

Abstract
It is unclear if HIV-1 variants lose the ability to prime naïve CD8+ cytotoxic T lymphocytes (CTL) during progressive, untreated infection. We conducted a comprehensive longitudinal analysis of viral evolution and its impact on primary and memory CD8+ T cell responses pre-seroconversion (SC), post-SC, and during combination antiretroviral therapy (cART). Memory T cell responses targeting autologous virus variants reached a nadir by 8 yr post-SC with development of AIDS, followed by a transient enhancement of anti-HIV-1 CTL responses upon initiation of cART. We show broad and high magnitude primary T cell responses to late variants in pre-SC T cells, comparable to primary anti-HIV-1 responses induced in T cells from uninfected persons. Despite evolutionary changes, CD8+ T cells could still be primed to HIV-1 variants. Hence, vaccination against late, mutated epitopes could be successful in enhancing primary reactivity of T cells for control of the residual reservoir of HIV-1 during cART.
Keywords: HIV-1, autologous viral variants, memory T cell responses, primary T cell responses, cART
Introduction
CD8+ cytotoxic T lymphocytes (CTL) are a crucial component of the immunological defense against human immunodeficiency virus type 1 (HIV-1) infection (Bangham, 2009). The appearance of CTL responses is associated with the early decline of plasma viremia during HIV-1 infection (Koup et al., 1994). Depletion of CD8+ T cells in the non-human primate model results in enhanced disease progression, supporting the protective role of these cells (Schmitz et al., 1999). Long term control of both HIV-1 and simian immunodeficiency virus (SIV) infections is associated with a broad and high magnitude CTL response (Pandrea et al., ; Saez-Cirion et al., 2007), particularly to Gag epitopes (Berger et al., 2011; Kiepiela et al., 2007; Rolland et al., 2008). However, despite the establishment of HIV-1-specific CTL responses during early HIV-1 infection, they fail to control persistent, chronic HIV-1 infection. A hallmark of HIV-1 pathogenesis is the ability of the virus to escape host CTL responses through mutations within and adjacent to CTL epitopes during the early and chronic phases of the infection (Allen et al., 2002; Allen et al., 2000; Leslie et al., 2004; O'Connor et al., 2002). HIV-1 prophylactic and therapeutic vaccine strategies have yet to establish protective CD8+ CTL responses to overcome the propensity of the virus to undergo escape mutations.
The breadth and magnitude of CD8+ T cell responses are thought to be critical indicators in the control of HIV-1 infection as well as prevention of AIDS (Baker et al., 2009; Deeks and Walker, 2007). Therefore, the induction of strong and broadly reactive memory CTL responses is believed to be necessary to respond to the diverse viral sequences generated during the course of infection. Indeed, the failure of the STEP prophylactic vaccine trial has been linked in part to induction of limited CTL responses and failure to cross-react with circulating viral strains (Barouch and Korber, 2010; Corey, McElrath, and Kublin, 2009). There is also a correlation between the larger number of T cell responses targeting different SIV proteins and control of viral load in non-human primates (Liu et al., 2009; Reynolds et al., 2008). Importantly, the plasticity of the T cell receptor (TCR) and its ability to recognize wild-type and variant epitopes are suggested to be critical in viral control (Ladell et al., 2013). Moreover, the ability of TCR usage to modulate the protective role of MHC class I alleles has been recently described to be more important than the magnitude, polyfunctionality and avidity of T cell responses (Chen et al., 2012). The continuous debate highlighted above emphasizes the importance of understanding the mechanisms underlying the ability of CD8+ T cell responses to control HIV-1 replication for the design of successful vaccine strategies.
Mechanisms promoting the establishment of escape mutations and thus evasion of clearance of HIV-1-infected cells by CTL include interaction defects between a viral epitope and its cognate MHC class I molecule or between an MHC class I molecule/epitope complex and its T cell receptor (TCR) (Leslie et al., 2004) and impairment at the level of epitope processing (Allen et al., 2004; Draenert et al., 2004; Yokomaku et al., 2004). It is imperative to overcome these complications of natural infection in order to design HIV-1 vaccines capable of eliciting antiviral immunity targeting the diverse viral strains circulating during the course of infection and spreading from person to person. We have previously shown the generation of broadly reactive and polyfunctional primary CD8+ T cell responses from HIV-1 naïve adults against HIV-1 and other viral epitopes (Colleton et al., 2009). Optimal priming of CD8+ T cells requires maturation of DC with CD40L and IFN-γ. This maturation protocol generates IL-12-producing DC (Fan et al., 2007) with increased expression of activation and costimulatory molecules. These factors are involved in activation of memory antigen-specific CD8+ T cell memory responses and are likely involved in priming of CD8+ T cells (Prlic, Williams, and Bevan, 2007). These in vitro, T cell memory and priming models represent a promising approach to evaluate the immunogenicity of potential HIV-1 epitopes for vaccine design (Malaspina et al., 2011).
To better understand the relationship between viral evolution and its impact/effect on primary and memory responses, we examined autologous HIV-1 gag, env, and nef sequences derived at multiple points up to approximately 12 years post-seroconversion (SC) in a participant in the Multicenter AIDS Cohort Study (MACS) (Detels et al., 2012; Kaslow et al., 1987). The breadth and magnitude of primary T cell responses were compared to memory, recall T cell responses observed early and late during infection as well as during cART. Our results reveal memory T cell responses specific for prior and contemporaneous HIV-1 variants. We further demonstrate the development of strong primary responses in pre-SC naïve T cells to naturally evolving HIV-1 variant sequences in an entirely autologous system. These primary CD8+ T cell responses were of similar breadth to memory responses and of higher magnitude, thus supporting the use of such models in immunotherapy of HIV-1 infection in persons on cART.
Results
Clinical and virologic characteristics of the study participant
Study subject 8 enrolled in the MACS in November, 1984, 3.2 years prior to SC to HIV-1. He was negative for both hepatitis B and C viruses throughout the period of study. PBMC and plasma samples were collected biannually from the time of enrollment. Within the first 3 years after SC (early post-SC: 0–2.8 years), the number of CD4+ T cells decreased and the number of CD8+ T cells increased, with an inversion in the CD4:CD8 T cell ratio (Fig. 1). Viral load increased sharply to 7.8 × 104 RNA copies/ml at the first SC visit (0.3 years) and then reached a set point ranging from 23,092 to 39,608 RNA copies/ml up to 2.8 years. For the next 4.4 years (late post-SC: 3.3–7.8 years), the numbers of CD4+ T cells decreased, reaching 200 cells/mm3 6.1 years post-SC (Stage 3 HIV-1 infection, AIDS) (Schneider et al., 2008) while CD8+ T cells continued to increase. This decline in CD4+ T cells was associated with a rise in viral load that began approximately 3.8 years post-SC. A decline in viral load to a nadir of 80,470 copies/ml at 7.8 years post-SC was observed after development of AIDS and before initiation of cART at 8.3 years post SC. Overall, we observed a negative correlation between HIV-1 viral load and CD4+ T cell counts (p=0.001), as well as a positive correlation between viral load and CD8+ T cell counts (p=0.0004) before cART. Imposition of cART led to a decline in viral load to <200 RNA copies/ml during the first 1.8 years (early ART: 8.3–10.1 years). HIV-1 plasma viremia was maintained between <20 and 119 copies/ml through the next 10 years (late post-ART: >12.4 years). During this period of viral suppression, an increase in the CD4+ T cell count was observed with levels ranging between 266 and 587 cells/mm3, with a concurrent decrease in CD8+ T cell counts (Fig. 1).
Figure 1. Clinical course of HIV-1 infection in the study participant.

Plasma viral load (red line), CD4+ (green line) and CD8+ T (blue line) cell counts in subject 8 are shown. The study participant developed AIDS as per CDC guidelines at year 6.2 post-SC. cART was administered at 8.4 years post-SC. Years post-SC (x-axis) include early, late, early cART and late cART time points defined as follow: 0–2.8 years post-SC, 3.3–7.8 years post-SC, 8.3–10.1 years post-SC, and >12.4 years post-SC, respectively. SC, seroconversion.
Taken together, these data demonstrate a typical course of HIV-1 infection from SC through development of AIDS, and recovery on cART. Consequently, we chose to use this participant in our longitudinal analysis of viral evolution and immunological responses to autologous HIV-1.
Dynamics of viral evolution and epitope variants
We examined the longitudinal changes in HIV-1 genes from subject 8 to define the effects of immune pressure during chronic, untreated infection and during ART. We sequenced 12, 16, and 9 time points that spanned >10 years of infection for gag-p17/- p24, env, and nef genes, respectively. We next determined the pairwise diversity and divergence from the founder virus population at each time point. As expected (Shankarappa et al., 1999), viral diversification and divergence in each HIV-1 gene gradually increased with time (Fig. 2).
Figure 2. Dynamics of genetic diversity and divergence of HIV-1 in the study participant.

(○) Divergence and (●) diversity of (A) gag p17, (B) gag p24, (C) env and (D) nef are plotted (y-axis) for each gene during the course of HIV-1 infection. The study participant developed AIDS at year 6.2 post-SC and cART was administered at 8.4 years post-SC (vertical lines). Trends in pairwise distance diversity and divergence from the founder virus are plotted through time from seroconversion to 10.1 years post-SC. SC, seroconversion.
Diversity accumulated linearly in gag p17 (Fig. 2A) and gag p24 (Fig. 2B) and then shrunk, with the peak being about 4.9 years post-SC. Notably, diversification of env (Fig. 2C) and nef (Fig. 2D) proceeded faster than that of gag. A linear increase in diversity was observed until 6.1 years post-SC in env, when X4 viruses first appeared, and then diversity began to shrink (Shankarappa et al., 1999). Diversification of the nef gene followed a different pattern, with diversification appearing to slow appreciably before 4 years of infection, and, following a transient contraction, increased slowly thereafter. No clear plateau in divergence from the founder strain was noted in nef (Fig. 2C).
In summary, the circulating viral populations within our study participant show increasing divergence from the founder virus. Interestingly, we observed a significant correlation between viral load and viral divergence (p<0.01) and diversity (p=0.05) up through 6.6 years post-SC, wherein viral load began a rapid decline prior to cART.
Autologous HIV-1 peptide binding affinity to MHC class I
The strength of peptide binding to MHC class I is a key determinant in CTL epitope immunogenicity (Goulder and Watkins, 2008). We therefore identified known HLA A*0201-and B*0702-restricted HIV-1 CTL epitopes in autologous sequences from subject 8 based on previously established immunologic activity (Yusim K, 2004). We also predicted additional HLA A*0201- and B*0702-restricted epitopes using the BioInformatics and Molecular Analysis Section (BIMAS) computational model (Parker, Bednarek, and Coligan, 1994). Accordingly, we identified 12 peptide families consisting of the Gag, Env, or Nef founder epitopes and their corresponding autologous variants that evolved during infection (Table 1). Epitope variants are referred to in Table 1 in order of their evolution in our study participant.
Table 1.
Evolving HIV-1 peptide sequences and their HLA-matched predicted and experimental affinity binding scores
| MHC Class I Binding | ||||||
|---|---|---|---|---|---|---|
| HIV-1 protein* (epitope position) |
Sequencea | HLAb restriction |
netMHCpanc | logIC50d | Correlation coefficient |
p value |
| Gag | ||||||
| p17 77–85 | SLFNTVATL | A*0201 | 139.2 | 3.81 | 0.9777 | 0.0002 |
| SLFNTVAAL | potential | 41.9 | 3.84 | |||
| SLFNTVATP | potential | 13108 | 5.24 | |||
| SLFSTVAT`L | potential | 106.9 | 3.66 | |||
| SLFNTIATL | potential | 55.12 | 3.92 | |||
| SLYNTVATL | A*0201 | 223.2 | 3.86 | |||
| p24 16–24 | SPRTLNAWV | B*0702 | 36.4 | 4.18 | 0.0357 | NSe |
| SPRTLDAWV | undefined | 102.5 | 4.38 | |||
| SPRALNAWV | undefined | 24.4 | 4.07 | |||
| PPRTLNAWV | undefined | 1221 | 4.22 | |||
| SPRPLNAWV | undefined | 20 | 4.18 | |||
| SPRTLSAWV | undefined | 36.9 | 6.08 | |||
| p24 19–27 | TLNAWVKVV | A*0201 | 113.7 | 4.16 | 0.8439 | 0.034 |
| TLDAWVKVV | potential | 85.6 | 4.30 | |||
| ALNAWVKVV | potential | 42.7 | 3.93 | |||
| PLNAWVKVV | potential | 2854.1 | 4.81 | |||
| TLSAWVKVV | potential | 105.4 | 4.18 | |||
| p24 143–151 | RMYSPTSIL | potential | 173.1 | 4.34 | 0.9738 | 0.0132 |
| RMYSPISIL | potential | 87.7 | 3.83 | |||
| RMYSPASIL | potential | 281 | 4.78 | |||
| RMYSPVSIL | potential | 123.8 | 3.92 | |||
| Env gp160/120 | ||||||
| 298–307 | RPNNNTRKSI | B*0702 | 14.7 | 3.56 | 0.0901 | NS |
| RPNNNTRRSI | undefined | 9.3 | 3.99 | |||
| RSNNNTRKSI | undefined | 3120.83 | 4.33 | |||
| RPNNNTRKST | RS9 B*07 | 130 | 4.04 | |||
| RPNNSTRKSI | undefined | 12.6 | 3.87 | |||
| RPNNDTRKSI | undefined | 23.9 | 3.85 | |||
| RPNNNTRKRI | undefined | 49.7 | 4.32 | |||
| RPNNNTGKRI | undefined | 77.4 | 4.36 | |||
| RPSNNTRKRI | undefined | 34.4 | 4.17 | |||
| RPTNNTRKSI | undefined | 16.6 | 3.75 | |||
| RPNNNTRKCI | undefined | 67.8 | 4.63 | |||
| RPNNNTRKSL | RS9 B*07 | 5.4 | 3.69 | |||
| 311–320f | IGPGRAFYAT | undefined | 30864.5 | 5.64 | 0.815 | NS |
| IGSGRAFYAT | undefined | 29119.6 | 6.56 | |||
| IGPGRAFYAA | undefined | 19736.5 | 4.48 | |||
| IGPGIAFYAT | undefined | 27013.2 | 5.91 | |||
| 341–349 | TLEQVVKKL | potential | 10358.5 | 5.34 | 0.824 | 0.001 |
| ALEQVVKKL | potential | 4739.2 | 4.93 | |||
| MLEQVVKKL | potential | 2643.9 | 4.99 | |||
| TLAQVVKKL | potential | 1205.9 | 4.55 | |||
| TLEQVVEKL | potential | 3291.8 | 5.11 | |||
| ILEQVVKKL | potential | 4295.9 | 5.35 | |||
| TLEQVVNKL | potential | 3678.9 | 5.14 | |||
| TLDKVVKKL | potential | 3003.6 | 4.96 | |||
| TLGRVAKKL | potential | 8819.8 | 6.03 | |||
| TLGQVVEKL | potential | 677.1 | 4.45 | |||
| TLDKVVEKL | potential | 431.5 | 4.36 | |||
| TLGKVVKKL | potential | 4549.2 | 4.85 | |||
| Nef | ||||||
| 68–76 | FPVRPQVPL | B*0702 | 8.6 | 3.83 | 0.9766 | 0.0015 |
| FSVRPQVPL | potential | 2500.2 | 4.85 | |||
| FPARPQVPL | potential | 5.2 | 3.66 | |||
| SPVRPQVPL | potential | 8.4 | 3.67 | |||
| FPIRPQVPL | potential | 7.9 | 3.62 | |||
| 77–85 | RPMTWKGAL | potential | 2.9 | 3.85 | 0.0733 | NS |
| RPMTWKAAL | potential | 2.7 | 3.86 | |||
| RPMTYKGAL | potential | 2.6 | 3.89 | |||
| RPMTRKAAL | potential | 2.1 | 3.82 | |||
| RPMTCKGAL | potential | 2.9 | 3.74 | |||
| RPMTYKAAL | B*0702 | 2.4 | 4.04 | |||
| RPITYKAAL | potential | 3.2 | 3.87 | |||
| 128–137 | TPGPGTRYPL | undefined | 20.3 | 3.57 | 0.9076 | <0.0001 |
| TPGPGIRFPL | undefined | 43.7 | 3.4 | |||
| TPGPGIRYPL | B7 | 34.8 | 3.78 | |||
| TPGPGIRYPV | undefined | 142.3 | 3.74 | |||
| TPGPGIRFPI | undefined | 150.3 | 3.77 | |||
| TSGPGTRFPL | undefined | 5462.9 | 4.85 | |||
| IPGPGRHPL | undefined | 16.8 | 3.51 | |||
| B7 | ||||||
| TPGPGVRYPL | supertype | 30.3 | 3.64 | |||
| TPGPGPRYPL | undefined | 30.3 | 3.77 | |||
| TPGPGTRFPL | undefined | 23.7 | 3.46 | |||
| TPGPGPRFPL | undefined | 35.9 | 3.66 | |||
| TPGPGIRYPM | undefined | 36.1 | 3.5 | |||
| TPGPGPRYPV | undefined | 117.7 | 3.82 | |||
| TPGPGPRYPM | undefined | 33.2 | 3.57 | |||
| TKGPGIRFPL | undefined | 12856.5 | 5.41 | |||
| 136–145 | PLTFGWCFKLg | undefined | 4173.8 | 6.48 | 0.00 | NS |
| PLTLGWCFKL | undefined | 5994.1 | 6.10 | |||
| PITFGWCFKL | undefined | 16105.3 | 6.35 | |||
| PVTFGWCFKL | undefined | 19876.4 | 6.48 | |||
| PLCFGWCFKL | undefined | 3451.6 | 4.86 | |||
| PVCFGWCFKL | undefined | 18878.7 | 5.4 | |||
| PMCFGWCFKL | undefined | 2898.1 | 5.3 | |||
| PLCFGWCFKP | undefined | 36444.9 | 5.34 | |||
| 180–189 | VLVWRFDSSL | undefined | 212.2 | 4.4 | 0.8315 | 0.0042 |
| VLVWKFDSSL | undefined | 179.6 | 4.37 | |||
| VLVWKSDSSL | undefined | 401.3 | 4.76 | |||
| VLVWKFDSNL | undefined | 197.4 | 4.31 | |||
| VLVWKFDSKL | undefined | 330.1 | 4.82 | |||
| VLVWKFDSRL | undefined | 290.0 | 4.44 | |||
| VLVWKFDSHL | undefined | 154.6 | 4.22 | |||
The following families and their variants were used in memory and priming T cell assays: SLFNTVATL, TLNAWVKVV, RMYSPTSIL, TLEQVVKKL, FPVRPQVPL, RPMTWKGAL, and TPGPGTRYPL. The following families and their variants were used in memory T cell assays only: SPRTLNAWV, IGPGRAFYAT, PLTFGWCFKL, and VLVWRFDSSL (except VLVWKFDSNL). All variants of the RPNNTRKSI Env family were used in memory T cell assays except the followings: RSNNNTRKSI, RPNNNTGKRI, RPSNNTRKRI, RPNNNTRKCI. All these variants in addition to RPNNNTRKSL were used in priming assays except RPNNNTRKCI.
These sequence variants of HIV-1 epitopes are listed in order of time of appearance during the course of infection.
These are known or potential MHC class I epitopes defined by the HIV sequence database as HLA A*0201 or B*0702. A potential epitope is a single peptide having Cterminal anchor residues and internal anchors matching one or more motifs associated with the submitted HLA, but are not found in the HIV sequence database.
This is the IC50 (nM) prediction binding score of peptides under investigation. Strong binders have an IC50 threshold of 50 and weak binders have a threshold of 500.
LogIC50 is used to refer to the experimental (in vitro) binding of peptides to HLA A*0201 or B*0702. The followings are the binding affinity based on the log IC50 high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0.
NS: Pearson correlation, between predicted and experimental MHC binding, is not significant, p value >0.05.
These autologous sequences are variants of the known, optimal HLA A*0201-restricted Env gp160311–320 epitope (RGPGRAFVTI), but are not defined as potential or known epitopes based on the Los Alamos HIV Database.
LTFGWCFKL can bind all five HLA-A2 supertypes alleles: A*0201, A*0202, A*0203, A*0206 and A*6802.
In vitro fluorescence polarization binding assay was performed for each variant (Buchli et al., 2005). We also evaluated the predicted MHC-restricted binding affinity of variant epitope sequences within Gag, Env, and Nef by netMHCpan (Hoof et al., 2009; Nielsen et al., 2007). Lower experimental logIC50 values and netMHCpan scores (IC50) correspond to actual and predicted higher peptide:MHC binding affinity, respectively. Significant positive correlations were found between observed and predicted MHC class I binding in the following families: SLFNTVATL (Gag p1777–85), TLNAWVKVV (Gag p2419–27), RMYSPTSIL (Gag p24143–151), TLEQVVKKL (Env gp160/120341–349), FPVRPQVPL (Nef68–76), TPGPGTRYPL (Nef128–137), and VLVWRFDSSL (Nef180–189) (Table 1).
To evaluate the relationship between MHC binding and T cell responses to autologous epitope variants, we first determined the frequency of epitope variants with different binding capacities during three distinct periods post-SC: early post-SC (0–2.8 yrs), late post-SC (3.25–7.8 yrs), and at early cART (8.3–10.1yrs) (Fig. S1). Three binding affinity categories of peptide variants to MHC class I molecules are referred to based on the log IC50 obtained by the FP assay: high affinity binders < 3.7, medium affinity binders < 4.7, low affinity binders < 5.5 and no affinity binders < 6.0. Results in Fig. 3 show : 1) medium binders constituted >97% of the Gag epitope pool at all three time points, 2) within Env contemporaneous epitopes, medium binders made up only 4% of the epitope pool early post-SC, but increased to 19.7% and 45.1% during late post-SC and early ART, respectively; frequency of high and low binders gradually decreased with this increase in medium binders, and 3) the frequency of Nef variants with medium binding affinity ranged from 50% to 70% through time with no trend in the fluctuation of low and high binder frequencies.
Figure 3. Proportion of HIV-1 epitope variants exhibiting differential MHC class I affinity.
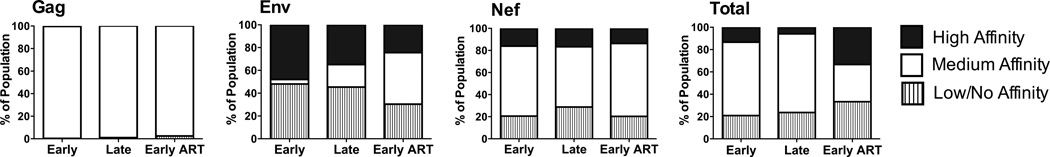
Synthetic 9 mer or 10 mer HIV-1 peptides representing autologous contemporaneous variants of the study participant were synthesized and evaluated for binding to soluble HLA A*0201 or B*0702 molecules by fluorescence polarization (FP)-based assay to determine the logIC50 for each variant. Variants were separated into three binding categories based on experimental logIC50 values: high affinity (< 3.7), medium affinity (3.7–4.7), and low affinity (>4.7). The longitudinal changes in the proportion of these variants within Gag, Env, and Nef variants, as well as in all epitopes evaluated, are shown for early post-SC, late-post-SC, and early ART time points. SC, seroconversion.
Taken together, peptide variants with medium binding affinity were more frequent than variants with low or high binding affinity during the course of the study. We observed 3 distinct patterns of evolutionary changes in epitope families (Fig. S1): 1) a complete switch in the dominant form of the epitope (e.g., RMYSPTSIL and RPMTWKGAL families), 2) no change in the dominant form of the epitope or a transient change in dominance that eventually resulted in the founder variant being more dominant (e.g., SLYNTVATL, TLNAWVKVV, SPRTLNAWV, and FPVRPQVPL families), or 3) multiple variants at each time point with no observable dominant variant (e.g., RPNNNTRKSI, TLEQVVKKL, TPGPTRYPL, PLTFGWCFKL, VLVWRFDSSL families).
Impact of peptide:MHC binding affinity on the breadth of memory T cell responses to autologous contemporaneous HIV-1 peptide sequences
To better understand the impact of CTL epitope variation and the frequency of different binders on corresponding T cell responses, we longitudinally evaluated HIV-1 specific T cell responses targeting autologous known and predicted CTL epitope variants. Autologous PBMC were used as responder cells in IFN-γ ELISPOT to measure memory (recall) T cell responses stimulated by DC-loaded with peptide sequences. We first calculated the percentage of naturally occurring Gag, Env, and Nef peptide variants yielding positive in vitro T cell responses at early post-SC, late post-SC, or at ART time points as a function of the total number of peptide sequences tested at that time point (Fig. 4) (Liu et al., 2011). Viewing the data in this fashion allowed us to evaluate the overall breadth and efficiency of memory responses during progressive HIV-1 infection. The total percent of epitope variants inducing HIV-1-specific memory T cell responses peaked early post-SC, then declined and remained minimal shortly after establishment of the viral set point. This early total T cell response was reflected in specific targeting of Gag, Env, and Nef peptide variant sequences (Fig. 4 A–C). Overall, we observed a trimodal pattern in the breadth of T cell memory (Fig. 4D). Following this early peak in T cell reactivity, there was a decline in CTL responses to all three HIV-1 proteins. This pattern of immune reactivity was associated with an early decrease in plasma viral load to 23,092 copies of HIV-1 RNA, followed by relatively steady levels of HIV-1 RNA for ~3.0 years. The greatest breadth, i.e. percent positive memory responses, to Gag, Env and Nef variants was observed at early ART (Fig. 4 A–C). This was associated with a decline in viral load to less than 200 copies/ml of plasma. During late cART, the total breadth of memory T cell responses declined for both Gag (Fig. 4A) and Nef (Fig. 4C) epitope variants. Memory responses to Env variants, however, remained relatively constant (Fig. 4B). Interestingly, there was no significant difference in the percent of positive responses induced by variants with high, medium or low affinity to MHC class I at any time point tested post-SC (Fig. 4E). Thus, our data suggest that HIV-1 epitope variants with different MHC class I binding capabilities can induce equally broad memory T cell responses. Overall, we observed significant correlations between the frequency of variants with high, medium, and low/no affinity and the percentage each of these contributed to the overall breadth of the autologous memory response early post-SC (p=0.003, r2=0.601), late post-SC (p<0.0001, r2=0.827), early ART (p<0.0001, r2=0.815), and at late ART (p<0.0001, r2=0.816).
Figure 4. Viral load and the proportion of positive of HIV-specific T cell memory responses pre- and post-therapy.

Synthetic peptide sequences representing autologous variants of subject 8 were loaded onto mature autologous DC and used to stimulate cryopreserved autologous PMBC. PBMC were added to wells at a responder-to-stimulator ratio of 10:1. Epitope-specific T cell responses were measured by IFN-γ ELISPOT assay. Results reported represent mean values of duplicates and expressed as spot forming cells (SFC)/106 PBMC. The proportion (percent) of positive memory-specific T cell responses is defined as the number of in vitro T cell responses directed against naturally occurring peptide sequences in subject 8 per total number of variant sequences tested during the corresponding time multiplied by 100. The proportion (percent) positive of memory T cell responses (y-axis) is plotted against time in years post-SC (x-axis). The viral load at respective time points during infection is also shown. Viral load (red line) and total proportion/or percent of positive T cell-specific memory responses (black line) to Gag (A), Env (B), Nef (C) and total number of peptides tested (D) per tested sequence are shown against time post-SC. (E) Gag, Env and Nef epitope variants classified by fluorescence polarization (FP) assay as high, medium or low binding affinity (including those classified as low and non-binders by the FP assay) to MHC class I are plotted (x-axis) against the proportion/percent positive memory responses they stimulate as measured by IFN-γ ELISPOT assay (y-axis). Affinity binding categories are based on the values of log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0. SC, seroconversion; SFC, spot-forming cells.
Our results indicate a typical course of progressive HIV-1 infection whereby an early increase in anti-HIV-1 CTL activity is associated with a decrease in HIV-1 viremia. As the untreated virus infection resulted in inexorable immune dysfunction, T cell reactivity no longer controlled viral replication. Initiation of cART was associated with control of viral replication and a temporal enhancement of anti-HIV-1 CTL responses that eventually declined to a low level, steady state (Casazza et al., 2005; Kalams et al., 1999; Rinaldo et al., 2000).
The changing magnitude of T cell memory responses targeting autologous HIV-1 variant sequences
We next assessed the longitudinal change in T cell responses to autologous Gag, Env, and Nef epitope variants (Table 1). Mature, autologous DC were loaded with synthetic peptides representing contemporaneous Gag, Env, and Nef CTL epitope variants and these peptides were used to stimulate PBMC in IFN-γ ELISPOT. Because sequencing data were not available for late cART time points, we used epitope variants reflecting circulating sequences during early cART in the late cART T cell memory assays. T cell memory responses to Gag families (SLFNTVATL, SPRTLNAW, TLNAWVKVV and RMYSPTSIL) ranged from 0 to 800 SFC/106 PBMC (Fig. 5). Early post-SC we observed modest IFN-γ recall responses to 3 out of the 4 Gag families (SLFNTVATL, SPRTLNAW and TLNAWVKVV) (Fig. 5A–C). These T cell responses decreased to low or undetectable levels late post-SC. More robust memory T cell responses were observed early following cART against the tested contemporaneous sequences of the same Gag families. In contrast, there were little or no memory responses to the RMYSPTSIL variants early post-SC, which moderately increased late post-SC (Fig. 5D). SLFNTVATL, TLNAWVKVV (known HLA A*0201 epitopes), and SPRTLNAWV (known HLA-B*0702 epitope) naturally circulated at all time points. The magnitude of T cell responses to these variants consistently increased early following cART. These responses were either reduced or completely lost during late therapy. T cell responses targeting Gag variants circulating at early cART were statistically significant and higher than responses targeting variants circulating at early and late post-SC, and during late cART (Fig. 5E).
Figure 5. Longitudinal development and changes in magnitude and breadth of Gag-specific T cell memory responses associated with the emergence of viral variants.

As previously described, synthetic 9 mer or 10 mer HIV-1 peptide sequences representing autologous contemporaneous variants of the study participant were sequenced, synthesized and loaded into mature autologous DC. DC are used to stimulate cryopreserved autologous PMBC at a responder-to-stimulator ratio of 10:1. Epitope-specific T cell responses are measured by IFN-γ ELISPOT assay. Gag-specific IFN-γ responses (y-axis) to contemporaneous variants sequences of (A) SLFNTVATL (p17 77–85), (B) SPRTLNAWV (p24 16–24), (C) TLNAVWVKVV (p24 19–27), and (D) RMYSPTSIL (p24 143–151) are shown across time. Blue histograms, early variants emerging at 0–2.8 years post-SC and tested with autologous PBMC; green histograms, late variants emerging at 3.3–7.8 years post-SC and tested with autologous PBMC; red histograms, early cART variants emerging at 8.3–10.1 years post-SC and tested with autologous PBMC. Epitope-specific T cell responses labeled as late cART represent sequences evolving at early cART and assumed to linger through (>10.1 years post-SC). (E) Total Gag-specific memory responses emerging at early (0–3 years post-SC), late (3.25–7.75 years post-SC), early cART (8–10.25 years post-SC) and late cART (>10.5 years post-SC). SC, seroconversion, SFC, spot-forming cells. Standard error bars are shown when applicable.
We observe strong T cell responses targeting the tested variants of the RPNNNTRKSI evolving late post-SC (Fig. 6A). RPNNNTRKSI, a known HLA-B*0702 lingering throughout the course of infection, did not induce memory responses early post-SC; however, increasing responses were detected late post-SC and following cART. Low to no memory responses were produced against the tested TLEQVVKKL sequences arising late post-SC (Fig. 6B). Variant sequences of this family of peptides are identified as potential epitopes by the Los Alamos database (Table 1). As observed with Gag variants, therapy maintained T cell responses to early and late variants with clear increase in T cell reactivity as tested by IFN-γ ELISPOT assay. A statistically significant mean difference was observed between T cell responses targeting early and late Env variants of RPNNNTRKSI and TLEQVVKKL families of peptides (Fig. 6C).
Figure 6. Longitudinal development and changes in magnitude and breadth of Env-specific T cell memory responses targeting naturally evolving viral variants.

As previously described for Gag, synthetic Env peptide sequences representing contemporaneous variants of the study participant were sequenced, synthesized and loaded into mature autologous DC. Similarly, DC are used to stimulate cryopreserved autologous PMBC at a responder-to-stimulator ratio of 10:1. Epitope-specific T cell responses are measured by IFN-γ ELISPOT assay. Env-specific IFN-γ responses (y-axis) to contemporaneous (A) RPNNNTRKSI (298–307) and (B) TLEQVVKKL (341–349) variants are shown across time. Blue histograms, early variants emerging at 0–2.8 years post-SC and tested with autologous PBMC; green histograms, late variants emerging at 3.3–7.8 years post-SC and tested with autologous PBMC; red histograms, early ART variants emerging at 8.3–10.1 years post-SC and tested with autologous PBMC. Epitope-specific T cell responses labeled as late ART represent sequences evolving at early ART and assumed to linger through (>10.1 years post-SC). (C) Pairwise-comparison between Env-specific T cell memory responses emerging at early (0–2.8 years post-SC), late (3.3–7.8 years post-SC), early cART (8.3–10.1 years post-SC) and late cART (>10.1 years post-SC). SC, seroconversion, SFC, spot-forming cells. Standard error bars are shown when applicable.
Variants of Nef families (FPVRPQVPL, RPMTWKGAL, TPGPGTRYPL, PLTFGWCFKL and VLVWRFDSSL) were next tested for their ability to induce T cell memory responses (Fig. 7). FPVRPQVPL, a founder sequence and a known B*0702 epitope (Table 1), was frequently present during the course of infection and stimulated low memory responses at all studied time points with a moderate increase following the administration of cART (Fig. 7A). The founder sequences of the RPMTWKGAL family induced specific-memory responses in vitro. The switch observed in the dominant form of the estimated founder sequence RPMTWKGAL to become RPMTWKAAL was associated with maintenance of the ability of the latter to induce memory responses until early cART (Fig. 7B). These responses were lost during late therapy. RPMTWKAAL is also identified as a known B*0702 epitope whereas the remaining variants are identified as potential epitopes (Table 1).
Figure 7. Longitudinal development and changes in magnitude and breadth of Nef-specific T cell memory responses associated with the emergence of viral variants.

As previously described for Gag and Env, synthetic contemporaneous variants of Nef peptide sequences were loaded into mature autologous DC isolated from our study participant and epitope-specific T cell responses are measured by IFN-γ ELISPOT assay. Nef-specific IFN-γ responses (y-axis) to contemporaneous variants sequences of (A) FPVRPQVPL (68–76), (B) RPMTWKGAL (77–85), (C) TPGPGTRYPL (128–137), and (D) PLTFGWCFKL (136–145), and (E) VLVWRFDSSL (180–189) are shown across time. Blue histograms, early variants emerging at 0–2.8 years post-SC and tested with autologous PBMC; green histograms, late variants emerging at 3.3–7.8 years post-SC and tested with autologous PBMC; red histograms, early cART variants emerging at 8.3–10.1 years post-SC and tested with autologous PBMC. Epitope-specific T cell responses labeled as late cART represent sequences evolving at early cART and assumed to linger through (>10.1 years post-SC). (F) Pairwise-comparison between Nef-specific memory responses emerging at early (0–3 years post-SC), late (3.25–7.75 years post-SC), early cART (8–10.25 years post-SC) and late cART (>10.5 years post-SC) time points. SC, seroconversion, SFC, spot-forming cells. Standard error bars are shown when applicable.
Peptide variants of the TPGPTRYPL, PLTFGWCFKL and VLVWRFDSSL families were phylogenetically similar in that multiple variants existed concurrently (Fig S1). The majority of TPGPGTRYPL variants arising early post-SC induced low to moderate memory responses (Fig. 7C); responses to the variants lingering to late time points were reduced. Early and late cART time points were associated with maintenance of moderate responses to contemporaneous sequences and loss of response to one of the studied founder sequences (TPGPGTRYPL). Responses to TPGPGIRYPL, a HLA B*07 epitope (Table 1), were reduced with time whereas the TPGPGVRYPL variant, a B*07 supertype, was not able to stimulate memory responses early post-SC. LTFGWCFKL (Table 1) is reported by the Los Alamos database to bind all five HLA-A2 supertype alleles: A*0201, A*0202, A*0203, A*0206 and A*6802. Memory responses to two 10mers containing variants of LTFGWCFKL (PLCFGWCFKL and PMCFGWCFKL) induced no-to-low memory responses; the latter recovered during therapy with eventual loss at late cART time points (Fig. 7D). VLVWKFDSKL and VLVWKFDSRL emerged at late and early cART time points respectively and were the only variants in this family to induce moderate to high memory responses (Fig. 7E). Pairwise comparisons showed statistical differences in mean memory responses between early and early cART time points as well as between late and early ART time points (Fig. 7F).
We evaluated the impact of MHC class I binding affinity on the magnitude of memory T cell responses during the course of infection (Fig. S2). Our data show that Gag, Env and Nef peptide variants with high or medium affinity induce similar responses early post-SC while low binders as classified by the FP-assay induce statistically significant higher responses as compared to high binders (Fig. S2A). Later during infection (i.e. late, early cART, and late cART), variants with high, medium and low binding affinity stimulated similar IFN-γ responses (Fig. S2 B–D).
Being a known or potential HLA-A*0201 or HLA-B80702, the magnitude of T cell responses elicited by the tested peptide variants with high and medium binding affinity was generally similar at different time point post-SC. Low affinity binders of Gag, Env and Nef variants tested in vitro variably induced T cell responses with a clear increasing magnitudes following cART.
Broad and high magnitude primary T cell responses to naturally evolving variants in HIV-1-positive and healthy donors
We have previously demonstrated the ability of mature monocyte-derived DC from HIV-1-naïve donors to induce broadly reactive primary CD8+ T cell responses (Colleton et al., 2009). Little is known about the ability to prime anti-HIV T cell immunity specific for variants that evolved during natural infection. Thus, we next determined the capacity of DC isolated from subject 8 during cART to prime autologous pre-SC T cells. Autologous DC matured with CD40-L and IFN-γ (Fan et al., 2007; Huang et al., 2008) were loaded with Gag (Fig. 8A), Env (Fig. 8B) and Nef (Fig. 8C) peptide sequences that emerged at early, late, or cART time points post-SC. These DC were then used in a 21-day in vitro priming assay. The magnitude of primary T cell responses was tested in an IFN-γ ELISPOT assay.
Figure 8. In vitro primary T cell responses induced in HIV-positive subject to naturally evolving viral variant sequences in an entirely autologous system.

Mature DC isolated from subject 8 and HLA-matched HIV-1 naïve donors were loaded with 10 µg/ml of subject 8 variant sequences detected during the course of infection. DC were used to prime autologous PBMC isolated from healthy donors and from the study participant prior to seroconversion. The magnitude of T cell primary responses is tested by IFN-γ ELISPOT assay. In vitro primary T cell responses (y-axis) of PBMC isolated prior to seroconversion to (A) Gag (SLFNTVATL, TLNAWVKVV, RMYSPTSIL), (B) Env (TLEQVVKKL, RPNNNTRRSI) and (C) Nef (FPVRPQVPL, RPMTWKGAL, TPGPGTRYPL) viral sequences emerging during early (blue histograms), late (green histograms) and/or ART (red histograms) time points are shown. The mean values ±SD of SFC/106 PBMC to BMLF1 control peptides are 2217±660 (data not shown). (D) Proportion of positive primary T cell responses (y-axis) to contemporaneous viral sequences of Gag, Env and Nef (x-axis) is defined as the number of peptide variants tested within each protein and inducing a positive IFN-γ response per total number of variants tested. SC, seroconversion; SFC, spot-forming cells.
Of the 59 peptides tested, HIV-naïve, pre-SC T cells were primed to 9/15 (60%) Gag, 10/17 (58.8%) Env, and 13/27 (48.4%) Nef variants. Among these 32 sequences eliciting primary responses, moderate (>=100 SFC) to strong (>=500SFC) primary IFN-γ responses were noted to known HLA-A*0201 or HLA-B*0702 epitopes SLYNTVATL (p17), naturally evolving at late time points; TLNAWVKVV (p24) identified as a founder sequence; FPVRPQVPL (Nef), a founder sequence; TPGPGVRYPL emerging early post-SC and TPGPGIRYPL (Env), a founder sequence (Table 1).
The peptide families containing SLYNTVATL, TLNAWVKVV and FPVRPQVPL, harbor variant sequences with potential CTL epitopes (Table 1). Variants of three out of the five remaining families of peptides tested in our priming model: i.e. RMYSPTSIL (Gag), TLEQVVKKL (Env), and RPMTWKGAL (Nef) are also defined as potential MHC class I -restricted epitopes (Table 1). In summary, a total of 16 out of 32 variants induced strong primary responses (>500 SFC) (Fig.8).
To determine the breadth of primary T cell responses, the proportion of variants inducing positive primary responses was defined as the number of variant sequences inducing a positive primary T cell response /total number of variants tested within a respective family. The proportion of variants that stimulated primary responses was not dependent on the HIV-1 protein from which these variants were derived, as we observed no significant differences between primary responses targeting Gag, Env, and Nef peptide variants (Fig. 8D). When combined, variants of Gag, Env and Nef with low, binding affinity induced primary T cell responses with comparable breadth and magnitude as variants with high or medium affinity (Fig. S3).
Taken together, our results indicate that substantial primary T cell responses are induced by our model to a diverse number of HIV-1 peptide variants of the virus proteome. Being a known, potential, or undefined HLA-matched epitope, with high, medium, or low binding affinity did not influence the ability of our priming model to stimulate robust T cell responses in an entirely autologous system.
To evaluate the person-to-person variation in T cell priming capacity of the HIV-1 variant peptides from subject 8, we used PBMC from 2 healthy, HIV-1 naïve, HLA matched (HLA-A2/B7) donors. SLYNTVATL (Gag p17), TLNAWVKVV (Gag p24), FPVRPQVPL (Nef) and their naturally evolving variants were tested. T cells from donor 0038 responded to all SLYNTVATL (6/6) variants, 4 out of 5 TLNAWVKVV variants and all (5/5) FPVRPQVPL variants. Similarly, donor 2256 responded to all variants except one within the FL9 family (Fig. 9). We found no significant difference between the magnitude of the IFN-γ primary responses stimulated by peptide variants of our HIV-positive study participant and those of HIV-naïve volunteers.
Figure 9. In vitro primary T cell responses induced in healthy HIV-1 negative donors to naturally evolving HIV-1 viral variants.

Mature DC isolated from HIV-1 naïve donors HLA A*0201 matched with subject 8 were loaded with naturally evolving variants detected during his course of infection, and used to prime autologous PBMC. The magnitude of T cell primary responses was tested by IFN-γ ELISPOT. SLFNTVATL, TLNAWVKVV and FPVRPQVPL variants (x-axis) of subject 8 were used in our priming model to stimulate primary T cell responses (y-axis) of PBMC derived from donor 0038 and donor 2256. SFC, spot-forming cells.
Discussion
We report the evolution of HIV-1 in an infected subject across time and the impact of HIV-1 variants and respective MHC class I binding affinities on the breadth and the magnitude of T cell memory responses. Our data show that with higher sequence diversity, the proportion of T cell memory responses targeting contemporaneous variant peptide sequences of the proteome increases. It has been previously reported that the continuous presence of antigen is important for the maintenance of CTL responses (Shin et al., 2007; Wherry et al., 2004). Moreover, Liu et al. (Liu et al., 2011) recently reported in cART-naïve setting that a balance is likely needed between the presence of antigen and prolonged antigen stimulation. Along similar lines, our results suggest the dynamic ability of evolving viral sequences to continuously exist and stimulate specific T cell responses.
T cell responses directed against individual known or optimal peptide sequences (Gag: SLFNTVATL, SLYNTVATL, TLNAWVKVV; Env: RPNNTRKSI; Nef: FPVRPQVPL, RPMTYKAAL, TPGPGVRYPL) evolving early during infection were temporally enhanced during early cART, followed by a decline or complete loss in late cART. We have previously observed such a temporal effect on CTL activity to consensus HIV-1 epitopes (Rinaldo et al., 2000). This observation was not limited to optimal epitopes as similar trends were observed against potential epitopes and previously undefined epitopes (Table 1). Moreover, our results show a clear correlation between the frequency of autologous peptide variants during infection and the breadth but not the magnitude of these responses. These results are in agreement with the lack of correlation between the magnitude of CTL responses and HLA binding affinity (Bihl et al., 2006; Tenzer et al., 2009). Importantly, our data indicate that the breadth of T cell responses targeting contemporaneous epitopes representing different parts of the HIV-1 proteome is comparable early post-SC, late post-SC, or during cART regardless of the experimental binding capacity to the cognate MHC class I molecules.
Even though a small repertoire of Gag, Env, and Nef peptide families were used in this study, all of the tested autologous HIV-1 epitope variants maintained a functionally detectable or predicted level of binding to MHC class I. Further assessment of peptide binding to MHC and its relation to T cell memory responses will need to categorize percent recognized (T cell reactive) and percent non-recognized (T cell non-reactive) variants in relation to viral peptides exhibiting a broader range of MHC binding. This is especially important since a number of mechanisms such as T cell escape, change in the antigen load and T cell exhaustions (Turnbull et al., 2009) could lead to decline of T cell responses during HIV-1 infection.
The minimum impact of MHC class I affinity observed in this study suggests that the breadth of recognition of peptide variants could be partially due to the promiscuity of TCR (Buseyne and Riviere, 2001). Limited TCR diversity within cognate clonotypes has been suggested to facilitate immune escape through loss of CD8+ T cell recognition (Davenport, Price, and McMichael, 2007; Price et al., 2004). Recently, dominant HIV-specific CD8+ T cell clonotypes were found to persist in vivo for long periods of time while being able to cross-recognize naturally occurring epitope variants (van Bockel et al., 2011). A high clonotypic turnover of HIV-specific CD8+ T cells was described following intiation of HAART or upon appearance of viral mutations; moreover, authors showed new cognate clonotypes during decreased or limited antigen load (Janbazian et al., 2012). These clonotypes became dominant and were characterized by high functional capabilities. While we don’t have data on the evolution of HIV-specific CD8+ T cell repertoire clonal composition in our participant or the functional profiles of the detected T cell responses, we believe that we might be dealing with a similar reconstitution post cART.
In our study, T cell responses were detected to 81% (63/78) of the total tested variants in the study participant, 8 of which are classified as optimal epitopes and 29 as potential epitopes as described by the Los Alamos HIV database. Post-STEP trial emphasis has been on the benefit of maximizing the T cell coverage, i.e. breadth, of the contemporaneous forms of the virus (McElrath et al., 2008). Even though correlation between the breadth of the CTL responses and the containment of virus in vivo has been controversial (Addo et al., 2003; Frahm et al., 2004; Liu et al., 2007), we and others (Rolland et al., 2008) believe that the probability of inducing protective anti-HIV CTL responses is likely to be enhanced by broader range of immunogenic epitopes presented professionally by APC. We acknowledge the limitation of monitoring viral sequences longitudinally from one subject in addition to the limited repertoire of peptide epitopes tested in this study; hence, we might have missed important motifs implicated in better MHC binding and consequently effective T cell responses.
The genetic diversity of HIV-1 and the propensity of the virus to undergo escape mutations are major hurdles for long-term immune control of HIV-1. With the current lack of a preventive HIV-1 vaccine, alternatives are needed to improve the quality of the immune responses targeting the virus and thus reducing its transmission. It has been proposed that the magnitude and the quality of the memory T cell pool is affected by the activation of naïve CD8+ T cells by licensed DC and that excessive antigenic stimulation leads to terminal differentiation and impairment of these memory CD8+ T cells (Zehn, Lee, and Bevan, 2009). We have previously demonstrated the potent capability of mature monocyte-derived DC from uninfected donors to induce a broad spectrum of primary CD8+ T cell responses targeting epitopes in Gag, Env, and Nef (Colleton et al., 2009). Our in vitro model was successful at priming HIV-naïve CD8+ T cells to consensus HIV-1 epitopes as well as other viral peptides in vitro. A series of events have been described to be involved in triggering the stimulation of the TCR on naïve CD8+ T cells. The TCR and signaling by co-stimulatory and cytokine receptors drive the magnitude and the quality of the response (Prlic, Williams, and Bevan, 2007).
Consequently, we have proposed the enhancement of the primary responses of naïve CD8+ T cells to a broad array of HIV-1 epitopes while patients are on ART for better control of viral replication and disease (Rinaldo, 2009). In this study, we confirm the ability of our in vitro model to stimulate autologous pre-SC naïve T cells from our study participant against 54% (32/59) of tested autologous peptide variants that emerged at different stages pre- and post-therapy. Moderate to strong primary responses were induced to variants from a variety of known and potential Gag, Env, and Nef epitopes in an entirely autologous system. Moreover, the time post-SC at which the epitope variant evolved did not affect the magnitude of the primary T cell responses. The elicited primary responses were not exclusive to already defined known and potential epitopes. A number of variant sequences of Env and Nef were not defined by the Los Alamos HIV database, yet had predicted and experimental HLA-binding affinity. Taken together, this in vitro priming model stimulates primary T cell responses regardless of the nature (i.e. known or undefined), origin (HIV-1 protein), and MHC binding affinity (high, medium or low binders) of the tested HIV-1 epitope variants. Even though cART has been successful at controlling HIV-1 viral load, immune recovery is still a challenge (Corbeau and Reynes, 2011). We propose by-passing this obstacle by using a DC immunotherapy aimed at inducing a primary T cell response from naïve precursors in subjects on cART. This is especially important due to de novo priming of new clonotypes occurring during persistent viral infections (Vezys et al., 2006) and vaccination (Rosario et al., 2010).
As a proof-of-concept, we demonstrated the ability of our priming model to robustly prime autologous T cells derived from 2 HIV-1 naïve volunteers to a broad range of Gag and Nef peptide variants derived from the study subject. 25% and 75 % of these peptides were known and potential HLA A*0201 and HLA B*0702 epitopes, respectively, as defined by the Los Alamos HIV Immunology Database. Our model was able to generate primary responses targeting immunodominant (SL9, FL9) and subdominant (TV9) epitopes and their variants. It is well established that immunodominant HIV-specific CTL responses can exert selection pressure on the virus during primary infection (Allen et al., 2000; Barouch et al., 2003). Even though subdominant epitopes stimulate T cells less frequently, the elicited responses are protective (Bihl et al., 2006; Borrow et al., 1997; Draenert et al., 2004; Jones et al., 2004). Our model does not differentiate between viral sequences based on immunodominance. A significant difference was observed between the magnitude of these in vitro primary responses generated by cells of our normal donors. The reason for failure to detect similar in vitro primary responses between the 2 normal donors could be due to differences in T cell precursor frequencies. Importantly, we found no difference between the magnitudes of primary responses of our 2 healthy donors targeting HIV-1 evolving variant sequences derived from our study participant when compared to those targeting the same variants using pre-SC or HIV-naïve cells from our study participant. These findings show that, prior to HIV infection and immune dysfunction, the naïve T cell repertoire found within subject 8 was capable of recognizing and responding to primary stimulation against the variants that evolved after seroconversion. The mutations that evolved after infection, particularly those that appeared late post-SC, may have evolved to specifically evade T cell recognition. This would have implications in immunotherapy approaches, which aim to induce primary responses against these “late evolving” viral variants. Indeed, our induction of primary responses against these variants prior to SC supports that immunotherapies implemented during cART could be successful at inducing CTL specific for autologous viral variants. While we acknowledge the need for further exploration of the specific TCR clonotypic repertoire associated with emerging viral variants and its ability to control HIV-1 replication, we believe that the present work highlight the importance of primary responses against HIV-1 variants in a continuous environment of competition between HIV-1 and virus-specific T cell responses. A recent study on mutations within the KK10 epitope at different positions suggest the capacity of KK10-specific CD8+ T cell response to attract an array of crossreactive clonotypes from existing repertoire to control HIV-1 infection (Ladell et al., 2013). We believe that the described primary responses are similarly capable of assembling T cell clonotypes to control the ability of emerging viral variants to affect TCR recognition.
With the clonal exhaustion of CD8+ T cells as a result of chronic HIV-1 infection and the pressure exerted on memory T cells to recognize wild-type and mutant variants, we and others (Appay, Douek, and Price, 2008) believe that an effective T cell response would have to generate a long lasting protection environment. Engineering DC to prime naïve T cells to immune escape variants (Rinaldo, 2009) could be a potential approach to overcome the challenges faced by circulating memory T cells.
Materials and Methods
Study participants
Human subject approval was obtained for this study from the institutional review board of the University of Pittsburgh. The participant in this study (subject 8) is enrolled in the Multicenter AIDS Cohort Study (MACS), a natural history study of HIV-1 infection in men who have sex with men (Detels et al., 2012; Kaslow et al., 1987). The subject’s MHC class I alleles as determined by high resolution PCR genotyping (Tissue Typing Laboratory, University of Pittsburgh Medical Center) are HLA-A*0201 A*2402 B*0702 B*4001 C*0304 C*00702. Subject 8 was HIV-1 seronegative upon enrollment in the MACS in November, 1984, and seroconverted to HIV-1 between October 1987 and May 1988. HIV-1 seropositivity was confirmed by a positive enzyme-linked immunosorbent assay (ELISA) for the presence of HIV-1 p24 and a Western blot with bands corresponding to at least 2 of either Gag, Pol, and Env (Kaslow et al., 1987). Blood specimens, and epidemiologic and clinical data were obtained at semiannual visits (Shankarappa et al., 1999). Participant 8 progressed to AIDS as defined by the CDC (<200 CD4+ T cells/mm3) 6.2 years post-SC. cART was initiated July, 1996 at 8.4 years post-SC. This treatment regimen, consisting of Retrovir (zidovidine azidothymidine, GlaxoSmithKline), Epivir (lamiduvidine 3TC, GlaxoSmithKline) and Invirase (saquinovir mesylate, Roche), was administered for 1 year. At 9.4 years post-SC, Viracept (nelfinavir mesylate, Agouron), Sustiva (efavirenz, Bristol-Meyers Squibb) and Ziagen (abacavir sulfate, GlaxoSmithKline) replaced the previous regimen. This treatment was finally replaced with Atrilpa (efavirenz/emtricitabine/tenofovir, Bristol-Meyers Squibb) at 20.5 years post SC and was maintained for the period of study. Blood was also obtained from 2 anonymous, healthy HIV-1 negative donors (Central Blood Bank, Pittsburgh, PA) who were typed as HLA-A2/B7 by flow cytometry for use in the T cell priming study.
Clinical and virologic characteristics
At each clinic visit pre- or post-SC and during cART, plasma samples and peripheral blood mononuclear cells (PBMC) were collected and stored at −80°C and −140 °C, respectively. Plasma samples and PBMC were used to determine HIV-1 load and CD4+ and CD8+ T cell counts, respectively. T cell phenotypes were determined by flow cytometry as previously described (Giorgi et al., 1990; Schenker et al., 1993). For HIV-1 plasma viremia, RNA was extracted from plasma using a COBAS© Ampliprep Instrument (Roche Diagnostics, Indianapolis, IN) and amplified by RT-PCR on a COBAS© Taqman© 48 Analyzer (Roche Diagnostics) using the COBAS© Ampliprep/COBAS© Taqman HIV-1 Test. This assay is capable of detecting from 20 to 106 HIV-1 RNA copies/ml of plasma. Negative, low positive and high positive controls were used in each RNA extraction and RT-PCR assay as per manufacturer’s instructions.
Sequencing and phylogenetic analysis
env C2-V5 sequences were reported previously (Shankarappa et al., 1999). gag and nef sequences were obtained using the procedures and primers as previously described (Liu et al., 2006; Malhotra et al., 2007; Shriner et al., 2004). Briefly, viral sequences were derived from 12, 16, and 9 time points for gag-p17 and -p24, env-gp120, and nef, respectively. Sequences spanned 8 years of untreated HIV-1 infection and included at least one post-ART time point. Viral sequencing was not performed during late ART due to inability to detect viral RNA in plasma.
Sequences bearing open reading frames (~90% of all sequences determined) were first aligned with the Pileup program in the GCG suite (Genetics Computer Group, Madison, WI) and then manually edited (Liu et al., 2006; Liu et al., 2007). Both viral divergence from the founder strain and viral diversity were estimated at each time point. To estimate viral diversity, we determined the mean and standard deviation for pairwise nucleic acid distances between all sequences obtained at each time point. To estimate viral divergence, we compared sequences from each visit to a founder sequence that was approximated as the consensus sequence found at the initial virus-positive time point. The mean and standard deviation for all pairwise comparisons were then calculated.
Synthetic peptides
Peptide sequences representing autologous Gag, Env, and Nef MHC class I -associated variants evolving in our study participant during infection were synthesized (SynBioSci, Livermore, CA) and used in T cell priming and memory T cell functional assays. Epstein-Barr virus (EBV) BMLF1280–288 (GL9, GLCTLVAML) (provided by the NIH AIDS Research Program) was used as a control peptide sequence in the functional assays. The Los Alamos HIV Molecular Immunology Database was referred to for defining MHC Class I epitopes based on being known as optimal epitopes or based on affinity, T cell receptor usage, computational epitope prediction or functional studies. For ease of interpretation, the naturally evolving peptide sequences of subject 8 are referred to in order of their detection post-SC. Consequently, we defined early post-SC (0–2.8 yr post-SC), late post-SC (3.25–7.8 yr), early cART (8.3–10.1yr) and late cART (12.4–13.1 or >12.4yr) time points.
In silico and in vitro analysis of peptide binding affinity
HLA peptide binding predictions of the sequences under investigation were scored using the netMHCpan 2.4 server (www.cbs.dtu.dk/services/NetMHCpan) (Hoof et al., 2009; Nielsen et al., 2007). In silico (predicted) IC50 values were computed with binding affinity of peptides for MHC class I. Strong binders were assigned as having an IC50 threshold of 50 nM and weak binders as having a threshold of 500nM. A fluorescence polarization (FP)–based assay was used to experimentally screen and identify high affinity binding peptides to HLA molecules (Pure Protein LLC, Oklahoma City, OK). Briefly, the assay utilizes fluorescently labeled control peptides and recombinant soluble HLA-A*0201 and B*0702 molecules to test for the competitive binding between a labeled reference peptide and the peptide or the peptide mix being investigated. The binding affinity of the competitor peptide is expressed as the concentration inhibiting 50% (IC50) of the binding of the labeled peptide (Buchli et al., 2005). The followings are the binding affinity categories based on the log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0 (Buchli et al., 2005).
Generation of dendritic cells (DC)
Monocyte-derived DC were generated from subject 8 and two healthy HIV-1 negative adults as previously described (Colleton et al., 2009) with minor modifications. Briefly, CD14+ monocytes were positively selected using anti-CD14 monoclonal antibody (mAb) coated magnetic beads (StemCell Technologies, Vancouver, Canada) or Percoll density separation (Sigma-Aldrich, Saint Louis, MO). DC were generated by culture of the purified monocytes with 1,000U/ml recombinant GM-CSF (Bayer Healthcare, Montville, NJ) and 1,000 U/ml recombinant IL-4 (R & D Systems, Minneapolis, MN). On day 5, immature DC were treated for 48 h with recombinant CD40L (0.5 µg/ml; Enzo, Farmingdale, New York) for use in memory T cell assays or CD40L (0.5 µg/ml) and interferon γ (IFN-γ; 1000 U/ml; R&D) for use in priming assays.
In vitro priming
In vitro priming of autologous PBMC was performed as previously described with minor modifications (Colleton et al., 2009). Briefly, DC were derived from PBMC obtained from subject 8 during virus suppressive cART and the HIV-1 negative, adult volunteers. Autologous DC matured with CD40L and IFN-γ were incubated with 10 µg/ml of HIV-1 peptides in IMDM medium (Gemini Bio-Products, West Sacramento, CA) containing 10% heat-inactivated fetal calf serum (FCS) (Gemini Bio-Products) for 2 hr at 37°C in a 5% CO2 atmosphere. The DC were harvested and re-suspended with autologous PBMC at a responder-to-stimulator (T:DC) ratio of 10:1. Autologous PBMC from subject 8 were obtained from cryopreserved cells prior to SC. After 3 days, the co-cultures were fed with fresh IMDM/10% FCS supplemented with recombinant IL-15 (2.5ng/ml; PeproTech, Rocky Hill, NJ), IL-2 (50 U/ml; Chiron, Emeryville, CA) and IL-7 (10 ng/ml; Miltenyi, Auburn, CA). This was repeated at 2–3 day intervals thereafter for 2 weeks. PBMC were then re-stimulated with autologous DC loaded with the same set of peptides as in the primary stimulation at a responder-to-stimulator (T:DC) ratio of 10:1. These cells were harvested after a total of 21 days of culture and used in IFN-γ ELISPOT assays.
IFN-γ ELISPOT assay
ELISPOT assays were performed as previously described (Colleton et al., 2009; Huang et al., 2008). Briefly, 96 well plates were coated overnight with 1 µg/ml antihuman IFN-γ mAb 1-D1K (Mabtech, Stockholm, Sweden) at 4°C. DC matured with CD40L were loaded with 10 µg/ml HIV-1 peptides or control EBV peptide in IMDM/10% FCS for 2 hr at 37°C. Control wells contained T cells in the presence of mature DC. Autologous PBMC in IMDM/10% FCS were added to wells at a responder-to-stimulator ratio of 10:1 and were incubated for 18 hr at 37 °C in a 5% CO2 atmosphere. Following incubation, wells were washed with PBS/0.05% Tween-20 (Fisher Scientific, Pittsburgh, PA) and were treated with biotinylated anti-IFN-γ mAb (1 µg/ml; Mabtech, Stockholm, Sweden). Plates were washed with PBS/0.05% Tween 20 and incubated with an avidin-peroxidase complex (Vectastain ABC Kit, Vector Laboratories, Burlingame, CA) for 45 min at room temperature. Plates were washed with 0.05% Tween 20/PBS and PBS alone to remove unbound complexes followed by peroxidase staining with diaminobenzidine solution (Sigma, St Louis, MO) for 5 min at RT. IFN-γ spot-forming cells (SFC) were enumerated using an AID ELISPOT reader (Cell Technology, Columbia, MD). Results reported represent mean values of duplicates and are expressed as spot forming cells/106 PBMC. T cell responses were considered positive after subtraction of the mean number of spots stimulated by DC alone from the mean number of spots induced by peptide-loaded DC. Mature DC loaded with the control EBV peptide stimulated the following SFC/106 PBMC: 494±122 (early post-SC), 927±66 (late post-SC), 1100±264 (early cART) and 997±62 (late cART).
Statistical analysis
The following statistical analyses were carried out: Pearson correlation between 1) predicted (IC50) and experimental (logIC50) MHC binding, 2) viral load and divergence and diversity, 3) MHC binding and magnitude of T cell responses, 4) MHC binding and percent positive T cell responses, 5) the frequency of occurrence of variants and the percent positive T cell responses. In addition, a two-tailed paired-T test was used to compare the proportion of positive primary responses in our study participant as well as to compare subject 8 primary responses targeting naturally evolving sequences to those observed against the same sequences when testing our priming model with cells from two healthy donors. Bonferroni posthoc comparisons were applied to compare memory responses targeting contemporaneous Gag, Env and Nef variant sequences at early, late, early ART or late ART time intervals. Statistical analyses were conducted using GraphPad Prism software (version 4) and SPSS software (version 17.0).
Supplementary Material
The frequency (y-axis) of epitope variants of Gag (A), Env (B) and Nef (C) naturally occurring during the course of infection is plotted against the corresponding time of emergence (x-axis) post-SC. The frequency of a specific epitope variant is calculated as the number of contemporaneous variant over a specific time point divided by the total number of variants circulating at the same time post-SC. Years post-SC (x-axis) include early, late, early cART time points defined as follow: 0–2.8 years post-SC, 3.3–7.8 years post-SC, 8.3–10.1 years post-SC, and >12.4 years post-SC, respectively. High, medium and low (including those classified as low and non-binders by the FP assay) affinities are defined based on the log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0. SC, seroconversion.
Synthetic 9 mer or 10 mer HIV-1 peptides representing autologous contemporaneous variants within the study participant were loaded into mature autologous DC. These DC were used to stimulate cryopreserved autologous PMBC at a responder-to-stimulator ratio of 10:1. Gag, Env and Nef-specific T cell responses were measured by IFN-γ ELISPOT. Results represent mean values of duplicates and are expressed as spot forming cells (SFC) /106 PBMC (y-axis) targeting evolving Gag, Env or Nef variants (x-axis) at early (A), late (B), early cART (C) and late cART (D). The right panels of A, B, C and D represent the magnitude of memory T cell responses targeting all variants (Gag, Env and Nef) with high, medium or low binding affinity. Black fill: high binding affinity variant, white fill: medium binding affinity variant, shaded fill: low binding affinity variant (including those classified as low and non-binders by the FP assay). Affinity binding categories were based on the values of log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0.Early: 0–2.8 years post-SC, late: 3.3–7.8 years post-SC, early ART: 8.3–10.1 years post-SC post-SC. SC, seroconversion.
Mature DC isolated from subject 8 were loaded with variant HIV-1 peptides and used to prime autologous PBMC isolated from the study participant prior to SC. The magnitude of primary T cell responses was determined by IFN-γ ELISPOT. In vitro primary T cell responses (y-axis) of PBMC isolated prior to seroconversion were plotted against variants (x-axis) of (A) Gag (SLFNTVATL, TLNAWVKVV, RMYSPTSIL), (B) Env (RPNNNTRRSI, TLEQVVKKL) and (C) Nef (FPVRPQVPL, RPMTWKGAL, TPGPGTRYPL) emerging during early, late and/or ART time points are shown. (D) The magnitude of IFN-γ responses (y-axis) to contemporaneous viral peptides of combined Gag, Env and Nef with high, medium and low binding affinity (x-axis). Black histograms: high binding affinity variant, empty histograms: medium binding affinity variant, dashed histograms: low (including those classified as low and non-binders by the FP assay) binding affinity variant. Affinity binding categories are based on the values of log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0. SC, seroconversion; SFC, spot-forming cells.
RESEARCH HIGHLIGHTS.
Loss of anti-HIV T cell responses doesn’t relate to MHC class I binding of HIV-1 epitope variants.
CD8 T cells from before HIV-1 infection can be primed in vitro to autologous HIV epitope variants.
Priming of CD8 T cells to autologous HIV-1 during ART could be an effective therapeutic approach.
ACKNOWLEDGMENTS
We thank Raj Shankarappa for HIV sequencing and analysis, John Mellors for access to Taqman® assay and Kelly Gordon for technical assistance. This work was supported in part by NIH grants R01-AI-40388, R37-AI-41870, U01-AI-35041 and T32-AI065380.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, Feeney ME, Rodriguez WR, Basgoz N, Draenert R, Stone DR, Brander C, Goulder PJ, Rosenberg ES, Altfeld M, Walker BD. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol. 2003;77(3):2081–2092. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TM, Altfeld M, Yu XG, O'Sullivan KM, Lichterfeld M, Le Gall S, John M, Mothe BR, Lee PK, Kalife ET, Cohen DE, Freedberg KA, Strick DA, Johnston MN, Sette A, Rosenberg ES, Mallal SA, Goulder PJ, Brander C, Walker BD. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol. 2004;78(13):7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TM, Jing P, Calore B, Horton H, O'Connor DH, Hanke T, Piekarczyk M, Ruddersdorf R, Mothe BR, Emerson C, Wilson N, Lifson JD, Belyakov IM, Berzofsky JA, Wang C, Allison DB, Montefiori DC, Desrosiers RC, Wolinsky S, Kunstman KJ, Altman JD, Sette A, McMichael AJ, Watkins DI. Effects of cytotoxic T lymphocytes (CTL) directed against a single simian immunodeficiency virus (SIV) Gag CTL epitope on the course of SIVmac239 infection. J Virol. 2002;76(20):10507–10511. doi: 10.1128/JVI.76.20.10507-10511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TM, O'Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, Dunphy E, Liebl ME, Emerson C, Wilson N, Kunstman KJ, Wang X, Allison DB, Hughes AL, Desrosiers RC, Altman JD, Wolinsky SM, Sette A, Watkins DI. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407(6802):386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- Appay V, Douek DC, Price DA. CD8+ T cell efficacy in vaccination and disease. Nat Med. 2008;14(6):623–628. doi: 10.1038/nm.f.1774. [DOI] [PubMed] [Google Scholar]
- Baker BM, Block BL, Rothchild AC, Walker BD. Elite control of HIV infection: implications for vaccine design. Expert Opin Biol Ther. 2009;9(1):55–69. doi: 10.1517/14712590802571928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangham CR. CTL quality and the control of human retroviral infections. Eur J Immunol. 2009;39(7):1700–1712. doi: 10.1002/eji.200939451. [DOI] [PubMed] [Google Scholar]
- Barouch DH, Korber B. HIV-1 vaccine development after STEP. Annu Rev Med. 2010;61:153–167. doi: 10.1146/annurev.med.042508.093728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch DH, McKay PF, Sumida SM, Santra S, Jackson SS, Gorgone DA, Lifton MA, Chakrabarti BK, Xu L, Nabel GJ, Letvin NL. Plasmid chemokines and colony-stimulating factors enhance the immunogenicity of DNA priming-viral vector boosting human immunodeficiency virus type 1 vaccines. J Virol. 2003;77(16):8729–8735. doi: 10.1128/JVI.77.16.8729-8735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger CT, Frahm N, Price DA, Mothe B, Ghebremichael M, Hartman KL, Henry LM, Brenchley JM, Ruff LE, Venturi V, Pereyra F, Sidney J, Sette A, Douek DC, Walker BD, Kaufmann DE, Brander C. High-Functional-Avidity Cytotoxic T Lymphocyte Responses to HLA-B-Restricted Gag-Derived Epitopes Associated with Relative HIV Control. J Virol. 2011;85(18):9334–9345. doi: 10.1128/JVI.00460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihl F, Frahm N, Di Giammarino L, Sidney J, John M, Yusim K, Woodberry T, Sango K, Hewitt HS, Henry L, Linde CH, Chisholm JV, 3rd, Zaman TM, Pae E, Mallal S, Walker BD, Sette A, Korber BT, Heckerman D, Brander C. Impact of HLA-B alleles, epitope binding affinity, functional avidity, and viral coinfection on the immunodominance of virus-specific CTL responses. J Immunol. 2006;176(7):4094–4101. doi: 10.4049/jimmunol.176.7.4094. [DOI] [PubMed] [Google Scholar]
- Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3(2):205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. Development and validation of a fluorescence polarization-based competitive peptide-binding assay for HLA-A*0201--a new tool for epitope discovery. Biochemistry. 2005;44(37):12491–12507. doi: 10.1021/bi050255v. [DOI] [PubMed] [Google Scholar]
- Buseyne F, Riviere Y. The flexibility of the TCR allows recognition of a large set of naturally occurring epitope variants by HIV-specific cytotoxic T lymphocytes. Int Immunol. 2001;13(7):941–950. doi: 10.1093/intimm/13.7.941. [DOI] [PubMed] [Google Scholar]
- Casazza JP, Betts MR, Hill BJ, Brenchley JM, Price DA, Douek DC, Koup RA. Immunologic pressure within class I-restricted cognate human immunodeficiency virus epitopes during highly active antiretroviral therapy. J Virol. 2005;79(6):3653–3663. doi: 10.1128/JVI.79.6.3653-3663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat Immunol. 2012;13(7):691–700. doi: 10.1038/ni.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleton BA, Huang XL, Melhem NM, Fan Z, Borowski L, Rappocciolo G, Rinaldo CR. Primary human immunodeficiency virus type 1-specific CD8+ T-cell responses induced by myeloid dendritic cells. J Virol. 2009;83(12):6288–6299. doi: 10.1128/JVI.02611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeau P, Reynes J. Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection. Blood. 2011;117(21):5582–5590. doi: 10.1182/blood-2010-12-322453. [DOI] [PubMed] [Google Scholar]
- Corey L, McElrath MJ, Kublin JG. Post-step modifications for research on HIV vaccines. AIDS. 2009;23(1):3–8. doi: 10.1097/QAD.0b013e32830e6d6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport MP, Price DA, McMichael AJ. The T cell repertoire in infection and vaccination: implications for control of persistent viruses. Curr Opin Immunol. 2007;19(3):294–300. doi: 10.1016/j.coi.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27(3):406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Detels R, Jacobson L, Margolick J, Martinez-Maza O, Munoz A, Phair J, Rinaldo C, Wolinsky S. The multicenter AIDS Cohort Study, 1983 to. Public Health. 2012;126(3):196–198. doi: 10.1016/j.puhe.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, Holmes EC, Chang SC, Feeney ME, Addo MM, Ruiz L, Ramduth D, Jeena P, Altfeld M, Thomas S, Tang Y, Verrill CL, Dixon C, Prado JG, Kiepiela P, Martinez-Picado J, Walker BD, Goulder PJ. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med. 2004;199(7):905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Huang XL, Kalinski P, Young S, Rinaldo CR., Jr Dendritic cell function during chronic hepatitis C virus and human immunodeficiency virus type 1 infection. Clin Vaccine Immunol. 2007;14(9):1127–1137. doi: 10.1128/CVI.00141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm N, Korber BT, Adams CM, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel AG, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, StJohn A, Khatri A, Davis K, Mullins J, Goulder PJ, Walker BD, Brander C. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J Virol. 2004;78(5):2187–2200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi JV, Cheng HL, Margolick JB, Bauer KD, Ferbas J, Waxdal M, Schmid I, Hultin LE, Jackson AL, Park L, et al. Quality control in the flow cytometric measurement of T-lymphocyte subsets: the multicenter AIDS cohort study experience. The Multicenter AIDS Cohort Study Group. Clin Immunol Immunopathol. 1990;55(2):173–186. doi: 10.1016/0090-1229(90)90096-9. [DOI] [PubMed] [Google Scholar]
- Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8(8):619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XL, Fan Z, Borowski L, Rinaldo CR. Maturation of dendritic cells for enhanced activation of anti-HIV-1 CD8(+) T cell immunity. J Leukoc Biol. 2008;83(6):1530–1540. doi: 10.1189/jlb.1107795. [DOI] [PubMed] [Google Scholar]
- Janbazian L, Price DA, Canderan G, Filali-Mouhim A, Asher TE, Ambrozak DR, Scheinberg P, Boulassel MR, Routy JP, Koup RA, Douek DC, Sekaly RP, Trautmann L. Clonotype and repertoire changes drive the functional improvement of HIV-specific CD8 T cell populations under conditions of limited antigenic stimulation. J Immunol. 2012;188(3):1156–1167. doi: 10.4049/jimmunol.1102610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NA, Wei X, Flower DR, Wong M, Michor F, Saag MS, Hahn BH, Nowak MA, Shaw GM, Borrow P. Determinants of human immunodeficiency virus type 1 escape from the primary CD8+ cytotoxic T lymphocyte response. J Exp Med. 2004;200(10):1243–1256. doi: 10.1084/jem.20040511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalams SA, Goulder PJ, Shea AK, Jones NG, Trocha AK, Ogg GS, Walker BD. Levels of human immunodeficiency virus type 1-specific cytotoxic T-lymphocyte effector and memory responses decline after suppression of viremia with highly active antiretroviral therapy. J Virol. 1999;73(8):6721–6728. doi: 10.1128/jvi.73.8.6721-6728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaslow RA, Ostrow DG, Detels R, Phair JP, Polk BF, Rinaldo CR., Jr The Multicenter AIDS Cohort Study: rationale, organization, and selected characteristics of the participants. Am J Epidemiol. 1987;126(2):310–318. doi: 10.1093/aje/126.2.310. [DOI] [PubMed] [Google Scholar]
- Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13(1):46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68(7):4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity. 2013;38(3):425–436. doi: 10.1016/j.immuni.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10(3):282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457(7225):87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Cao J, Zhao H, Genowati I, Wong K, McLaughlin S, McSweyn MD, Diem K, Stevens CE, Maenza J, He H, Nickle DC, Shriner D, Holte SE, Collier AC, Corey L, McElrath MJ, Mullins JI. Selection on the human immunodeficiency virus type 1 proteome following primary infection. J Virol. 2006;80(19):9519–9529. doi: 10.1128/JVI.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin J, Zhao H, Tebit DM, Troyer RM, McSweyn M, Ghosh AK, Shriner D, Arts EJ, McElrath MJ, Mullins JI. Evolution of human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitopes: fitness-balanced escape. J Virol. 2007;81(22):12179–12188. doi: 10.1128/JVI.01277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, McNevin JP, Holte S, McElrath MJ, Mullins JI. Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS One. 2011;6(1):e15639. doi: 10.1371/journal.pone.0015639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina A, Rinaldo CR, Sekaly RP, Flores J, D'Souza PM. "In vitro systems to characterize the immune response to HIV-1 and HIV-1 vaccine candidates", NIAID Workshop Report, Bethesda, August 4, 2010. Vaccine. 2011;29(29–30):4647–4653. doi: 10.1016/j.vaccine.2011.04.035. [DOI] [PubMed] [Google Scholar]
- Malhotra U, Li F, Nolin J, Allison M, Zhao H, Mullins JI, Self S, McElrath MJ. Enhanced detection of human immunodeficiency virus type 1 (HIV-1) Nef-specific T cells recognizing multiple variants in early HIV-1 infection. J Virol. 2007;81(10):5225–5237. doi: 10.1128/JVI.02564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet. 2008;372(9653):1894–1905. doi: 10.1016/S0140-6736(08)61592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2(8):e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DH, Allen TM, Vogel TU, Jing P, DeSouza IP, Dodds E, Dunphy EJ, Melsaether C, Mothe B, Yamamoto H, Horton H, Wilson N, Hughes AL, Watkins DI. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med. 2002;8(5):493–499. doi: 10.1038/nm0502-493. [DOI] [PubMed] [Google Scholar]
- Pandrea I, Gaufin T, Gautam R, Kristoff J, Mandell D, Montefiori D, Keele BF, Ribeiro RM, Veazey RS, Apetrei C. Functional cure of SIVagm infection in rhesus macaques results in complete recovery of CD4+ T cells and is reverted by CD8+ cell depletion. PLoS Pathog. 2011;7(8):e1002170. doi: 10.1371/journal.ppat.1002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–175. [PubMed] [Google Scholar]
- Price DA, West SM, Betts MR, Ruff LE, Brenchley JM, Ambrozak DR, Edghill-Smith Y, Kuroda MJ, Bogdan D, Kunstman K, Letvin NL, Franchini G, Wolinsky SM, Koup RA, Douek DC. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21(6):793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Prlic M, Williams MA, Bevan MJ. Requirements for CD8 T-cell priming, memory generation and maintenance. Curr Opin Immunol. 2007;19(3):315–319. doi: 10.1016/j.coi.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Reynolds MR, Weiler AM, Weisgrau KL, Piaskowski SM, Furlott JR, Weinfurter JT, Kaizu M, Soma T, Leon EJ, MacNair C, Leaman DP, Zwick MB, Gostick E, Musani SK, Price DA, Friedrich TC, Rakasz EG, Wilson NA, McDermott AB, Boyle R, Allison DB, Burton DR, Koff WC, Watkins DI. Macaques vaccinated with live-attenuated SIV control replication of heterologous virus. J Exp Med. 2008;205(11):2537–2550. doi: 10.1084/jem.20081524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo CR. Dendritic cell-based human immunodeficiency virus vaccine. J Intern Med. 2009;265(1):138–158. doi: 10.1111/j.1365-2796.2008.02047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo CR, Jr, Huang XL, Fan Z, Margolick JB, Borowski L, Hoji A, Kalinyak C, McMahon DK, Riddler SA, Hildebrand WH, Day RB, Mellors JW. Anti-human immunodeficiency virus type 1 (HIV-1) CD8(+) T-lymphocyte reactivity during combination antiretroviral therapy in HIV-1-infected patients with advanced immunodeficiency. J Virol. 2000;74(9):4127–4138. doi: 10.1128/jvi.74.9.4127-4138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, Goulder PJ, Walker BD, Brander C, Mullins JI. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS One. 2008;3(1):e1424. doi: 10.1371/journal.pone.0001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario M, Bridgeman A, Quakkelaar ED, Quigley MF, Hill BJ, Knudsen ML, Ammendola V, Ljungberg K, Borthwick N, Im EJ, McMichael AJ, Drijfhout JW, Greenaway HY, Venturi V, Douek DC, Colloca S, Liljestrom P, Nicosia A, Price DA, Melief CJ, Hanke T. Long peptides induce polyfunctional T cells against conserved regions of HIV-1 with superior breadth to single-gene vaccines in macaques. Eur J Immunol. 2010;40(7):1973–1984. doi: 10.1002/eji.201040344. [DOI] [PubMed] [Google Scholar]
- Saez-Cirion A, Lacabaratz C, Lambotte O, Versmisse P, Urrutia A, Boufassa F, Barre-Sinoussi F, Delfraissy JF, Sinet M, Pancino G, Venet A. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci U S A. 2007;104(16):6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenker EL, Hultin LE, Bauer KD, Ferbas J, Margolick JB, Giorgi JV. Evaluation of a dual-color flow cytometry immunophenotyping panel in a multicenter quality assurance program. Cytometry. 1993;14(3):307–317. doi: 10.1002/cyto.990140311. [DOI] [PubMed] [Google Scholar]
- Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283(5403):857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Schneider E, Whitmore S, Glynn KM, Dominguez K, Mitsch A, McKenna MT. Revised surveillance case definitions for HIV infection among adults, adolescents, and children aged <18 months and for HIV infection and AIDS among children aged 18 months to <13 years--United States, 2008. MMWR Recomm Rep. 2008;57(RR-10):1–12. [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73(12):10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med. 2007;204(4):941–949. doi: 10.1084/jem.20061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner D, Rodrigo AG, Nickle DC, Mullins JI. Pervasive genomic recombination of HIV-1 in vivo. Genetics. 2004;167(4):1573–1583. doi: 10.1534/genetics.103.023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, Chang CH, Harndahl M, Weimershaus M, Gerstoft J, Akkad N, Klenerman P, Fugger L, Jones EY, McMichael AJ, Buus S, Schild H, van Endert P, Iversen AK. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol. 2009;10(6):636–646. doi: 10.1038/ni.1728. [DOI] [PubMed] [Google Scholar]
- Turnbull EL, Wong M, Wang S, Wei X, Jones NA, Conrod KE, Aldam D, Turner J, Pellegrino P, Keele BF, Williams I, Shaw GM, Borrow P. Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J Immunol. 2009;182(11):7131–7145. doi: 10.4049/jimmunol.0803658. [DOI] [PubMed] [Google Scholar]
- van Bockel DJ, Price DA, Munier ML, Venturi V, Asher TE, Ladell K, Greenaway HY, Zaunders J, Douek DC, Cooper DA, Davenport MP, Kelleher AD. Persistent survival of prevalent clonotypes within an immunodominant HIV gag-specific CD8+ T cell response. J Immunol. 2011;186(1):359–371. doi: 10.4049/jimmunol.1001807. [DOI] [PubMed] [Google Scholar]
- Vezys V, Masopust D, Kemball CC, Barber DL, O'Mara LA, Larsen CP, Pearson TC, Ahmed R, Lukacher AE. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection. J Exp Med. 2006;203(10):2263–2269. doi: 10.1084/jem.20060995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004;101(45):16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomaku Y, Miura H, Tomiyama H, Kawana-Tachikawa A, Takiguchi M, Kojima A, Nagai Y, Iwamoto A, Matsuda Z, Ariyoshi K. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J Virol. 2004;78(3):1324–1332. doi: 10.1128/JVI.78.3.1324-1332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusim KSJ, Honeyborne I, Calef C, Goulder PJ, Korber BT. Enhanced Motif Scan: A Tool to Scan for HLA Anchor Residues in Proteins, pp. 25–36. in HIV Immunology and HIV/SIV Vaccine Databases 2003. In: Korber BT, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI, editors. Los Alamos, New Mexico: Los Alamos National Laboratory, Theoretical Biology and Biophysics; 2004. Published by: LA-UR 04-8162. [Google Scholar]
- Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458(7235):211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The frequency (y-axis) of epitope variants of Gag (A), Env (B) and Nef (C) naturally occurring during the course of infection is plotted against the corresponding time of emergence (x-axis) post-SC. The frequency of a specific epitope variant is calculated as the number of contemporaneous variant over a specific time point divided by the total number of variants circulating at the same time post-SC. Years post-SC (x-axis) include early, late, early cART time points defined as follow: 0–2.8 years post-SC, 3.3–7.8 years post-SC, 8.3–10.1 years post-SC, and >12.4 years post-SC, respectively. High, medium and low (including those classified as low and non-binders by the FP assay) affinities are defined based on the log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0. SC, seroconversion.
Synthetic 9 mer or 10 mer HIV-1 peptides representing autologous contemporaneous variants within the study participant were loaded into mature autologous DC. These DC were used to stimulate cryopreserved autologous PMBC at a responder-to-stimulator ratio of 10:1. Gag, Env and Nef-specific T cell responses were measured by IFN-γ ELISPOT. Results represent mean values of duplicates and are expressed as spot forming cells (SFC) /106 PBMC (y-axis) targeting evolving Gag, Env or Nef variants (x-axis) at early (A), late (B), early cART (C) and late cART (D). The right panels of A, B, C and D represent the magnitude of memory T cell responses targeting all variants (Gag, Env and Nef) with high, medium or low binding affinity. Black fill: high binding affinity variant, white fill: medium binding affinity variant, shaded fill: low binding affinity variant (including those classified as low and non-binders by the FP assay). Affinity binding categories were based on the values of log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0.Early: 0–2.8 years post-SC, late: 3.3–7.8 years post-SC, early ART: 8.3–10.1 years post-SC post-SC. SC, seroconversion.
Mature DC isolated from subject 8 were loaded with variant HIV-1 peptides and used to prime autologous PBMC isolated from the study participant prior to SC. The magnitude of primary T cell responses was determined by IFN-γ ELISPOT. In vitro primary T cell responses (y-axis) of PBMC isolated prior to seroconversion were plotted against variants (x-axis) of (A) Gag (SLFNTVATL, TLNAWVKVV, RMYSPTSIL), (B) Env (RPNNNTRRSI, TLEQVVKKL) and (C) Nef (FPVRPQVPL, RPMTWKGAL, TPGPGTRYPL) emerging during early, late and/or ART time points are shown. (D) The magnitude of IFN-γ responses (y-axis) to contemporaneous viral peptides of combined Gag, Env and Nef with high, medium and low binding affinity (x-axis). Black histograms: high binding affinity variant, empty histograms: medium binding affinity variant, dashed histograms: low (including those classified as low and non-binders by the FP assay) binding affinity variant. Affinity binding categories are based on the values of log IC50: high affinity < 3.7, medium < 4.7, low < 5.5, and no affinity < 6.0. SC, seroconversion; SFC, spot-forming cells.