproteasome,10-12 and performed on a compound library at Genomics Research Institute (GRI), the University of Cincinnati (UC). The UC/GRI compound library containing structural information for about 300,000 compounds was provided by Procter & Gamble (P&G) and belonged to a consortium including the University of Kentucky as a member. The virtual screening procedure utilized to screen the chemical compound library is essentially similar to that we used to identify small-molecule inhibitors of other proteins.13, 14 First of all, the ~300,000 compounds were first screened by performing rigid docking using FRED (OpenEye Scientific Software),15 leading to identification of top-25,000 compounds. The subsequent energy-minimization and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) binding energy calculations using Amber9 software16 led to identification of the top-1,000 compounds. Further structural analysis on possible interactions with key amino-acid residues (including Thr1, Ser21, Ser27, Gly47, Ala49, and Asp324) led to selection of the top-90 compounds that could inhibit the immunoproteasome.
The computationally selected 90 compounds were tested for their inhibitory activity against the CT-L activity of the immunoproteasome. The identified active compounds were also tested for their inhibitory activity against the CT-L activity of the constitutive proteasome. For the initial activity screening, the selected compounds were dissolved in DMSO and used at a concentration of 5 μM. Epoxomicin (1 μM), which reached the 100% inhibition, was used as a positive control. 20S human immunoproteasome and constitutive proteasome (Boston Biochem) were 2-fold diluted in an assay buffer (20 mM Tris/Cl, pH 8.0, 0.5 mM EDTA, 0.035% SDS).17 Specifically, selected compounds were preincubated with 50 ng/well of the immunoproteasome or constitutive proteasome in a 96-well plate at room temperature for 90 minutes. 100 μM of Suc-LLVY-AMC, a fluorogenic peptide substrate for the CT-L activity, was then added to the wells. Fluorogenic signals of the free AMC (Ex:360, Em:460)18 were recorded for 90 min. The initial reaction velocities (RFU/min) of each compound were calculated as a percentage of the positive control. All enzyme activity assays were carried out in triplicate.
According to the activity assays, nine of the tested compounds (1 to 9) showing the significant inhibition against the immunoproteasome are depicted in Figure 1 for their molecular structures and listed in Table 1 for their activity data. So, the hit rate of the virtual screening was 10%. Based on the activity data in Table 1, compounds 1 to 3 at 5 μM inhibited the CT-L activity of the immunoproteasome by about 36% to 85%, whereas these compounds at 5 μM inhibited the constitutive proteasome by only 2% to 20%. This indicates that compounds 1 to 3 are highly selective inhibitors for the immunoproteasome. In particular, compounds 1 and 2 at 5 μM inhibited the immunoproteasome by 85% to 62%, respectively. These most potent two compounds, along with compound 3, were tested further for the dose-dependent inhibition (Figure 2) in order to determine their IC50 values (Table 1) against the immunoproteasome. As shown in Table 1, the IC50 values for compounds 1 to 3 are 1.7 μM, 4.9 μM, and 22 μM, respectively. These compounds, particularly compounds 1 and 2, are promising immunoproteasome inhibitors with non-peptide scaffolds.
Figure 1.
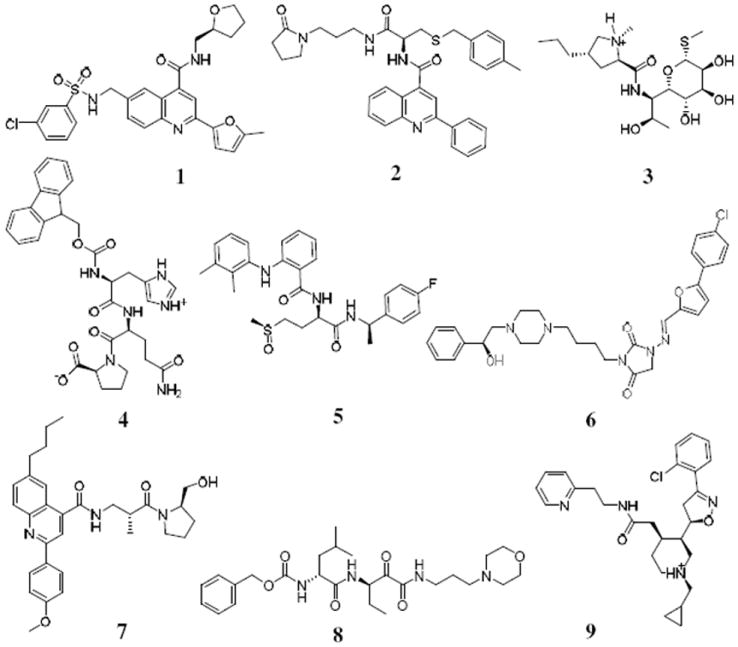
Molecular structures of the identified new inhibitors of the immunoproteasome.
Table 1.
The calculated binding energies (ΔGbind, in kcal/mol) of nine compounds (1 to 9) with the immunoproteasome (IP) and their inhibitory activities against the immunoproteasome and constructive proteasome (CP).
| Compound | ΔGbinda | % Inhibition of IP activity [IC50]b,c | % Inhibition of CP activityb |
|---|---|---|---|
| 1 | -26.5 | 85 [1.7 μM] | 20 |
| 2 | -25.9 | 62 [4.9 μM] | 15 |
| 3 | -24.0 | 36 [22 μM] | 2 |
| 4 | -21.7 | 35 | 24 |
| 5 | -26.0 | 31 | 33 |
| 6 | -15.1 | 25 | 16 |
| 7 | -22.6 | 21 | 2 |
| 8 | -23.7 | 14 | 6 |
| 9 | -24.4 | 13 | 14 |
The binding energies were estimated from the MM-GBSA calculations. The MM-GBSA binding energies were empirically scaled according to scaling factor obtained by fitting to a training set of 99 protein-ligand complexes.19
The % inhibition was determined in the presence of the inhibitor at 5 μM.
The determined IC50 values are given in brackets.
Figure 2.
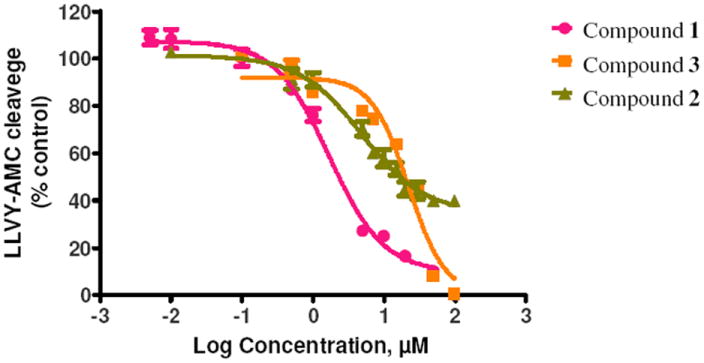
Dose-dependent inhibition of the immunoproteasome CT-L activity (initial velocity) by compounds 1 to 3: plots of the remaining CT-L activity vs the inhibitor concentration.
Depicted in Figure 3 are the energy-minimized structures of the immunoproteasome binding with compounds 1 and 2. As shown in Figure 3A, compound 1 has favorable hydrophilic interactions with amino-acid residues Thr1, Ser21, Ser27, and Gly47, including strong hydrogen bonds with the NH group of Ser21 backbone, hydroxyl group of Ser27 side chain, and carbonyl oxygen of Gly47 backbone. As shown in Figure 3B, compound 2 has favorable hydrophilic interactions with Ser21, Ser27, Ala49, and Asp324, including hydrogen bonds with the NH group of Ser21 backbone and carboxylate oxygen of Asp324 side chain. Notably, the H⋯O distance with the hydroxyl oxygen of Ser27 side chain is as long as 3.6 Å in the energy-minimized structure. Further, molecular dynamics (MD) simulation was carried out to examine the dynamically stable binding structures using the same computational protocol (starting from the energy-minimized structures) as we used in our previous computational studies on immunoproteasome-ligand binding.10,11 For each inhibitor (compound 1 or 2) binding with immunoproteasome, we obtained a stable MD trajectory for 1 ns and saved 1000 snapshots (one snapshot per ps) for structural analysis. Based on the MD trajectory with compound 2, the H⋯O distance with the hydroxyl oxygen of Ser27 side chain is shorter than 2.5 Å for ~15% of the snapshots. So, when 2.5 Å is used as the cutoff for the H⋯O distance in the hydrogen bonding, we may say that the hydroxyl oxygen of Ser27 side chain has ~15% hydrogen bond with compound 2 (as indicated in Figure 3B) and ~93% hydrogen bond with compound 1 (as indicated in Figure 3A).
Figure 3.
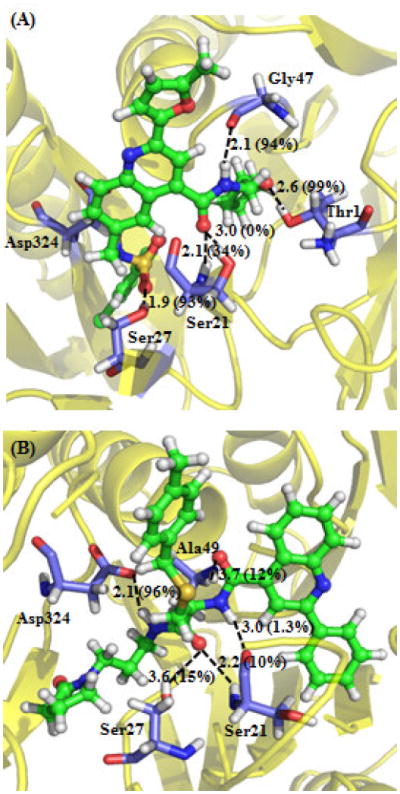
Binding structures of the immunoproteasome interacting with compounds 1 (A) and 2 (B). Indicated in the Figures are the key distances (Å) of the protein-ligand interactions in the energy-minimized structures. Indicated in parentheses is the percent of the snapshots with the H⋯O distance shorter than 2.5 Å in the MD-simulated binding structure.
Notably, Ser21 and Ser27 are common residues of the immunoproteasome that have favorable interactions with both compounds 1 and 2. Ser21 and Ser27 in the immunoproteasome become Thr21 and Ala27, respectively, in the constitutive proteasome. The other residues of the immunoproteasome interacting with compounds 1 and 2 are essentially the same as the corresponding ones of the constitutive proteasome. So, the selectivity of these new inhibitors for the immunoproteasome over the constitutive proteasome is likely associated with the favorable interaction between the inhibitors and the hydroxyl group of Ser27 side chain in the immunoproteasome. For the purpose of verification of this point, we also modeled constitutive proteasome binding with compounds 1 and 2 in the same way as we did for immunoproteasome with the same compounds, and we concluded that each compound binds with both proteins in the similar orientation, but without a hydrogen bond with Ala27 of constitutive proteasome (because Ala27 does not have a hydroxyl group on the side chain; data not shown).
Unlike currently available proteasome inhibitors in clinic, these new immunoproteasome inhibitors are non-peptide scaffold-based. In addition, these non-peptide scaffold-based compounds are expected to reversibly inhibit proteasomes due to the lack of reactive electrophilic pharmacophors, although further experimental tests need to be done in the future in order to examine the reversibility of the inhibition, the compound stability and the anti-cancer activities. Therefore, these non-peptide scaffolds may serve as a new platform for future rational drug design and discovery targeting the immunoproteasome. For example, potentially more potent and selective inhibitors may be designed to enhance the favorable interactions with all of the amino-acid residues mentioned above. Particularly, enhancement of the favorable interaction with Ser27 of the immunoproteasome is expected to improve both the potency and selectivity of the immunoproteasome inhibitors.
Acknowledgments
The research was supported in part by the NIH (grants RC1MH088480 and R01CA128903), Kentucky Science & Engineering Foundation (grant KSEF-925-RDE-008). The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldberg AL. Nature. 2003;426:895. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 2.Kisselev AF, Goldberg AL. Chem Biol. 2001;8:739. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 3.McBride A, Ryan PY. Exp Rev anticancer therapy. 2013;13:339. doi: 10.1586/era.13.9. [DOI] [PubMed] [Google Scholar]
- 4.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Cur Cancer Drug Targ. 2011;11:239. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. New Engl J Med. 2003;348:2609. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 6.Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, Chanan-Khan A, Buadi F, Reu FJ, Somlo G, Zonder J, Song K, Stewart AK, Stadtmauer E, Kunkel L, Wear S, Wong AF, Orlowski RZ, Jagannath S. Blood. 2012 doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rock KL, Goldberg AL. Annu Rev Immunol. 1999;17:739. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 8.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, Shenk KD, Bennett MK. Blood. 2009;114:3439. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- 9.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discov Today. 2010;15:40. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Lei B, AbdulHameed MDM, Hamza A, Wehenkel M, Muzyka JL, Yao X-J, Kim K-B, Zhan C-G. J Phys Chem B. 2010;114:12333. doi: 10.1021/jp1058098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei B, Hamza A, Zhan CG. Theo Chem Acc. 2012;131:1203. [Google Scholar]
- 12.Wei D, Lei B, Tang M, Zhan CG. J Am Chem Soc. 2012;134:10436. doi: 10.1021/ja3006463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, AbdulHameed MDM, Hamza A, Zhan C-G. Bioorg Med Chem Lett. 2012;22:1629. doi: 10.1016/j.bmcl.2011.12.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamza A, Zhao X, Tong M, Tai HH, Zhan CG. Bioorg Med Chem. 2011;19:6077. doi: 10.1016/j.bmc.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Biopolymers. 2003;68:76. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- 16.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho YK, Bargagna-Mohan P, Wehenkel M, Mohan R, Kim KB. Chem Biol. 2007;14:419. doi: 10.1016/j.chembiol.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kisselev AF, Goldberg AL. Methods Enzymol. 2005;398:364. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- 19.Naim M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, Drabik P, Sulea T, Bayly CI, Jakalian A, Purisima EO. J Chem Inf Model. 2007;47:122. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]