

Abstract
Cellulosomes are discrete multienzyme complexes used by a subset of anaerobic bacteria and fungi to digest lignocellulosic substrates. Assembly of the enzymes onto the noncatalytic scaffold protein is directed by interactions among a family of related receptor-ligand pairs comprising interacting cohesin and dockerin modules. The extremely strong binding between cohesin and dockerin modules results in dissociation constants in the low picomolar to nanomolar range, which may hamper accurate off-rate measurements with conventional bulk methods. Single-molecule force spectroscopy (SMFS) with the atomic force microscope measures the response of individual biomolecules to force, and in contrast to other single-molecule manipulation methods (i.e. optical tweezers), is optimal for studying high-affinity receptor-ligand interactions because of its ability to probe the high-force regime (>120 pN). Here we present our complete protocol for studying cellulosomal protein assemblies at the single-molecule level. Using a protein topology derived from the native cellulosome, we worked with enzyme-dockerin and carbohydrate binding module-cohesin (CBM-cohesin) fusion proteins, each with an accessible free thiol group at an engineered cysteine residue. We present our site-specific surface immobilization protocol, along with our measurement and data analysis procedure for obtaining detailed binding parameters for the high-affinity complex. We demonstrate how to quantify single subdomain unfolding forces, complex rupture forces, kinetic off-rates, and potential widths of the binding well. The successful application of these methods in characterizing the cohesin-dockerin interaction responsible for assembly of multidomain cellulolytic complexes is further described.
Keywords: Bioengineering, Issue 82, biophysics, protein unfolding, atomic force microscopy, surface immobilization
Introduction
Cellulosomes are large multienzyme complexes displayed on the surface of anaerobic cellulolytic bacteria (e.g. C. thermocellum) that have evolved to efficiently depolymerize plant cell wall lignocellulose into soluble oligosaccharides1. A central attribute of cellulosomes is the high-affinity cohesin-dockerin interaction. In the most prominent paradigm, a highly conserved 60-75 amino acid type I dockerin module is displayed at the C-terminal end of the various bacterial enzymes. The dockerin module directs assembly of synergistic combinations of enzymes onto the noncatalytic scaffold protein ('scaffoldin'), which comprises a polyprotein of cohesin domains that are specific for the type I dockerin module. At higher levels, cellulosome architecture can become very complex, incorporating alternative cohesin and dockerin pairs (e.g. type II, type III) that anchor the structures to the cell surface and allow for the assembly of branched structures containing multiple scaffoldins2. The various cohesin-dockerin types, despite having related structures, exhibit differential binding specificities suppressing cross reactivity with unintended scaffoldins or components from other cellulosome-producing bacterial species. While bioinformatic approaches have successfully identified thousands of unique cellulosomal components at the genetic level, comparatively few protein structures are known, and the mechanisms at work in cohesin-dockerin specificity determination remains an active area of structural biology research.
Since the invention of the atomic force microscope (AFM) by Binnig et al.3, various AFM operational modes have been developed and continuously improved, including noncontact imaging, oscillation mode imaging4, and single molecule force spectroscopy (SMFS)5,6. SMFS has evolved into a widely used technique to directly probe individual proteins7-11, nucleic acids12-15, and synthetic polymers16-19. In a typical SMFS experiment to investigate receptor-ligand binding20,21, an AFM cantilever tip is modified with one of the binding partners, while a flat glass surface is modified with the complementary binding partner. The modified cantilever is brought into contact with the surface allowing the partners to bind. The base of the cantilever is then withdrawn at constant speed and the force is measured using the optical lever deflection method. The resultant force-distance data traces exhibit sawtooth-like peaks if binding was established. In cases where the binding partners are fused to multiple protein domains, each peak in the force-distance trace can be correlated to the unfolding of a single protein domain or folded subdomain, while the last peak corresponds to rupture of the protein binding interface. The specific positions of the force-resistant elements can be used as a fingerprint to identify the various protein domains of interest. This method can be used to interrogate important amino acids involved in protein folding and stabilization. Many models have been reported in the literature to treat the characteristic force extension behavior observed in SMFS experiments. The most commonly used models include the freely jointed chain (FJC) model22, the worm-like chain (WLC) model18,23-25, and the freely rotating chain (FRC) model25,26.
In our prior work11, we used single-molecule force spectroscopy to investigate the interaction of cohesin and dockerin modules. Here, we present an experimental protocol for glass surface and cantilever functionalization with enzyme-dockerin and CBM-cohesin protein constructs. We also present an AFM-based SMFS protocol including data acquisition and analysis procedures. The described protocol can easily be generalized to other molecular systems, and should prove particularly useful to researchers interested in high-affinity receptor ligand pairs.
Protocol
A schematic of the pulling geometry used in this work to probe the cohesin-dockerin interaction is shown in Figure 1A. The protein immobilization protocol reported here for cantilever and cover glass functionalization is a modified version of the procedure published previously27. The proteins were expressed from plasmid vectors in E. coli using conventional methods. The proteins were designed with a solvent-accessible thiol group, which was used in combination with maleimide chemistry to tether the protein via a stable thioether linkage to the cover glass surface and cantilever. The engineered cysteine residues in both the CBM-cohesin and xylanase-dockerin fusion proteins were located towards the N-terminal side of the proteins, away from the cohesin-dockerin binding interface11. A detailed overview of the chemical bonding employed in protein immobilization is shown in Figure 1B.
1. Sample Preparation
- Buffers
- Prepare Tris buffered saline supplemented with calcium (TBS): 25 mM TRIS, 75 mM NaCl, 1 mM CaCl2, pH 7.2
- Prepare sodium borate buffer: 50 mM Na2B4O7, pH 8.5 The process flow diagram showing sample preparation steps is shown in Figure 2. When handling cantilevers and cover glasses, self-locking tweezers are recommended.
- Aminosilanization of cover glass (approximately 1.5 hr)
- Place cover glass (24 mm diameter, 0.5 mm thickness) in a PTFE holder.
- Sonicate cover glass in 1:1 ethanol : ultrapure water (v/v) for 15 min.
- Rinse cover glass with ultrapure water.
- Place cover glass in piranha solution (1:1 H2SO4 (concentrated) : H2O2 (30%) (v/v)) for 30 min, then thoroughly rinse with ultrapure water. Dry cover glass under a gentle stream of N2. Caution: piranha solution is extremely corrosive. Eye and skin protection are required.
- Submerge cover glass in 45:5:1 ethanol : ultrapure water : 3-aminopropyl dimethyl ethoxysilane (v/v). Place on a shaker at RT for 60 min (approximately 50 rpm).
- Submerge cover glass sequentially in ethanol and ultrapure water (2x each). Dry cover glass under a gentle stream of N2.
- Bake cover glass in an oven (80 °C for 30 min).
- Silanized cover glasses may be stored under argon for up to 6 weeks.
- Aminosilanization of cantilevers (approximately 1.5 hr) NOTE: The presented protocol for tip functionalization is appropriate for silicon cantilever tips.
- Place cantilevers on a clean glass slide. Treat with UV-ozone for 15 min.
- Submerge cantilevers for 3 min in 1:1 ethanol : 3-aminopropyl dimethyl ethoxysilane (v/v) with a catalytic amount (0.25%, (v/v)) of ultrapure water.
- Rinse cantilevers with gentle stirring sequentially for 60 sec in beakers of toluene, ethanol, and ultrapure water. Carefully dry cantilevers on filter paper between rinses.
- Place levers on a clean glass slide and bake (80 °C for 30 min).
- Protein disulfide reduction (approximately 3 hr) All solutions should be prepared to obtain approximately 30 µl of diluted protein per cantilever and 20 µl of diluted protein per cover glass. Protein solutions should be mixed with Tris(2-carboxyethyl)phosphine (TCEP) disulfide reducing gel in a ratio of 1:2 (v/v).
- Prepare aliquots of TCEP disulfide reducing beads in micro-tubes. It is recommended to cut micropipette tips with scissors to widen the hole diameter when pipetting the TCEP bead slurry.
- Rinse TCEP bead slurry with 1 ml TBS buffer, and centrifuge at 850 rcf for 3 min.
- Carefully remove and discard the supernatant with a micropipette.
- Repeat steps 1.4.2-1.4.3 2x.
- Apply concentrated protein solution (1-10 mg/ml) to the TCEP beads (1:2 protein : TCEP bead slurry (v/v)) and gently mix by stirring with micropipette tip. Avoid introducing air bubbles.
- Place protein/TCEP bead slurry mixture on a rotator for 2.5 hr.
- PEGylation of cover glasses and cantilevers (approximately 1.5 hr)
- Prior to modification with NHS-PEG-maleimide linkers, soak aminosilanized cantilevers and cover glasses in sodium borate buffer (pH 8.5) for 45 min to deprotonate primary amine groups on the surface.
- Ensure that the NHS-PEG-maleimide powder is warmed up to RT before opening the cap and weighing the appropriate amount for a 25 mM solution. Unused NHS-PEG-maleimide should be stored under argon at -20 °C. Approximately 30 µl of polymer solution per cantilever, and 90 µl per 2 cover glasses (sandwiched together) is required.
- After weighing the 5 kDa NHS-PEG-maleimide, add sodium borate buffer and vortex to obtain a 25 mM solution. Note: The solution should be used as quickly as possible due to the extremely short half-life of NHS at pH 8.5. Vortexing and transfer of the liquid onto the cantilevers/cover glasses should be completed within 1-2 min.
- Incubate cantilevers in 30 µl droplets of NHS-PEG-maleimide solution in a Petri dish. For cover glasses, place 90 µl of NHS-PEG-maleimide solution onto a single cover glass, and add a second cover glass on top creating a cover glass sandwich with NHS-PEG-maleimide solution in the middle.
- Incubate the cantilevers/cover glasses with the NHS-PEG-maleimide solution in a water-saturated atmosphere at RT for 1 hr.
- Protein conjugation (approximately 2 hr) Critical: Minimize the exposure of PEGylated cantilevers and cover glasses to air.
- Centrifuge TCEP-bead/ reduced protein solutions at 100 rcf for 1 min and collect the supernatant.
- Dilute protein solution with TBS. Aim for a protein concentration during surface conjugation in a range of 0.5-2 mg/ml. Set reduced and diluted protein solutions aside for a few minutes while rinsing cantilevers and cover glasses.
- Rinse cantilevers and cover glasses in three sequential beakers of ultrapure water.
- Carefully remove residual liquid from cover glasses by carefully touching the edges to a filter paper under a gentle stream of N2. Carefully remove residual liquid from cantilevers by touching to a filter paper. Apply diluted protein solution immediately.
- Mount cover glasses in an appropriate sample holder that is compatible with the AFM instrument.
- Incubate PEGylated cover glasses and cantilevers with respective diluted protein solutions at RT for 1-2 hr.
- Rinse cantilevers in three sequential beakers with TBS to remove unbound proteins. Pipette rinse cover glasses at least 10x.
- Store cantilevers and cover glasses under TBS prior to measurement.
2. Data Acquisition
In this work, a custom-built AFM28 controlled by an MFP-3D AFM controller from Asylum Research with custom written Igor Pro software was used. Cantilever deflection is measured via the optical beam deflection method29. The sample preparation and data analysis protocols provided here are applicable regardless of the exact AFM model used. However, the AFM model should be suitable for measuring in liquids and support an accessible speed range on the z-piezo of approximately 200-5,000 nm/sec.
Mount the functionalized cantilever and glass surface on the AFM. During the whole procedure, the surface should stay covered with buffer. When mounting the cantilever, minimize exposure to air. Upon correct adjustment of the laser beam, let the system equilibrate for at least 30 min to reduce any drift effects and readjust if necessary.
Record a thermal noise spectrum with the cantilever far away from the surface, i.e. in the absence of damping effects.
Use a minimally invasive method like the acoustic approach to find the surface without damaging the cantilever tip prior to measurement. If possible, manually approach the surface with the cantilever and use headphones to listen to the thermal noise on the raw deflection output from the AFM controller. As soon as the cantilever draws near the surface, a distinct change in sound is audible. Note: The cantilever tip should now be within 2-5 µm of the surface. The nature of the sound change is dependent on the cantilever used. The resonance frequency of the cantilever used in this work is approximately 25 kHz in water, above the human audible range. Due to damping effects near the surface, the resonance is shifted towards lower frequencies bringing the cantilever resonance into the audible range. Hence, an apparent increase in frequency and sound intensity is perceived. In cases where an audio output of the deflection signal is not available, the surface can be approached with the z-piezo while an active feedback on the deflection signal is enabled. As soon as the deflection signal increases by a defined amount due to indentation of the surface, the approach is stopped.
Determine the inverse optical lever sensitivity, (InvOLS) which represents the tip displacement distance (in nm) per volt deflection signal. Do this by indenting the surface with the cantilever tip. A deflection set point voltage corresponding to a cantilever tip displacement of approximately 3 nm is recommended.
Determine the spring constant of the cantilever by fitting a simple harmonic oscillator response function to the thermal noise spectrum, according to the equipartition theorem30,31.
Initialize an experimental routine. For this work, the following set of measurement parameters was used: approach speed: 3,000 nm/sec; indentation force: 180 pN; surface dwell time: 10 msec; retract velocities: 0.2, 0.7, 2.0, 5.0 µm/sec with sampling rates of 2,000, 5,000, 15,000, 20,000 Hz respectively; retract distance: 500 nm. Note: The sampling rate should not be set higher than 10 points/nm to avoid oversampling and to keep data sizes reasonable.
After each force-distance trace, actuate the x- and y-piezo stages to expose a new surface location to the cantilever in each force-distance curve. This technique samples a larger area of the cover glass surface during long-term measurements.
Use periodic rezeroing of the deflection stage (i.e. photodiode position) and height of the z-piezo chassis during long-term measurements in case the deflection signal drifts out of range, or contact is lost with the surface.
Upon completion of the measurement run, perform another InvOLS measurement with a significantly higher indentation force than used prior to measurements to obtain a more precise InvOLS value.
Record another thermal noise spectrum far away from the surface. Determine the spring constant at the end of the experimental run.
3. Data Analysis
The flow diagram in Figure 3 illustrates the process of data analysis. Perform all data manipulations using an appropriate software package such as Igor Pro or MATLAB. First convert the raw signal from the detector into units of force, and correct for offset and drift. Subsequently, use models of biopolymer elasticity to locate energy barriers in the unfolding pathways, and identify protein subdomains. Finally, kinetic and energetic parameters of the receptor-ligand interaction are obtained.
- Unit conversion and data corrections
- Multiply the raw deflection signal (volts) by the InvOLS (nm/volt) and spring constant (pN/nm) to convert the detector voltage into units of force.
- Offset the data such that the unloaded cantilever has a force value of zero pN by first averaging the force values from the last 10% of the force-distance trace (acquired farthest away from the surface), and then subtracting the average from all force values in the data trace.
- Offset each trace in the x-direction such that the first intercept with the distance axis occurs at a distance of 0 nm.
- The InvOLS is dependent on the laser spot position on the cantilever. Even small amounts of drift in the optical readout system may cause noticeable changes in the InvOLS when the footprint of the cantilever is comparable to the laser spot size. Correct for this by analysis of the noise on the deflection signal at zero force. Assuming constant ambient conditions, noise on the deflection signal is directly proportional to the InvOLS.
- Measure the route mean square (RMS) deflection value (noise level under zero force) of the last 10% of each force-distance trace.
- Plot the noise vs. the curve number and apply a suitable fit. Typically an exponential fit in the form of
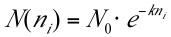 will work best, where N is the noise, ni is the curve number, and N0 and k are fit parameters. A linear fit may also be appropriate for certain data sets.
will work best, where N is the noise, ni is the curve number, and N0 and k are fit parameters. A linear fit may also be appropriate for certain data sets. - Determine a scaling factor (SF) for each curve: Equation 1:
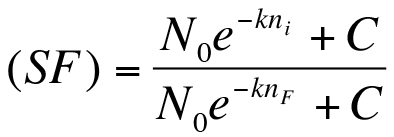 where, ni is the curve number, nf is the final curve number, and C is an offset.
where, ni is the curve number, nf is the final curve number, and C is an offset. - Next divide all the force values in each individual curve by the scaling factor. This procedure scales each curve by the ratio of the RMS noise value of the current curve to the RMS noise value of the final trace that was acquired immediately prior to the InvOLS measurement.
- Perform a deflection correction to transform the distance axis (z) to molecular extension (z*). This accounts for bending of the cantilever under force which shortens the distance between cantilever tip and sample from the value reported by the z-piezo sensor position. Equation 2:
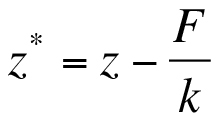 Where z is the measured z-sensor position, F the force acting on the cantilever and k the spring constant.
Where z is the measured z-sensor position, F the force acting on the cantilever and k the spring constant.
- Contour length analysis The contour length of a protein is the maximum stretched length of the polypeptide chain. The folding state of a protein refers to its geometry and end to end distance determined by secondary and tertiary structure. The contour length of a protein is directly related to its folding state9,25,32. The position of specific ruptures in force-extension traces varies widely due to polydispersity of PEG linkers, as well as external parameters such as temperature, buffer properties and loading rates. This complicates direct data analysis but can be overcome by transforming force-extension data into contour length space. This technique enables averaging over huge datasets, and allows automatic pattern recognition to be used to identify characteristic unfolding events. It is therefore possible to sort individual force traces depending on the type of interaction exhibited. The following previously described procedure25 is used to transform force-extension data into contour length space.
- Solving the WLC model (Equation 3)23 for the contour length L at a fixed persistence length p results in Equation 4, which provides the contour length L(x,u). Here, x is the distance and u=F*p/kBT, where kB is Boltzmann's constant and T is the temperature. Only real solutions can be considered. Additional constraints are x<L, F>0, L>0, x>0; Equation 3:
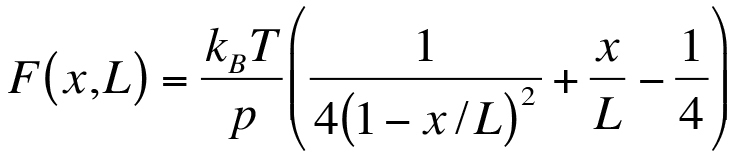 Equation 4:
Equation 4: 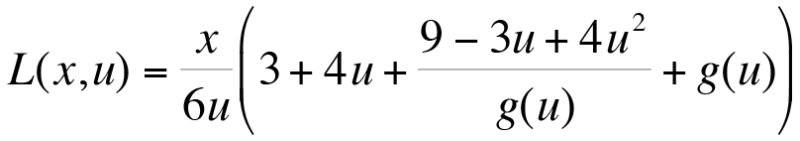 where,
where, 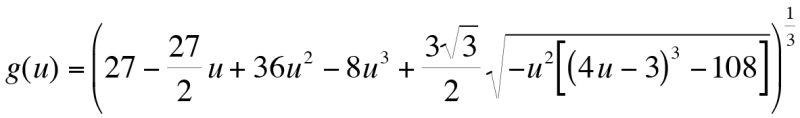
- Plot the transformed data points in a force vs. contour length plot. Apply a force threshold of approximately 10 pN to exclude noise. Unspecific interactions can be excluded by applying a long-pass length filter. Assemble a histogram of contour lengths.
- Cross-correlate33 the obtained histograms with a template histogram, and offset along the x-axis to correct for PEG polydispersity. Use the resulting correlation values to measure the similarity of individual data traces. Thereby, data traces can be sorted into predefined classes to simplify further analysis.
- Use a similar technique to find repeating features in a single trace by autocorrelation, e.g. for multiple Ig-domain unfolding.
- Sort traces manually to investigate other unfolding events.
- Loading Rate Analysis Extract kinetic and energetic information about receptor-ligand dissociation by applying suitable models to the force spectrum, i.e. the rupture-force vs. ln(loading-rate) plot.
- For a given pulling speed, determine the rupture force and loading rate for rupture events of interest:
- Perform a line fit to a force-time trace in the vicinity of the rupture event of interest. Determine the loading rate from the slope of the line fit to the peak. Repeat this procedure for every trace showing the rupture event of interest.
- Determine the most probable rupture force by applying a Gaussian fit to a histogram of the rupture forces. Alternative fit functions are possible.
- Determine the most probable loading rate.
- Repeat steps 3.3.3.1 - 3.3.3.3 for all pulling speeds.
- Plot the most probable rupture forces against the natural logarithm of the most probable loading rates to obtain the force spectrum.
- Apply a suitable theoretical model to the force spectrum to extract kinetic and energetic parameters (Figure 4C). In many cases, the linear Bell-Evans model20,34 can be used and will yield good estimates for koff, the dissociation rate in the absence of force, and Dx, the distance to the transition state along the reaction coordinate, as shown in Equation 5. Equation 5:

Representative Results
We used the described procedure to investigate a type I cohesin-dockerin pair from C. thermocellum. Upon successful binding of the cohesin-dockerin pair, the recorded force distance traces showed characteristic peak patterns. A typical trace is shown in Figure 4a. Every peak in the trace represents the unfolding of one protein subdomain with the last peak corresponding to the dissociation of the receptor-ligand complex.
For the CBM-cohesin-dockerin-xylanase complex investigated in this work, the initial rise in force corresponds to stretching of the PEG linker molecules. The subsequent series of up to three descending force dips reflects the unfolding of the xylanase domain. The final peak represents the rupture of the cohesin-dockerin binding interface.
All recorded force-distance traces were transformed to force-contour length space. The resulting barrier position histogram is shown in Figure 4B. The data show a contour length increment of approximately 89 nm. The xylanase domain consists of 378 amino acids, 260 of which are located C-terminally from the engineered cysteine residue. From the crystal structure, the folded length of the domain is assumed to be 6 nm. Further assuming a length per stretched amino acid of 0.365 nm35, the measured 89 nm increment can be unambiguously assigned to the unfolding of the xylanase domain. This is consistent with previously published results11.
To probe the energy landscape of the cohesin-dockerin interaction, we analyzed a total of 186 data traces obtained with four different pulling speeds (0.2, 0.7, 2.0, and 5.0 µm/sec). The resulting force spectrum is shown in Figure 4C. Fitting Equation (5) to the data yields values for koff and Dx of 3.13 x10-5/sec and 0.70 nm, respectively. These values are in good agreement with previously published results11.
 Figure 1.Schematic of biomolecule immobilization. (A) Xylanase-dockerin fusion proteins are attached to the glass slide via PEG linkers. The cantilever is similarly modified with a cohesin protein fused to a cellulose binding module (CBM). (B) Depiction of chemical bonding employed in cover glass and cantilever functionalization. Click here to view larger image.
Figure 1.Schematic of biomolecule immobilization. (A) Xylanase-dockerin fusion proteins are attached to the glass slide via PEG linkers. The cantilever is similarly modified with a cohesin protein fused to a cellulose binding module (CBM). (B) Depiction of chemical bonding employed in cover glass and cantilever functionalization. Click here to view larger image.
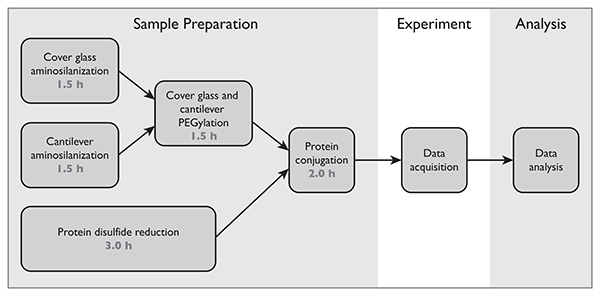 Figure 2.Process flow diagram showing sample preparation steps followed by data acquisition and analysis.Click here to view larger image.
Figure 2.Process flow diagram showing sample preparation steps followed by data acquisition and analysis.Click here to view larger image.
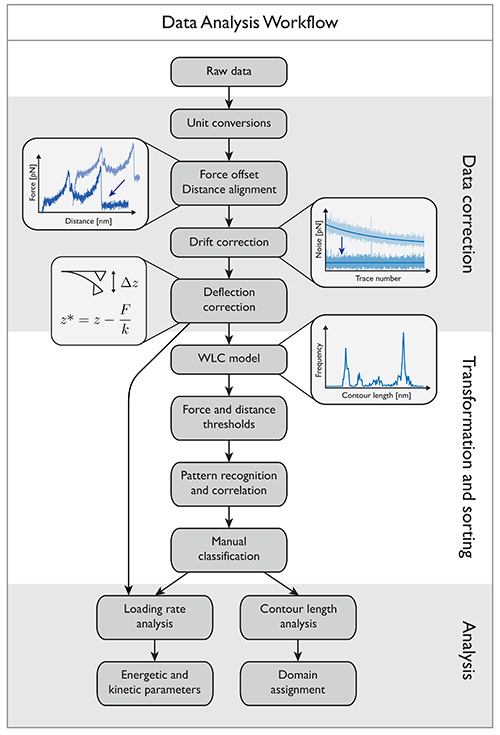 Figure 3. Data analysis workflow diagram showing the processing steps involved in converting the raw detector signals into force-extension traces. These traces are further analyzed to obtain information about receptor-ligand binding. The final results provide energetic and kinetic parameters about specific domains. Click here to view larger image.
Figure 3. Data analysis workflow diagram showing the processing steps involved in converting the raw detector signals into force-extension traces. These traces are further analyzed to obtain information about receptor-ligand binding. The final results provide energetic and kinetic parameters about specific domains. Click here to view larger image.
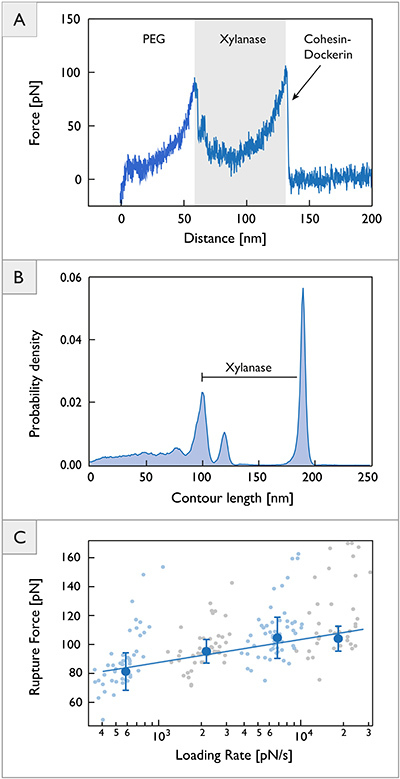 Figure 4.Single molecule force spectroscopy data on cohesin-dockerin. (A) Typical unfolding trace showing PEG linker stretching, xylanase unfolding, and rupture of the cohesin-dockerin binding interface. (B) Contour length histogram assembled from 314 force distance traces exhibiting energy barrier positions along the contour length. (C) Dynamic force spectrum obtained from 186 force-extension traces. Large blue circles represent the most probable rupture force at a given loading rate. The solid line represents a least squares fit to Equation 5. Rupture event populations are shown in the background. Error bars represent standard deviation obtained from Gaussian fits. Click here to view larger image.
Figure 4.Single molecule force spectroscopy data on cohesin-dockerin. (A) Typical unfolding trace showing PEG linker stretching, xylanase unfolding, and rupture of the cohesin-dockerin binding interface. (B) Contour length histogram assembled from 314 force distance traces exhibiting energy barrier positions along the contour length. (C) Dynamic force spectrum obtained from 186 force-extension traces. Large blue circles represent the most probable rupture force at a given loading rate. The solid line represents a least squares fit to Equation 5. Rupture event populations are shown in the background. Error bars represent standard deviation obtained from Gaussian fits. Click here to view larger image.
Discussion
To obtain meaningful data from single molecule force spectroscopy experiments, it is crucial to achieve well-defined and reproducible pulling geometries. The protocol used here results in site-specific immobilization of protein complexes in a defined pulling geometry.
The cantilevers used in this study were chosen due to their force sensitivity and high resonance frequency in water. Moreover, the small tip curvature of approximately 10 nm is advantageous for single molecule experiments due to reduced likelihood of multiple interactions. However, the small footprint (38x16 µm2) of the cantilever arm complicates the adjustment of the laser beam when the optical deflection method29 is used. The diameter of the focused laser beam in the setup used for this study is comparable to the width of the cantilever. As a result, obtaining a steady sum signal can be difficult. The laser drift on the cantilever can be partially compensated for using noise analysis across the data curves to correct the inverse optical lever sensitivity, as we have described. A new atomic force microscope with a shortened optical path and smaller laser spot size is currently under development in our group to improve data quality.
To obtain reliable information about rupture events, analysis of many traces is necessary. Since single molecule force spectroscopy measurements are subject to various fluctuations, averaging in force-extension space is not constructive. Barrier position histograms, however, once aligned in contour length space can be averaged since they are independent of fluctuations. As a result, even tiny features in the unfolding pathway are resolved. Conventional superposition of force extension traces does not achieve this kind of resolution.
In a force regime above 500 pN, a corrected WLC model accounting for electron cloud elasticity (QM-WLC) describes force-extension behavior better than the classical WLC model18. The freely rotating chain26 model (FRC) can also be used in a high force regime. With rupture forces up to 125 pN, the cohesin-dockerin interface shows one of the strongest receptor-ligand interactions reported in the literature. The WLC model was used in this work and in practice there was little difference between WLC, QM-WLC, and FRC models for analysis of cohesin-dockerin unfolding traces.
The conventional Bell-Evans20,34 model was used to analyze the force-loading rate dependency of the cohesin-dockerin binding interface. Recent works36,37 have extended the theoretical framework for the interpretation of single molecule experiments. These models treat nonlinear trends in the force spectra. Furthermore, they produce the free energy barrier height DG of the dissociation event. To observe distinct nonlinear trends in the force spectra, loading rates need to be varied over many orders of magnitude. Realizing extremely low loading rates is theoretically achievable using extremely slow z-piezo pulling speeds, however in practice this poses a challenge due to drift in the tip-substrate distance. Extremely high loading rates can also be difficult to obtain since increasing noise might obscure certain features in the recorded force-distance traces. Choice of the theoretical model must be balanced with these practical aspects of data acquisition while considering the specific proteins under investigation. In many cases the linear Bell-Evans model is entirely sufficient.
In conclusion, a complete experimental protocol for the study of receptor-ligand interactions using AFM-based single-molecule force spectroscopy has been presented. The positioning accuracy and force sensitivity of the atomic force microscope in conjunction with versatile biomolecule immobilization strategies provide an excellent toolbox for the investigation of receptor-ligand systems for structural biology studies.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors acknowledge funding from a European Research Council advanced grant to Hermann Gaub. Michael A. Nash gratefully acknowledges funding from Society in Science - The Branco Weiss Fellowship program. The authors thank Edward A. Bayer, Yoav Barak, and Daniel B. Fried at the Weizmann Institute of Science for generously providing the proteins used in this study. The authors thank Hermann E. Gaub, Elias M. Puchner, and Stefan W. Stahl for helpful discussions.
References
- Bayer EA, Belaich JP, Shoham Y, Lamed R. The cellulosomes: Multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 2004;58:521–554. doi: 10.1146/annurev.micro.57.030502.091022. [DOI] [PubMed] [Google Scholar]
- Bayer EA, Lamed R, White BA, Flint HJ. From Cellulosomes to Cellulosomics. Chem. Rec. 2008;8(6):364–377. doi: 10.1002/tcr.20160. [DOI] [PubMed] [Google Scholar]
- Binnig G, Quate CF. Atomic Force Microscope. Phys. Rev. Lett. 1986;56(9):930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- García R, Perez R. Dynamic atomic force microscopy methods. Surf. Sci. Rep. 2002;47(6):197–301. [Google Scholar]
- Engel A, Müller DJ. Observing single biomolecules at work with the atomic force microscope. Nat. Struct. Biol. 2000;7(9):715–718. doi: 10.1038/78929. [DOI] [PubMed] [Google Scholar]
- Li H, Cao Y. Protein Mechanics: From Single Molecules to Functional Biomaterials. Acc. Chem. Res. 2010;43(10):1331–1341. doi: 10.1021/ar100057a. [DOI] [PubMed] [Google Scholar]
- Florin E-L, Moy VT, Gaub HE. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264(5157):415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- Oberhauser A, Hansma P, Carrion-Vazquez M, Fernandez J. Stepwise unfolding of titin under force-clamp atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 2001;98(2):468–472. doi: 10.1073/pnas.021321798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchner EM, Gaub HE. Force and function: probing proteins with AFM-based force spectroscopy. Curr. Opin. Struct. Biol. 2009;19(5):605–614. doi: 10.1016/j.sbi.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Rief M, Gautel M, Oesterhelt F, Fernandez J, Gaub H. Reversible unfolding of individual titin immunoglobulin domains by AFM. Sci. 1997;276(5315):1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- Stahl SW, Nash MA, et al. Single-molecule dissection of the high-affinity cohesin–dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 2012;109(50):20431–20436. doi: 10.1073/pnas.1211929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland T, Ratner BD. Direct measurement of hydrogen bonding in DNA nucleotide bases by atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 1995;92(12):5297–5301. doi: 10.1073/pnas.92.12.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfill J, Kühner F, Blank K, Lugmaier RA, Sedlmair J, Gaub HE. B-S Transition in Short Oligonucleotides. Biophys. J. 2007;93(7):2400–2409. doi: 10.1529/biophysj.107.106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rief M, Clausen-Schaumann H, Gaub HE. Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 1999;6(4):346–350. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- Severin PMD, Zou X, Gaub HE, Schulten K. Cytosine methylation alters DNA mechanical properties. Nucleic Acids Res. 2011;39(20):8740–8751. doi: 10.1093/nar/gkr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler M, Balzer BN, Hugel T. Polymer Adhesion at the Solid-Liquid Interface Probed by a Single-Molecule Force Sensor. Small. 2009;5(24):2864–2869. doi: 10.1002/smll.200901237. [DOI] [PubMed] [Google Scholar]
- Giannotti MI, Vancso GJ. Interrogation of Single Synthetic Polymer Chains and Polysaccharides by AFM-Based Force Spectroscopy. Chem. Phys. Chem. 2007;8(16):2290–2307. doi: 10.1002/cphc.200700175. [DOI] [PubMed] [Google Scholar]
- Hugel T, Rief M, Seitz M, Gaub H, Netz R. Highly Stretched Single Polymers: Atomic-Force-Microscope Experiments Versus Ab-Initio Theory. Phys. Rev. Lett. 2005;94(4):10–1103. doi: 10.1103/PhysRevLett.94.048301. [DOI] [PubMed] [Google Scholar]
- Nash MA, Gaub HE. Single-Molecule Adhesion of a Stimuli-Responsive Oligo(ethylene glycol) Copolymer to Gold. ACS Nano. 2012;6(12):10735–10742. doi: 10.1021/nn303963m. [DOI] [PubMed] [Google Scholar]
- Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 1999;397(6714):50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- Morfill J, Neumann J, et al. Force-based Analysis of Multidimensional Energy Landscapes: Application of Dynamic Force Spectroscopy and Steered Molecular Dynamics Simulations to an Antibody Fragment–Peptide. Complex. J. Mol. Biol. 2008;381(5):1253–1266. doi: 10.1016/j.jmb.2008.06.065. [DOI] [PubMed] [Google Scholar]
- Ortiz C, Hadziioannou G. Entropic Elasticity of Single Polymer Chains of Poly(methacrylic acid) Measured by Atomic Force Microscopy. Macromolecules. 1999;32(3):780–787. [Google Scholar]
- Bustamante C, Marko JF, Siggia ED, Smith F. Entropic Elasticity of l-phage DNA. Science. 1994;265(5178):1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28(26):8759–8770. [Google Scholar]
- Puchner EM, Franzen G, Gautel M, Gaub HE. Comparing Proteins by Their Unfolding Pattern. Biophys. J. 2008;95(1):426–434. doi: 10.1529/biophysj.108.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livadaru L, Netz RR, Kreuzer HJ. Stretching Response of Discrete Semiflexible Polymers. Macromolecules. 2003;36(10):3732–3744. [Google Scholar]
- Zimmermann JL, Nicolaus T, Neuert G, Blank K. Thiol-based, site-specific and covalent immobilization of biomolecules for single-molecule experiments. Nat. Protoc. 2010;5(6):975–985. doi: 10.1038/nprot.2010.49. [DOI] [PubMed] [Google Scholar]
- Gumpp H, Stahl SW, Strackharn M, Puchner EM, Gaub HE. Ultrastable combined atomic force and total internal fluorescence microscope. Rev. Sci. Instrum. 2009;80(6) doi: 10.1063/1.3148224. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL, Clarke J. Scanning force microscope springs optimized for optical-beam deflection and with tips made by controlled fracture. J. Appl. Phys. 1994;76(1) [Google Scholar]
- Cook SM, Lang KM, Chynoweth KM, Wigton M, Simmonds RW, Schäffer TE. Practical implementation of dynamic methods for measuring atomic force microscope cantilever spring constants. Nanotechnology. 2006;17(9):2135–2145. [Google Scholar]
- Proksch R, Schäffer TE, Cleveland JP, Callahan RC, Viani MB. Finite optical spot size and position corrections in thermal spring constant calibration. Nanotechnology. 2004;15(9):1344–1350. [Google Scholar]
- Ainavarapu SRK, Brujic J, et al. Contour Length and Refolding Rate of a Small Protein Controlled by Engineered Disulfide Bonds. Biophys. J. 2007;92(1):225–233. doi: 10.1529/biophysj.106.091561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, Rief M. Detecting Molecular Fingerprints in Single Molecule Force Spectroscopy Using Pattern Recognition. Jap. J. Appl. Phys. 2007;46:5540–5542. [Google Scholar]
- Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997;72(4):1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, Rief M. Protein structure by mechanical triangulation. Proc. Natl. Acad. Sci. U.S.A. 2006;103(5):1244–1247. doi: 10.1073/pnas.0509217103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudko OK, Hummer G, Szabo A. Intrinsic Rates and Activation Free Energies from Single-Molecule Pulling Experiments. Phys. Rev. Lett. 2006;96(10) doi: 10.1103/PhysRevLett.96.108101. [DOI] [PubMed] [Google Scholar]
- Friddle RW, Noy A, De Yoreo JJ. Interpreting the widespread nonlinear force spectra of intermolecular bonds. Proc. Natl. Acad. Sci. U.S.A. 2012;109(34):13573–13578. doi: 10.1073/pnas.1202946109. [DOI] [PMC free article] [PubMed] [Google Scholar]