

Abstract
Background
Tumor metabolism is a crucial factor for the carcinogenesis of oral squamous cell carcinoma (OSCC).
Methods
Expression of IGF-R1, glycolysis-related proteins (GLUT-1, HK 2, PFK-1, LDHA, TKTL1), mitochondrial enzymes (SDHA, SDHB, ATP synthase) were analyzed in normal oral mucosa (n = 5), oral precursor lesions (simple hyperplasia, n = 11; squamous intraepithelial neoplasia, SIN I-III, n = 35), and OSCC specimen (n = 42) by immunohistochemistry and real-time polymerase chain reaction (qPCR) analysis in OSCC cell lines. Metabolism-related proteins were correlated with proliferation activity (Ki-67) and apoptotic properties (TUNEL assay) in OSCC. Specificity of antibodies was confirmed by western blotting in cancer cell lines.
Results
Expression of IGF-R1, glycolysis-related proteins (GLUT-1, HK 2, LDHA, TKTL1), and mitochondrial enzymes (SDHA, SDHB, ATP synthase) were significantly increased in the carcinogenesis of OSCC. Metabolic active regions of OSCC were strongly correlated with proliferating cancer (Ki-67+) cells without detection of apoptosis (TUNEL assay).
Conclusions
This study provides the first evidence of the expression of IGF-R1, glycolysis-related proteins GLUT-1, HK 2, PFK-1, LDHA, and TKTL1, as well as mitochondrial enzymes SDHA, SDHB, and ATP synthase in the multi-step carcinogenesis of OSCC. Both, hypoxia-related glucose metabolism and mitochondrial oxidative phosphorylation characteristics are associated with the carcinogenesis of OSCC. Acidosis and OXPHOS may drive a metabolic shift towards the pentose phosphate pathway (PPP). Therefore, inhibition of the PPP, glycolysis, and targeted anti-mitochondrial therapies (ROS generation) by natural compounds or synthetic vitamin derivatives may act as sensitizer for apoptosis in cancer cells mediated by adjuvant therapies in OSCC.
Keywords: Oral squamous cell carcinoma, Tumor metabolism, Glycolysis-related proteins, Mitochondrial oxidative phosphorylation, Carbohydrate-restricted diets, Targeted anti-mitochondrial therapy
Introduction
Cancer is regarded as an acquired genetic disease. The genetic model of multistep carcinogenesis describes the rise of malignant tumors from a single transformed cell (monoclonal theory of carcinogenesis) and subsequent development through morphologically and clinically detectable precancerous stages [1]. The carcinogenesis of oral squamous cell carcinoma (OSCC) is a highly complex multifocal process that occurs when squamous epithelium is affected by several genetic alterations [2]. Understanding the mechanistic basis await the availability of molecular tools to experimentally and selectively manipulate this multistep process with subsequent clinical implications for therapy of precursor lesions and OSCC.
OSCC is an aggressive tumor with low response to chemotherapy and basic resistance to most standard of care anticancer drugs [3,4]. Tumor metabolism [5] with a special focus on increased hypoxia/glycolytic activity is regarded as a crucial factor for the carcinogenesis of OSCC and is associated with radio- and, chemotherapy resistance, as well as tumor recurrence [6-9].
Cancer can be considered as integrated metabolic ecosystem and includes several pathways of carcinogenesis associated with metabolic phases of transformation [10]. Glycolysis [11], mitochondrial oxidative phosphorylation (OXPHOS) [12], and glutaminolysis have been shown to play key roles in tumor metabolism. Mitochondria have an important role in carcinogenesis due to their roles in mediating apoptosis [13]. They act as a major source of endogenous reactive oxygen species (ROS) that escape from the electron transport chain (ETC.) during OXPHOS [14]. Although glycolysis is a major characteristic of tumor cell metabolism this pathway alone cannot account for energy usage in all types of cancer cells. Finally, the dominant metabolic process can be either glycolysis or mitochondrial oxidative metabolism based on the tumor type [15]. Both metabolic phenotypes have been associated with subsequent nutritional consequences [16-19].
The generation of adenosine triphosphate (ATP) in glycolysis has a lower efficiency, but a faster rate than OXPHOS [11,20]. This enhanced rate of ATP generation has been postulated to be beneficial for rapidly proliferating cells. However, several studies have suggested that OXPHOS is the major source of cellular ATP in proliferating and non-proliferating [21] cancer cells [11,21-23].
A recent study by Vander Heiden [24] indicated that the induction of the Warburg effect in cancer cells is more the consequence of the activation of protooncogenes (e.g., Myc), transcription factors (e.g., hypoxia-inducible factor-1, HIF-1), and signaling pathways (e.g., PI3K), as well as the inactivation of tumor suppressors (e.g. p53) rather than the primary generation of much needed energy [11]. Moreover, it has been stated that tumor cells profit from the enhanced glycolytic activity in glycolytic intermediates, which are shunted into subsidiary pathways (e.g. by the pentose phosphate pathway [PPP]) to fuel metabolic pathways that generate de novo nucleotides, lipids, amino acids, and nicotinamide adenine dinucleotide phosphate (NADPH) [11,25,26]. Frezza et al.[12] showed that defects in mitochondrial enzymes or complexes within the electron transport chain are not frequently observed in cancer. Therefore, investigation of OXPHOS provides a clear rational for future anti-cancer therapy strategies in OSCC [27].
Today, it is estimated that more than 30% of all tumor entities may be due to dietary factors [17]. Studies have clearly linked diabetes and obesity to cancer [28]. Hyperinsulinemia leads to increased production of insulin-like growth factor-1 (IGF-1) [29], which activates insulin-like growth factor-1 receptor (IGF-1R). IGF-1R is a receptor tyrosine kinase (RTK) that stimulates protein synthesis by activating the mammalian target of rapamycin (mTOR), and in turn mTOR mediated upregulation of glycolytic enzymes may promote tumor development [30,31]. Therefore, the IGF-1R pathway is an emerging therapeutic target in oncology [32-34] but has not yet been described for the carcinogenesis of OSCC.
Hexokinase 2 (HK 2) is expressed in insulin-sensitive tissues such as muscle and adipose [11], is one of the rate-limiting enzymes of glucose catabolism in tumor cells, is upregulated in many cancers [35,36], and was recently described for OSCC [37]. Phosphofructokinase-1 (PFK-1) [38] is a key enzyme in glycolysis where it forms a rate-limiting step, but its expression has not been described for OSCC. Among glycolytic enzymes PFK-1 has been more extensively studied than other enzymes, which is likely to be due to its various regulatory mechanisms.
Recently, we have demonstrated glucose transporter 1 (GLUT-1) (solute carrier family 2 [facilitated glucose transporter], member 1 [SLC2A1]) [9], transketolase-like-1 (TKTL1) [7], and lactate dehydrogenase A (LDHA/LDH5) [39] as adverse prognostic factors for the survival of patients with OSCC. However, the expression of GLUT-1, HK 2, PFK-1, LDHA, and TKTL1 during a multi-step carcinogenesis has not been described yet.
More recently, characterization of OXPHOS in cancer was performed by describing succinate dehydrogenase SDHA, SDHB (respiratory complex II in mitochondria), and ATP synthase (respiratory complex V in mitochondria) [40,41]. None of these enzymes have yet been described for OSCC.
The purpose of this study was to examine the relationship between metabolism-related proteins [8] with a multistep carcinogenesis. This is the first study describing glycolysis-related PFK-1, OXPHOS-related SDHA, SDHB, and ATP synthase in OSCC.
Materials and methods
Patients and tumor specimen
The records of healthy individuals (normal oral mucosal tissues, n = 5), patients with oral precursor lesions (simple hyperplasia, n = 11; squamous intraepithelial neoplasia SIN I, n = 5; SIN II, n = 9; SIN III, severe dysplasia, n = 10; SIN III, carcinoma in situ, n = 11), and patients with invasive OSCC (n = 42) were retrospectively assessed from January 2009 to December 2013. The diagnosis of normal oral mucosal tissues, precursor lesions, and invasive squamous cell carcinoma was confirmed by the department of Pathology, University Hospital Tuebingen. The material was archival formalin-fixed, paraffin-embedded tissue from routine histopathological work-ups. The material has been stored with permission of the local ethics committee of the University Hospital Tuebingen (approval number: 562-2013BO2), after informed consent obtained from the patients prior to surgical resection. Tumor blocks of paraffin-embedded tissue were selected by experienced pathologists, evaluating the routine H&E stained sections. Sections from all available tissues underwent histopathological assessment, blinded to the prior histopathology report. Serial tissue sections (2 μm thickness) were cut from formalin-fixed paraffin-embedded (FFPE) blocks on a microtome and mounted from warm water onto adhesive microscope slides. First, we assessed H&E sections (Additional file 1: Figure S1) from each tissue section to differentiate between normal tissue, precursor lesions, tumor cell areas, stromal areas, and infiltrating immune cells. Oral precursor lesions were classified according to WHO criteria [1]. Tumor staging was performed according to the 7th edition of the TNM staging system by the UICC/AJCC of 2010 [42]. Grading of OSCC was defined according to WHO criteria [43].
Staining procedure and quantification of immunohistochemistry
The antibodies used for immunohistochemistry are shown in Additional file 2: Table S1. We stained for IGF-R1β, glycolysis-related proteins GLUT-1, HK 2, PFK-1, LDHA, TKTL1, mitochondrial enzymes SDHA, SDHB, ATP synthase, and proliferation characteristics Ki-67 in serial sections (Additional file 2: Table S1). Staining was performed on serial sections of 2 μm thickness as previously described [39].
Five representative high power fields (1 HPF = 0.237 mm2, original magnification: x200-fold) were analyzed for IGF-R1β, GLUT-1, HK 2, PFK-1, LDHA, TKTL1, SDHA, SDHB, and ATP synthase expression in normal tissue, oral precursor lesions, tumor tissue and averaged, respectively. The extent of the staining, defined as the percentage of positive staining areas of tumor cells in relation to the whole tissue area, was semi-quantitatively scored on a scale of 0 to 3 as the following: 0, <10%; 1, 10–30%; 2, 30–60%; 3, >60%. The intensities of the signals were scored as 1+ (weak), 2+ (intermediate), and 3+ (strong). Then, a combined score (0–9) for each specimen was calculated by multiplying the values of these two categories [44]. Cases were classified as negative, 0 points, positive, 1–9 points. Two observers blinded to the diagnosis performed scoring on identical sections marked by circling with a water-resistant pencil and finally with diamond-tipped pencil on the opposite side of the microscopic slide. Pictures were analyzed using a Canon camera (Krefeld, Germany). The photographed images were imported into the Microsoft Office Picture Manager.
In situ detection of apoptosis
Apoptotic cells and bodies were detected by the terminaldeoxynucleotidyl transferase-mediated deoxyuridinetriphosphate nick-end labeling (TUNEL) method (ApopTag® Plus Peroxidase In Situ Apoptosis Kit, Chemicon, Planegg-Muenchen, Germany). The TUNEL assay is regarded as the ‘gold standard’ in apoptosis detection and was performed as described previously [45-47].
Cell culture, western blot and densitometric quantification
BICR3 and BICR56 OSCC cell lines [9,48] were cultured in DMEM F-12 medium (Invitrogen, Belgium) containing 10% fetal calf serum (Sigma-Aldrich, Germany), 1% fungicide, and penicillin/streptomycin (Biochrom, Germany) at 37°C and 5% CO2.
IGF-R1β, HK 2, PFK-1, LDHA, SDHA, and SDHB antibody specificity was confirmed by western blot analysis in BICR3, BICR56 cell lines. Specificity of GLUT-1 pAb (clone A 3536) [9], TKTL1 mAb (clone JFC12T10) [49] and Ki-67 mAb (clone MIB-1) [50] have been previously demonstrated. Protein extraction from OSCC cell lines BICR3 and BICR56 was performed as described previously [51]. Normal human oral mucosal tissue protein was purchased from BioChain (Hayward, CA, USA) as control. The membranes were analyzed by immunoblotting using IGF-R1β, HK 2, PFK-1, LDHA, SDHA, SDHB, and ATP synthase antibodies (Additional file 2: Table S1), or IgG control antibodies (BD Pharmingen, Heidelberg), and monoclonal mouse anti-human GAPDH (Abcam, Cambridge, UK, dilution: 1:5000) specific primary antibody overnight at 4°C. Binding of the primary antibodies was detected with HRP-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, CA, USA) and visualized by the enhanced chemiluminescence method (GE Healthcare, Freiburg, Germany).
Quantification of western blot bands was carried out by using an automated densitometric quantification digitizing system (UN-SCAN-IT Gel software, version 6.1, Silk Scientific, Inc., Utah, USA) [39].
Real-time polymerase chain reaction (qPCR) analysis
To analyze gene expression of IGF-R1, GLUT-1, HK 2, PFK-1, TKTL1, SDHA, SDHB, and ATP synthase by RT-PCR, we extracted total cellular RNA and performed cDNA synthesis from OSCC cell lines (BICR3, BICR56) as previously described [52]. Gene expression of LDHA in OSCC cell lines has been shown previously [39]. The amount of total RNA was determined by measuring absorbance at 260 nm. The purity of the total RNA was established by confirming that the 260 nm: 280 nm ratio was within a 1.8-2.0 range, indicating that the RNA preparations were free of contaminants. Normal human oral keratinocyte cDNA (HOK cDNA) was purchased by ScienCell (Carlsbad, CA, USA) as control. The reference genes GAPDH and beta-actin were used for relative quantification and cDNA quality (integrity) control. To quantitate mRNA expression, qPCR with the LightCycler System (Roche Applied Science, Mannheim, Germany) was established as described before [53]. Commercial primer kits were purchased from Search LC (Heidelberg, Germany). Melt-curve analysis was be used to identify specific reaction products. The relative quantification value, fold difference, is expressed as 2-ΔΔCt as described previously [54].
Statistical analysis
Statistical analysis was performed with MedCalc Software, Version 13.1.1 (Mariakerke, Belgium). Data were analyzed using the non-parametric Mann–Whitney U Test or Kruskal-Wallis test when more than 2 groups were compared. Correlation analysis of TUNEL assay or Ki-67 with metabolism-related proteins was performed by the non-parametric Spearman Rho rank correlation coefficient. All p-values presented were 2-sided and p < 0.05 was considered statistically significant.
Results
Expression of IGF-R1β, glycolysis-related proteins GLUT-1, HK 2, PFK-1, LDHA, TKTL1, mitochondrial enzymes SDHA, SDHB, and ATP synthase in normal mucosa, oral precursor lesions and OSCC
Invasive OSCC of immunohistochemical stained serial sections was confirmed by H&E staining (Additional file 1: Figure S1). In comparison to normal tissue and hyperplasia a significantly (p < 0.05) increased expression of IGF-R1β (Figure 1), GLUT-1 (Figure 2), HK 2 (Figure 3), TKTL1 (Figure 4), LDHA (Figure 5), SDHA (Figure 6), SDHB (Figure 7), and ATP synthase (Figure 8) was observed in cancer cells of OSCC. Compared with SIN I-III PFK-1 expression (Figure 9) was significantly decreased in OSCC.
Figure 1.
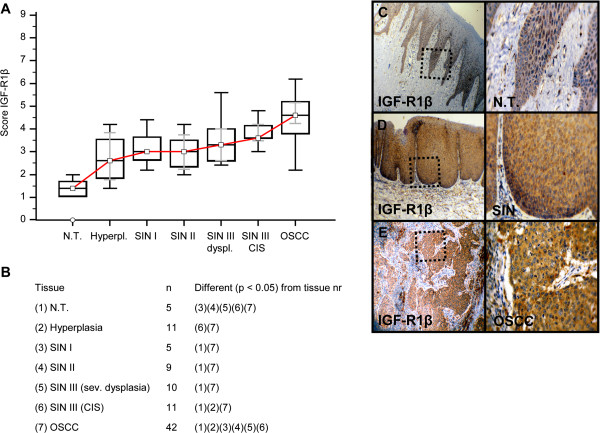
Immunohistochemical analysis and staining of IGF-R1β in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of IGF-R1β is observed in OSCC. IGF-R1β expression is significantly increased in OSCC compared with SIN I-III (p < 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates IGF-R1β expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). IGF-1R, insulin-like growth factor-1 receptor; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of IGF-R1β expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 2.
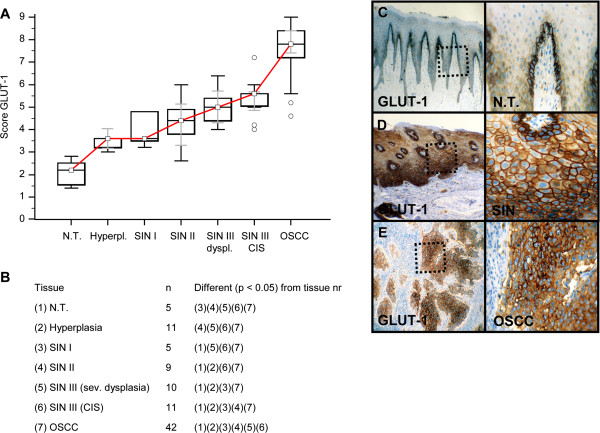
Immunohistochemical analysis and staining of GLUT-1 in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of GLUT-1 is observed in OSCC. GLUT-1 expression is significantly increased in OSCC compared with SIN I-III (p < 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates GLUT-1 expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). GLUT-1, glucose transporter-1; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of GLUT-1 expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 3.
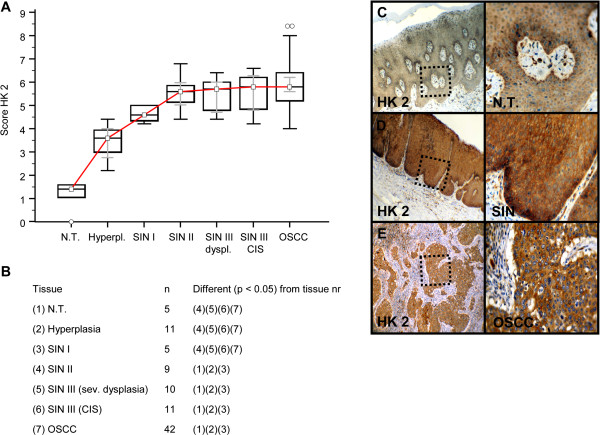
Immunohistochemical analysis and staining of HK 2 in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of HK 2 is observed in OSCC. HK 2 expression is significantly increased in OSCC compared with SIN I-III (p < 0.0397, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates HK 2 expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). HK 2, hexokinase 2; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of HK 2 expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 4.
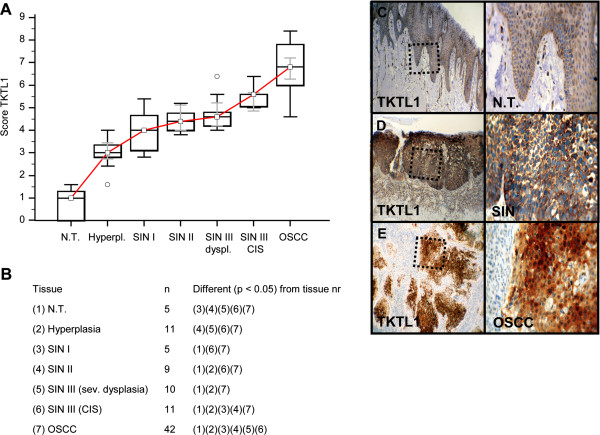
Immunohistochemical analysis and staining of TKTL1 in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of TKTL1 is observed in OSCC. TKTL1 expression is significantly increased in OSCC compared with SIN I-III (p < 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates TKTL1 expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). TKTL1, transketolase-like-1; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of TKTL1 expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 5.
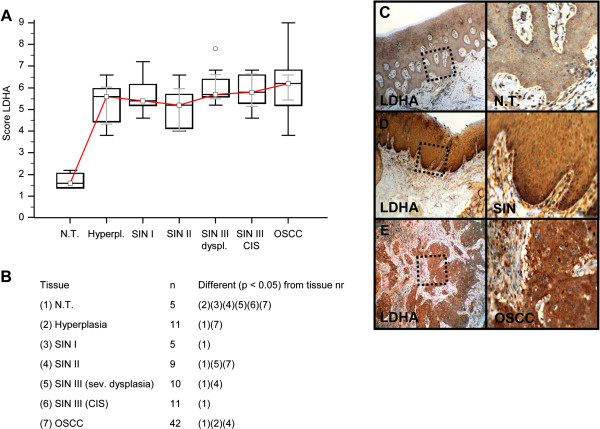
Immunohistochemical analysis and staining of LDHA in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of LDHA is observed in OSCC. No difference of LDHA expression in OSCC is observed in comparison with SIN I-III (p = 0.0822, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates LDHA expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). LDHA, lactate dehydrogenase; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of LDHA expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 6.
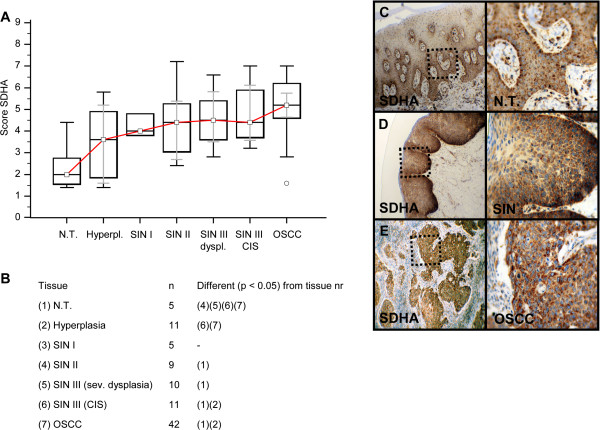
Immunohistochemical analysis and staining of SDHA in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of SDHA is observed in OSCC. SDHA expression is significantly increased in OSCC compared with SIN I-III (p = 0.0103, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates SDHA expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SDHA is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). SDHA, succinate dehydrogenase A; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of SDHA expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 7.
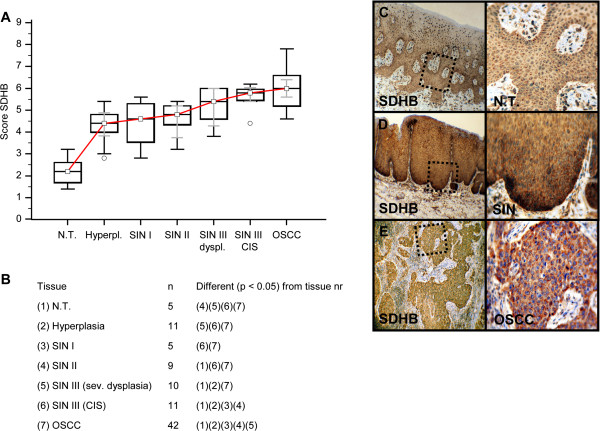
Immunohistochemical analysis and staining of SDHB in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of SDHB is observed in OSCC. SDHB expression is significantly increased in OSCC compared with SIN I-III (p = 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates SDHB expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SDHA is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). SDHB, succinate dehydrogenase B; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of SDHB expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 8.
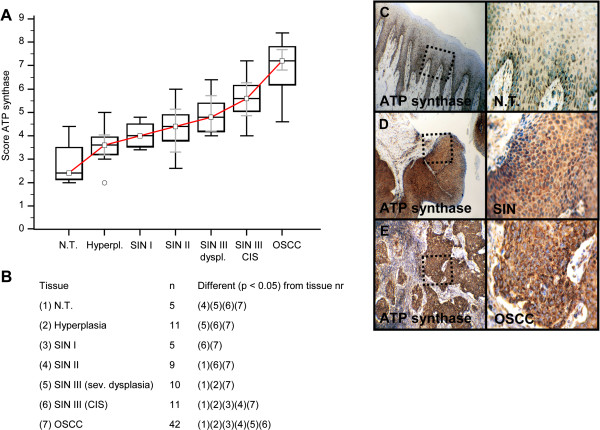
Immunohistochemical analysis and staining of ATP synthase in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison to normal tissue/hyperplasia a significantly (p < 0.05, Kruskal-Wallis Test; A and B) increased expression of ATP synthase is observed in OSCC. ATP synthase expression is significantly increased in OSCC compared with SIN I-III (p < 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates ATP synthase expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). ATP synthase is subdivided in severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of ATP synthase expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Figure 9.
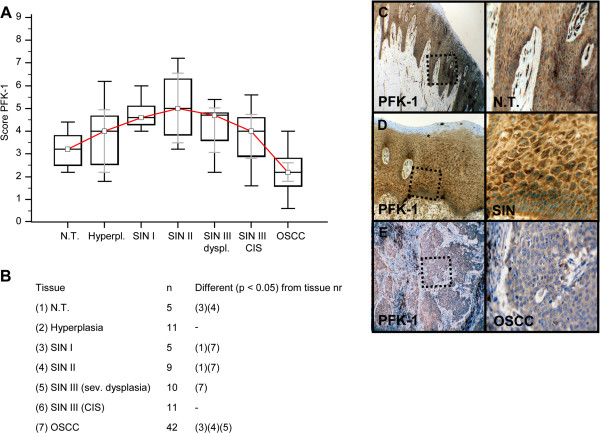
Immunohistochemical analysis and staining of PFK-1 in normal oral mucosal tissue, oral precursor lesions - hyperplasia, SIN I, SIN II, SIN III, and invasive OSCC. In comparison with normal tissue a significantly increased expression of PFK-1 expression is observed in SIN I and SIN II lesions (p < 0.05, Kruskal-Wallis Test; A and B). In comparison with SIN I, SIN II, and SIN III (sev. dysplasia) a significantly decreased expression of PFK-1 expression is observed in OSCC. PFK-1 expression is significantly decreased in OSCC compared with SIN I-III (p < 0.0001, Mann–Whitney U Test). Analysis refers to averaged scores. Red line indicates PFK-1 expression results during carcinogenesis. Grey lines show 95% confidence intervals. Analysis of significant statistically different single values is indicated in the table below (B). SIN III is subdivided in severe dysplasia and carcinoma in situ (CIS). PFK-1, phosphofructokinase-1; SIN, squamous intraepithelial neoplasia; N.T., normal tissue. Immunohistochemical staining shows representative images of PFK-1 expression in N.T. (C), SIN (D), and OSCC (E). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: x100-fold, left panel) which is also shown in larger magnification (x200-fold, right panel).
Correlation of proliferation activity (Ki-67) with metabolic markers (GLUT-1, HK 2, PFK-1, LDHA, TKTL1, SDHA, SDHB, and ATP synthase) in OSCC serial sections
For investigation of proliferating cancer cells and its relation to metabolic characteristics, we performed correlation analysis of Ki-67 with GLUT-1, HK 2, LDHA, TKTL1, SDHA, SDHB, and ATP synthase in OSCC. Evaluation of immunohistochemically stained FFPE slides were measured by observer related semi-quantitative scoring and showed a strong positive correlation of Ki-67+ expression with metabolic active cancer cells as observed in OSCC serial sections. Significant correlation of proliferating cancer cells was observed with GLUT-1, TKTL1 mitochondrial markers SDHA, SDHB, and ATP synthase: GLUT-1 (rho = 0.370, 95% CI = 0.0750 to 0.606, p = 0.0157), TKTL1 (rho = 0.460, 95% CI = 0.165 to 0.704, p = 0.0056), SDHA (rho = 0.485, 95% CI = 0.213 to 0.688, p = 0.0011), SDHB (rho = 0.657, 95% CI = 0.441 to 0.801, p < 0.001), and ATP synthase (rho = 0.413, 95% CI = 0.125 to 0.637, p = 0.0065). No significant correlation of proliferation activity was found with HK 2 (rho = 0.152, 95% CI = -0.159 to 0.436, p = 0.3365) and LDHA (rho = 0.153, 95% CI = -0.158 to 0.437, p = 0.3336). Moreover, a significant correlation of GLUT-1 with TKTL1 in OSCC was analyzed (rho = 0.419, 95% CI 0.131 to 0.641, p = 0.0058) as previously indicated by our work. There was a significant inverse correlation of PFK-1 with TKTL1 detected (rho = -0.475, 95% CI -0.619 to -0.301, p < 0.0001).
Correlation of apoptotic activity (TUNEL assay) with metabolic markers (GLUT-1, HK 2, PFK-1, LDHA, TKTL1, SDHA, SDHB, and ATP synthase) in OSCC serial sections
For the investigation of apoptotic properties in cancer cells caused by OXPHOS (due to putative increased free radical production) we performed TUNEL assays in OSCC. Although tumor cells show increased OXPHOS-related enzymes (SDHA, SDHB, ATP-synthase, Figures 6, 7 and 8) no apoptotic activity (AI < 10%) in cancer cells was observed in those highly metabolic active regions (Figure 10). In contrast, tumor-infiltrating leucocytes adjacent to the tumor demonstrated increased apoptotic activity (Figure 10).
Figure 10.
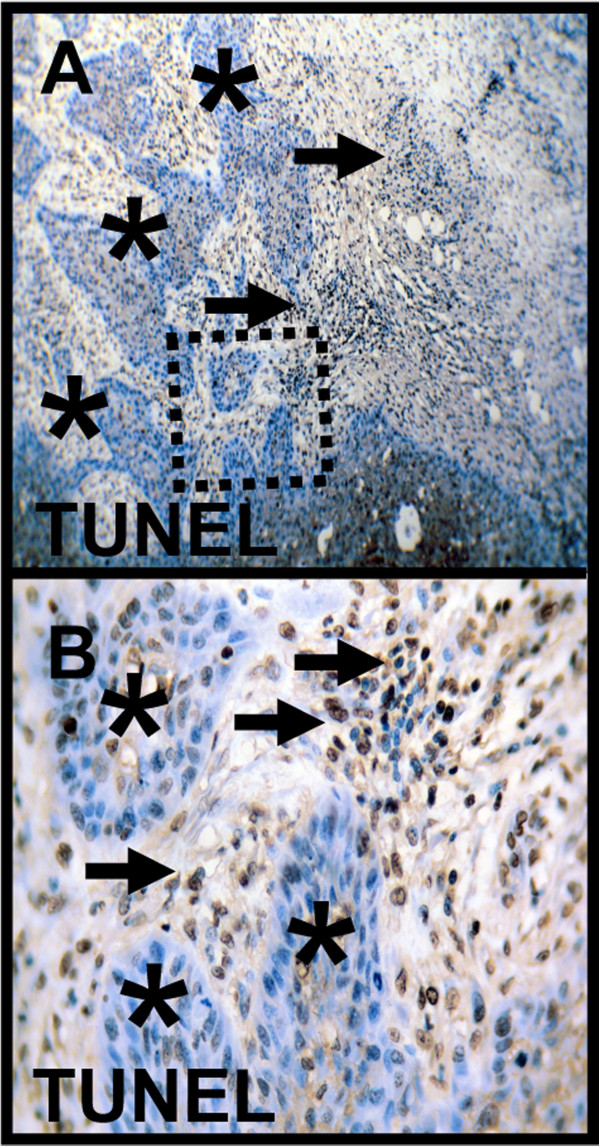
In situ detection of DNA fragmentation by TUNEL-staining in OSCC. In metabolic highly active regions as demonstrated by markers IGF-R1β, HK 2, TKTL1, PFK-1, LDHA, SDHA, SDHB, and ATP synthase cancer cells do not undergo apoptosis (serial section, asterisk, A). In contrast, tumor-infiltrating leucocytes adjacent to the tumor demonstrate increased apoptotic activity (arrow, B). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, the blue color shows the nuclear counterstaining by hematoxylin. Original magnification: x200-fold. TUNEL, terminaldeoxynucleotidyl transferase-mediated deoxyuridinetriphosphate nick-end labeling.
IGF-R1β, HK 2, PFK-1, LDHA, SDHA, SDHB, ATP synthase antibody specifity is confirmed by western blot analysis
Western Blot analysis of HK 2, IGF-R1β, PFK-1, SDHA, ATP synthase, LDHA, and SDHB in BICR3 and BICR56 OSCC cell lines confirmed immunohistochemical staining specifity of antibodies used in immunohistochemistry (Figure 11).
Figure 11.
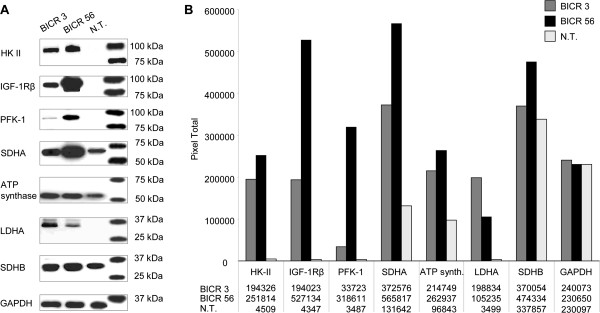
Western blot analysis in normal tissue, BICR3 and BICR56 OSCC cell lines. Western Blot of HK 2, IGF-R1β, PFK-1, SDHA, ATP synthase, LDHA, and SDHB in BICR3 and BICR56 OSCC cell lines confirm immunohistochemical staining specifity of antibodies (left panel, A). Western blot analysis shows increased HK 2 (~102 kDa), IGF-R1β (~97 kDa), PFK-1 (~85 kDa), SDHA (~70 kDa), ATP synthase (~53 kDa), LDHA (~37 kDa), and SDHB (~32 kDa) expression compared to normal tissue. GAPDH (loading control) is shown as a band of approximately 35 kDa. Densitometric quantification (B) of western blot protein bands (pixel total) is given in the right panel (B). IGF-1R, insulin-like growth factor-1 receptor; HK 2, hexokinase 2; PFK-1, phosphofructokinase-1; LDHA, lactate dehydrogenase A; SDH, succinate dehydrogenase; GAPDH, glycerinaldehyd-3-phosphat-dehydrogenase; N.T., normal tissue.
Analysis of IGF-R1, GLUT-1, HK 2, PFK-1, TKTL1, SDHA, SDHB, and ATP synthase gene expression
IGF-R1, GLUT-1, HK 2, PFK-1, TKTL1, SDHA, SDHB, and ATP synthase gene expression in OSCC cell lines was increased in comparison to normal human oral keratinocytes (Table 1).
Table 1.
Increased gene expression analysis (qPCR) of cancer metabolism-related proteins in OSCC cell lines compared with normal human oral keratinocytes (x-fold difference)
| IGF-1R | GLUT-1 | HK 2 | PFK-1 | TKTL1 | SDHA | SDHB | ATP synthase | |
|---|---|---|---|---|---|---|---|---|
| BICR3 |
12.6-fold |
22.4-fold |
13.6-fold |
2.8-fold |
3.3-fold |
14.7-fold |
7.7-fold |
10.3-fold |
| BICR56 | 18.2-fold | 25.3-fold | 11.9-fold | 8.4-fold | 4.9-fold | 16.0-fold | 6.6-fold | 14.1-fold |
Discussion
In our study, we investigated cancer metabolism-related proteins in the carcinogenesis of OSCC. For the first time, we found increased expression of mitochondrial enzymes (SDHA, SDHB, ATP synthase) in OSCC compared with normal oral mucosa. However, very few data is available describing a mitochondrial oxidative metabolism [27] in OSCC. Authors assume that OXPHOS is an important pathway for the generation of ATP [11,22,23] and ROS [18,55-58] during the carcinogenesis of OSCC. The TUNEL assay demonstrated that tumor cells do not undergo apoptosis and therefore, increased ROS generation by OXPHOS does not reach toxic levels. Based on our results and as currently stated by Whitaker-Menezes et al.[57] in the context of breast cancer we assume that mitochondria are the ‘Achilles heel’ and ‘powerhouse’ in the carcinogenesis of OSCC [23,56-59]. Increased levels of ROS in tumor cells are generated by altered metabolic activity, oncogene activation, and deregulated proliferation [60]. Oncogenic transformation promotes the production of excessive ROS, which would become toxic if not counteracted, while low levels of ROS can help to promote cell proliferation. This is the reason why many cancer cells may show an increased expression of antioxidant proteins [26] such as LDHA [39] and TKTL1 [7] as indicated by our observation, which contribute to the survival and success of the tumor. Indeed, this dependence on antioxidants can make cancer cells more vulnerable to the inhibition of these detoxifying systems than normal cells, which do not harbor such a high burden of oxidative stress [61-63]. On the other hand, an increase in ATP production by OXPHOS has been shown in response to hypoxic stress and protects cells from a critical energy crisis [64]. However, we do not know which metabolic pathway (glycolysis vs. OXPHOS) has been upregulated in carcinogenesis of OSCC as first.
In the literature, bioactive food components [5,17,65,66] have been demonstrated to mediate the reversal of a glycolytic phenotype in cancer cells, thus leading to growth inhibition and induction of apoptosis (Table 2). The reprogramming of energy metabolism [67-70] has been suggested for targeting of mitochondria [18,19,21,23,55,58,61-63,71-74] and subsequent induction of apoptosis [71] as a valid anti-cancer strategy [18] for which bioactive food components [19] have been suggested. Rapidly proliferating cells are more sensitive to radio-, and chemotherapy, which have been shown to be less effective in non-dividing cancer cells [75]. Activation of mitochondrial OXPHOS [58,61] and other mechanisms in cancer cells by natural compounds may induce apoptosis even in therapy resistant cancer cells [55]. Because OXPHOS is the predominant supplier of ATP in (proliferating and) non-proliferating cancer cells [21] targeted anti-mitochondrial therapies could be of interest for apoptosis induction in quiescent (non-proliferating) but metabolically active cancer cells, which rely on mitochondrial lipid β-oxidation [76]. Therefore, bioactive food components inducing apoptosis by ROS generation (Table 2) and other mechanisms play an emerging role in cancer therapy. According to other tumor entities several other natural compounds have been shown to activate ROS [58,61] in OSCC [77-81] and subsequent apoptosis in cancer cells and may therefore provide a clear rational to study them in further pre-clinical and clinical trials (Table 2). Moreover, phytochemicals [82] and vitamins have different hypoxia-inducible factor-1 (HIF-1) binding capacities (inhibitory activity: lycopene > curcumin > tocopherol > ascorbic acid) suggestive for their impact on the decrease in tumor hypoxia and antioxidative properties in normal tissue [83].
Table 2.
Bioactive food components (natural or synthetic compounds/vitamin derivatives) targeting mitochondria (ROS generation) and/or glycolysis that may act as sensitizer for chemoprevention and (neo-)adjuvant therapies in cancer treatment
| Tumor entity | In-vitro/in-vivo mechanism on tumor cells (OSCC/HNSCC/other tumor entities) | Pre-clinical (in-vitro/in-vivo) chemoprevention in cancer development | Clinical data | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compounds (polyphenols*, isothiocyanates, terpen/carotinoid** vitamins, derivates, fatty acids) |
OSCC [66,84]/HNSCC |
other |
Apoptosis↑ (ROS↑ [18,23], Caspasen↑) |
Glykolysis↓ [17] (mTOR↓ [31,85,86], HIF-1↓ [83], enzymes↓) |
PPP↓ [87] (e.g. TKTL1↓) |
|
Successful [88],*** approach in prospective clinical trials |
||||
| |
natural |
synthetic |
|
|
|
|
|
OSCC [66,84]/HNSCC |
other |
OSCC [66,84]/HNSCC |
Other |
| Curcumin* (turmeric) [83,85,88-102] |
X |
- |
Yes [92-95,101] |
Yes |
Yes [96] (ROS↑) [93] |
Yes (mTOR↓ [85,94,102]; HIF-1↓ [83,100]) |
n.d. |
Yes [92-94,101] |
Yes |
Yes (phase I) [95,97] |
Yes (phase II) |
| Resveratrol* (grapes) [85,103-114] |
X |
- |
Yes [104] |
Yes |
Yes, (ROS↑ [107]) |
Yes [109] (mTOR↓ [85,103,110,111]; HIF-1↓ [109]) |
n.d. |
Yes [107,112] |
Yes |
n.d. |
Yes |
|
EGCG* (green tea) [66,84,85,88,104,115-119] |
X |
- |
Yes [66,104,117] |
Yes |
Yes, (ROS↑ [117]) |
Yes (mTOR↓ [85]; HIF-1↓ [118,119]) |
n.d. |
Yes [66,104,116] |
Yes |
Yes (phase II) [66,84,115] |
Yes (phase II) |
| Ellagic acid*, (Pro-) Anthocyanins* (berrys) [66,84,120-127] |
X |
- |
Yes [66,84] |
Yes |
Yes, (ROS↑ [122-124]) |
Yes (mTOR↓ [124,125]; HIF-1↓ [126]) |
n.d. |
Yes [66,84,120,121,127] |
Yes |
Yes (phase II) [66,84] |
Yes |
| Genistein* (soyabeans) [85,88,128-133] |
X |
- |
Yes [128,129] |
Yes |
Yes, (ROS↑ [130]) |
Yes (mTOR↓ [85,131]; HIF-1↓ [132,133]) |
n.d. |
Yes [128,129] |
Yes |
n.d. |
Yes (phase II) |
| Apigenin* (parsley) [134-141] |
X |
- |
Yes [134-137] |
Yes |
Yes, (ROS↑ [135]) |
Yes (mTOR↓ [138]; HIF-1↓ [139,140]) |
n.d. |
Yes [134-136] |
Yes |
n.d. |
Yes |
| No [137] | |||||||||||
| Quercetin* (fruits/vegetables) [141-150] |
X |
- |
Yes [142-144,149,150] |
Yes |
Yes, (ROS↑ [144,145]) |
Yes (mTOR↓ [146,147]; HIF-1↓ [147,148]) |
n.d. |
Yes [142-144,149] |
Yes |
Yes [150] |
Yes |
| ITC, glucosinolates (cruciferous vegetables) [85,151-157] |
X |
- |
Yes [152-154,157] |
Yes |
Yes, (ROS↑ [155]) |
Yes (mTOR↓ [85,151]; HIF-1↓ [157]) |
n.d. |
Yes [152-154,157] |
Yes |
n.d. |
Yes (phase I) |
| Lycopene** (tomato) [83,158-169] |
X |
|
Yes [158,163,164] |
Yes |
Yes (ROS↑↓) [160-162] |
Yes (mTOR↓ [167-169]; HIF-1↓ [83]) |
n.d. |
Yes [158,163,164] |
Yes [163] |
n.d. [163] |
Yes (phase II) |
| Vit. A** (retinoids) [101,170-175] |
X |
- |
Yes [101,170,171,174,175] |
Yes |
Yes, (ROS↑ [172]) |
Yes (mTOR↓ [173]; HIF-1 n.d.) |
n.d. |
Yes [101] |
Yes |
Yes [174,175]; No (phase III) [170,171] |
Yes |
| Vit. D [176-181] |
X |
X [179] |
Yes [177,178] |
Yes |
Yes, (ROS↑ [181]) |
Yes (mTOR↓ [179]; HIF-1↓ [180]) |
n.d. |
Yes [177,178] |
Yes |
n.d. |
Yes |
| Vit. E (γ-T3) [83,176,182-185] |
X |
- |
n.d. |
Yes |
Yes, (ROS↑ [183]) |
Yes (mTOR↓ [184]; HIF-1↓ [182,185]) |
n.d. |
n.d. |
Yes |
n.d. |
n.d. |
| Vit. C + K [83,186-194] |
X |
X |
Yes [186] |
Yes |
Yes (ROS↑ [191]) |
Yes (mTOR↓ [192]; HIF-1↓ [83,187,193]) |
n.d. |
Yes [186] |
Yes |
n.d. |
Yes (Phase I/II) |
| Oxybenfotiamine [195] |
- |
X |
n.d. |
Yes |
Yes |
n.d. |
Yes |
n.d. |
n.d. |
n.d. |
n.d. |
| Benzoquinone (wheat germ extract) [196-198] |
X |
- |
Yes [197] |
Yes |
Yes (Caspasen↑ [197], ROS n.d.) |
Yes [198] (mTOR n.d.; HIF-1 n.d.) |
Yes [197,198] |
n.d. |
Yes |
Yes (Phase II/III) [197] |
Yes (Phase II/III) |
| PUFAs (n-3/n-6 family) [199-203] | X | - | Yes [200,201] | Yes | Yes (ROS↑ [202]) | Yes [199] (mTOR↓ [202]; HIF-1↓ [199,203]) | n.d. | Yes [200] | Yes | Yes (Phase II) [201] | Yes (Phase II) |
Most compounds may decrease glycolytic activity by targeting mTOR/HIF-1 pathway and increase apoptotic activity by ROS generation in cancer cells. Focused on OSCC most experience is available for polyphenols (flavonoids: EGCG, anthocyanins, in bold).
ROS, reactive oxygen species; Caspasen, cysteinyl-aspartate specific protease; OSCC, oral squamous cell carcinoma; HNSCC, head and neck squamous cell carcinoma; HIF, Hypoxia-inducible factor; mTOR, mammalian target of rapamycin; EGCG, epigallocatechin-3-gallate; ITC, isothiocyanate; γ-T3, gamma-tocotrienol; Vit., vitamin; PUFAs, polyunsaturated fatty acids; n.d., no data; ***chemopreventive outcome: decrease of precursor lesions or decrease of cancer biomarkers or decrease of secondary malignancies or increase in patient survival or increase in quality of life or reduction of toxic side effects of radio- and/or chemotherapy. The arrow indicates an increase (↑) or decrease (↓) in levels, phosphorylation status or activity of the different signals.
Lactate, pyruvate, gluthathione, and NADPH generated in glycolysis and/or the PPP effectively scavenge free radicals and ROS, thereby protecting the tumor cell from free radical-mediated DNA damage [26] (e.g. radiation therapy) or other ROS-inducing therapies by natural compounds leading to apoptosis. Most likely, modulation of one pathway will be not effective in most cases [17]. Therefore, synchronous [59] targeting of glycolysis (e.g. carbohydrate-restricted diets [16,204-217] or natural compounds, Table 2) with anti-mitochondrial therapies [18,19,21,23,55,58,61-63,71-74] increasing ROS (Table 2) may act as sensitizer for adjuvant therapies in OSCC or could be useful for chemoprevention. Based on the literature a synergistic effect of a carbohydrate-restricted diet with an anti-mitochondrial therapy can be concluded, since carbohydrate-restricted diets may induce enhanced OXPHOS and lead to inhibition of mTOR [218], which is responsible for synthesis of glycolytic enzymes [30,31]. Specifically observed in patients with head and neck cancer a ketogenic diet decreased the in vivo production of lactate in tumor cells [213].
However, it must be stated that natural compounds like phytochemicals [65,219-225] and vitamins may also prevent ROS-mediated carcinogenicity in cancer chemoprevention. During carcinogenesis ROS may act as a double-edged sword [226]. ROS are important intermediates of cellular signaling that suppress and promote tumorigenesis at once. They make both mitochondrial DNA and nuclear DNA susceptible to damage, and mutations in these two DNA pools are reported to lead to carcinogenesis [227]. However, targeted anti-mitochondrial therapies inducing apoptosis probably require functional active mitochondria without mutations that may respond to radiotherapy/chemo-radiotherapy in OSCC [228].
With specific regard to SDHA and SDHB, vitamin E (α-tocopheryl succinat, target: respiratory complex II in mitochondria) [229] and resveratrol (target: respiratory complex V in mitochondria, ATP synthase) [230] were shown to induce apoptosis in cancer cells. Metformin has been demonstrated to block respiratory complex I in mitochondria [231] as an effective anti-cancer agent [232] and prevented the development of OSCC from carcinogen-induced premalignant lesions [233]. More recently, a synthetic modified thiamine analog oxybenfotiamine [195] specifically inhibits TKTL1 in the PPP [87], of which elevated levels have been detected in the carcinogenesis of OSCC [7]. Targeting the PPP [87] as a detoxifying system [26] may revise tumor hypoxia and resistance to radio- and chemotherapy [7,9]. Therefore, small molecules like oxybenfotiamine [195] provide new opportunities for targeted therapies in cancer and specifically OSCC. Nevertheless, the cytoprotective function of the PPP is not limited to defending against ROS but also expands to helping DNA damage repair [70].
However, it remains unclear whether phytochemicals are standardized effective for chemoprevention [2,17,65,66,84,88,115,120,219,221,223,234,235] in the treatment of precursor lesions or OSCC development as suggested for multistep carcinogenesis [2] but they provide a clear rational for further in-vitro, in-vivo, and clinical studies in the carcinogenesis of OSCC (Table 2) [2,84,88,115,120,219,234-236]. Polyphenols like flavonoids and anthocyanidins have been well investigated in pre-clinical and clinical trials for the treatment of oral precursor lesions and OSCC [84,115,234]. For example, in 1999 Li et al. have already been reported of the chemopreventive impact of green tea on oral leukoplakia with increased rate of partial regression (systemically, oral capsules with 1.2 g polyphenols, and topical tea extract in glycerine over a period of 6 months) [236].
Proliferating cells have intrinsic increased metabolic activities compared to non-proliferating cells [21,69]. This is supported by our data showing a significantly correlation of proliferating cancer cells with both glycolysis-related proteins (GLUT-1, TKTL1), and OXPHOS-related enzymes (SDHA, SDHB, ATP synthase). In this context glycolysis-related proteins may act as detoxifying system [26] (LDHA, TKTL1) of increased ATP producing (and ROS generating) OXPHOS-related proliferating cancer cells. These findings can be clinically addressed by differentiating cancer patients into metabolic responders and non-responders for malignancies such as SCC of the esophagus or head and neck squamous cell carcinoma [237-239].
As for OSCC, there are several reports for glycolysis [9] as the predominant energy metabolism pathway. Glycolysis is involved in aggressive tumor behavior because it causes radio-, and chemotherapy resistance, creates a tumor microenvironment favorable for tumor cell migration, induces angiogenesis, and contributes to the immunologic escape of tumors [26]. However, a previous study by Yi et al. demonstrated that inhibition of the glycolysis-related PFK-1 activity redirects the glucose flux through the PPP [240], thereby conferring a selective growth advantage on cancer cells. Our results are well in line with this hypothesis showing increased TKTL1 expression and decreased PFK-1 expression in OSCC (significant inverse correlation). Zhang et al.[8] presented a similar mechanism describing a metabolic shift from glycolysis into the PPP [67] in OSCC. The authors conclude that the highly robust nature of OSCC metabolism implies that a systematic medical approach targeting multiple metabolic pathways is needed to improve cancer treatment. Downregulation of PFK-1 as observed in our study can be explained by an increase of natural inhibitors such as ATP, which is generated by OXPHOS, and citrate (from the citric acid cycle) that inhibits PFK-1 expression [241]. Therefore, we assume a metabolic shift [8,67,240,241] of glucose from glycolysis towards the PPP mediated by the increased presence of PFK-1 inhibitors like ATP/citrate generated in OXPHOS (indicated by SDHA, SDHB, ATP synthase expression) during the carcinogenesis of OSCC.
If not provided by glycolysis, metabolites (pyruvate) for lactate production are available from amino acids [242]. Amino acid catabolism from the citric acid cycle (e.g. glutaminolysis) supports pyruvate anabolism leading to lactate and NADPH production [69,242]. NADPH, pyruvate, and lactate itself have been proven to scavenge free radicals, thus protecting cancer cells from apoptosis [26]. However, this hypothesis of lactate anabolism through amino acids catabolism requires further studies in OSCC. Glutamine metabolism is also a cancer cell metabolic pathway important for both ATP production and providing intermediates for macromolecular synthesis. However, Glucose, not glutamine, was described as the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells [243]. This result does not automatically exclude lactate generation by amino acid catabolism, as the glutamine pathway has not been described for OSCC and has yet to be revealed. Finally, focusing on combination strategies [116,158,186,244] (Table 2) with different signaling pathways (e.g. mTOR) [245] that have the potential to eradicate malignant and premalignant clones are warranted [245,246].
For the first time, our study provides evidence of increased IGF-1R in OSCC. The expression of IGF-1R has been described for in-vitro analysis of an OSCC cell line [247] but not for carcinogenesis of OSCC yet. The authors state that IGF-1R activation is associated with resistance of EGFR-tyrosine-kinase inhibitor (TKI) treatment. Therefore, targeting IGF-1R pathway, reversal of hyperinsulinemia and IGF by dietry recommendations [16,34,199,204-217,248] or metformin [232] may decrease resistance of EGFR-TKI as well as reduce the risk of cancer recurrence in tumor patients [34].
Conclusions
This study provides the first evidence of the expression of glycolysis-related proteins GLUT-1, HK 2, PFK-1, LDHA, TKTL1 and mitochondrial enzymes SDHA, SDHB, ATP synthase in the multi-step carcinogenesis of OSCC. It seems that both, hypoxia-related glucose metabolism and mitochondrial oxidative phosphorylation characteristics are associated with the carcinogenesis of OSCC. Acidosis and OXPHOS may drive a metabolic shift towards the PPP [67]. Therefore, inhibition of the PPP and glycolysis, as well as targeted anti-mitochondrial therapies (ROS generation) by natural compounds (polyphenol mix, selective vitamins) or synthetic vitamin derivatives (e.g. oxybenfotiamine) may act as sensitizer for apoptosis in cancer cells mediated by adjuvant therapies in OSCC. Summarizing in other words, targeting detoxifying systems (e.g. TKTL1, LDHA) make cancer cells or (oral) precursor lesions more vulnerable to apoptosis.
Abbreviations
ATP: Adenosine Triphosphate; GLUT-1: glucose transporter-1; HK 2: Hexokinase 2; HIF-1: Hypoxia-inducible factor-1; IGF-1R: Insulin-like growth factor-I receptor; IGF: Insulin-like growth factor; LDHA: Lactate dehydrogenase A; mTOR: Mammalian target of rapamycin; OSCC: Oral squamous cell carcinoma; OXPHOS: (Mitochondrial) Oxidative phosphorylation; PFK-1: Phosphofructokinase-1; PPP: Pentose phosphate pathway; RTK: Receptor tyrosine kinase; SDH: Succinate dehydrogenase; SIN: Squamous intraepithelial neoplasia; TKI: Tyrosine-kinase inhibitor; TKTL1: Transketolase-like-1.
Competing interests
The authors have no competing interests.
Authors’ contributions
MG and SR conceived the study, performed the coordination and drafted the manuscript. MC, ML and AM carried out immunohistochemistry studies, cell culture, and western blot analysis. TB analysed histopathological specimen and carried out immunohistochemistry studies. PT and MG performed qPCR analysis. PT and WK carried out the data collection and performed the statistical analyses. All authors read and approved the final manuscript.
Supplementary Material
Hematoxylin-Eosin (H&E) staining. H&E staining shows representative images of normal tissue (A), squamous intraepithelial neoplasia SIN I (B), SIN II (C), SIN III carcinoma in situ(D) and invasive OSCC (E). Original magnification: x100-fold. N.T., normal tissue.
Clonality, host species, dilution, and company of antibodies used for immunohistochemistry and western blot analysis [249-252].
Contributor Information
Martin Grimm, Email: dr.dr.martingrimm@googlemail.com.
Marcel Cetindis, Email: Marcel.Cetindis@med.uni-tuebingen.de.
Max Lehmann, Email: lax.mehmann@gmail.com.
Thorsten Biegner, Email: Thorsten.Biegner@med.uni-tuebingen.de.
Adelheid Munz, Email: adelheid.munz@med.uni-tuebingen.de.
Peter Teriete, Email: pteriete@sanfordburnham.org.
Wiebke Kraut, Email: wiebke.kraut@web.de.
Siegmar Reinert, Email: Siegmar.Reinert@med.uni-tuebingen.de.
Acknowledgements
We thank Julia Grimm for her technical assistance.
References
- Driemel O, Hertel K, Reichert TE, Kosmehl H. [Current classification of precursor lesions of oral squamous cell carcinoma principles of the WHO classification 2005] Mund Kiefer Gesichtschir. 2006;10(2):89–93. doi: 10.1007/s10006-006-0675-3. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Tanaka M, Tanaka T. Oral carcinogenesis and oral cancer chemoprevention: a review. Patholog Res Int. 2011;2011:431246. doi: 10.4061/2011/431246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Grimm M. Prognostic value of clinicopathological parameters and outcome in 484 patients with oral squamous cell carcinoma: microvascular invasion (V+) is an independent prognostic factor for OSCC. Clin Transl Oncol. 2012;14(11):870–880. doi: 10.1007/s12094-012-0867-2. [DOI] [PubMed] [Google Scholar]
- Perez-Sayans M, Suarez-Penaranda JM, Pilar GD, Barros-Angueira F, Gandara-Rey JM, Garcia-Garcia A. Hypoxia-inducible factors in OSCC. Cancer Lett. 2011;313(1):1–8. doi: 10.1016/j.canlet.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- PO D, Jorge CC, Oliveira DT, Pereira MC. Hypoxic condition and prognosis in oral squamous cell carcinoma. Anticancer Res. 2014;34(2):605–612. [PubMed] [Google Scholar]
- Grimm M, Schmitt S, Teriete P, Biegner T, Stenzl A, Hennenlotter J, Muhs HJ, Munz A, Nadtotschi T, Koenig K, Sanger J, Feyen O, Hofmann H, Reinert S, Coy JF. A biomarker based detection and characterization of carcinomas exploiting two fundamental biophysical mechanisms in mammalian cells. BMC Cancer. 2013;13(1):569. doi: 10.1186/1471-2407-13-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chai YD, Brumbaugh J, Liu X, Rabii R, Feng S, Misuno K, Messadi D, Hu S. Oral cancer cells may rewire alternative metabolic pathways to survive from siRNA silencing of metabolic enzymes. BMC Cancer. 2014;14(1):223. doi: 10.1186/1471-2407-14-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Munz A, Teriete P, Nadtotschi T, Reinert S. GLUT-1+/TKTL1+ co-expression predicts poor outcome in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;1:1. doi: 10.1016/j.oooo.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Alfarouk KO, Shayoub ME, Muddathir AK, Elhassan GO, Bashir AH. Evolution of tumor metabolism might reflect carcinogenesis as a reverse evolution process (Dismantling of Multicellularity) Cancers (Basel) 2011;3(3):3002–3017. doi: 10.3390/cancers3033002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. J Exp Med. 2012;209(2):211–215. doi: 10.1084/jem.20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol. 2009;19(1):4–11. doi: 10.1016/j.semcancer.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- Fine EJ, Segal-Isaacson CJ, Feinman RD, Herszkopf S, Romano MC, Tomuta N, Bontempo AF, Negassa A, Sparano JA. Targeting insulin inhibition as a metabolic therapy in advanced cancer: a pilot safety and feasibility dietary trial in 10 patients. Nutrition. 2012;28(10):1028–1035. doi: 10.1016/j.nut.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Keijer J, Bekkenkamp-Grovenstein M, Venema D, Dommels YE. Bioactive food components, cancer cell growth limitation and reversal of glycolytic metabolism. Biochim Biophys Acta. 2011;1807(6):697–706. doi: 10.1016/j.bbabio.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Fulda S. Modulation of apoptosis by natural products for cancer therapy. Planta Med. 2010;76(11):1075–1079. doi: 10.1055/s-0030-1249961. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Mandujano-Tinoco EA, Gallardo-Perez JC, Marin-Hernandez A, Moreno-Sanchez R, Rodriguez-Enriquez S. Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim Biophys Acta. 2013;1833(3):541–551. doi: 10.1016/j.bbamcr.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313(3):459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Gallardo-Perez JC, Marin-Hernandez A, Aguilar-Ponce JL, Mandujano-Tinoco EA, Meneses A, Moreno-Sanchez R. Oxidative phosphorylation as a target to arrest malignant neoplasias. Curr Med Chem. 2011;18(21):3156–3167. doi: 10.2174/092986711796391561. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Canc Res. 2012;18(20):5585–5594. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Kuo CY, Fan CC, Fang WC, Jiang SS, Lo YK, Wang TY, Kao MC, Lee AY. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013;4:e681. doi: 10.1038/cddis.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher EJ, Fierz Y, Ferguson RD, LeRoith D. The pathway from diabetes and obesity to cancer, on the route to targeted therapy. Endocr Pract. 2010;16(5):864–873. doi: 10.4158/EP10098.RA. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha X, Sun Q, Zhang H. mTOR upregulation of glycolytic enzymes promotes tumor development. Cell Cycle. 2011;10(7):1015–1016. doi: 10.4161/cc.10.7.15063. [DOI] [PubMed] [Google Scholar]
- Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18(1):40–50. doi: 10.1158/1078-0432.CCR-11-0998. [DOI] [PubMed] [Google Scholar]
- Emond JA, Pierce JP, Natarajan L, Gapuz LR, Nguyen J, Parker BA, Varki NM, Patterson RE. Risk of breast cancer recurrence associated with carbohydrate intake and tissue expression of IGF-1 receptor. Cancer Epidemiol Biomarkers Prev. 2014;23:1273–1279. doi: 10.1158/1055-9965.EPI-13-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey RB, Hay N. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24(2):213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19(1):17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Uchida M, Kwang-Lee K, Kitamura N, Yoshimura T, Sasabe E, Yamamoto T. Correlation of metabolism/hypoxia markers and fluorodeoxyglucose uptake in oral squamous cell carcinomas. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(4):464–471. doi: 10.1016/j.tripleo.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Wang G, Xu Z, Wang C, Yao F, Li J, Chen C, Sun S. Differential phosphofructokinase-1 isoenzyme patterns associated with glycolytic efficiency in human breast cancer and paracancer tissues. Oncol Lett. 2013;6(6):1701–1706. doi: 10.3892/ol.2013.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Alexander D, Munz A, Hoffmann J, Reinert S. Increased LDH5 expression is associated with lymph node metastasis and outcome in oral squamous cell carcinoma. Clin Exp Metastasis. 2013;30(4):529–540. doi: 10.1007/s10585-012-9557-2. [DOI] [PubMed] [Google Scholar]
- Kim HM, Kim Do H, Jung WH, Koo JS. Metabolic phenotypes in primary unknown metastatic carcinoma. J Transl Med. 2014;12:2. doi: 10.1186/1479-5876-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Kim DH, Jung WH, Koo JS. Expression of metabolism-related proteins in triple-negative breast cancer. Int J Clin Exp Pathol. 2014;7(1):301–312. [PMC free article] [PubMed] [Google Scholar]
- Sobin LH, Ch W. UICC. TNM Classification of Malignant Tumors. 7. Berlin: Springer Verlag; 2010. [Google Scholar]
- Hamilton SR, Aaltonen LA. Pathology and Genetics. Tumours of the Digestive System. Third. Lyon: IARC Press; 2000. [Google Scholar]
- Walker RA. Quantification of immunohistochemistry–issues concerning methods, utility and semiquantitative assessment I. Histopathology. 2006;49(4):406–410. doi: 10.1111/j.1365-2559.2006.02514.x. [DOI] [PubMed] [Google Scholar]
- Grimm M, Kim M, Rosenwald A, von Raden BH, Tsaur I, Meier E, Heemann U, Germer CT, Gasser M, Waaga-Gasser AM. Tumour-mediated TRAIL-Receptor expression indicates effective apoptotic depletion of infiltrating CD8+ immune cells in clinical colorectal cancer. Eur J Cancer. 2010;46(12):2314–2323. doi: 10.1016/j.ejca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119(3):493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari B, Coates PJ, Greenstein BD, Hall PA. In situ end-labelling detects DNA strand breaks in apoptosis and other physiological and pathological states. J Pathol. 1993;170(1):1–8. doi: 10.1002/path.1711700102. [DOI] [PubMed] [Google Scholar]
- Edington KG, Loughran OP, Berry IJ, Parkinson EK. Cellular immortality: a late event in the progression of human squamous cell carcinoma of the head and neck associated with p53 alteration and a high frequency of allele loss. Mol Carcinog. 1995;13(4):254–265. doi: 10.1002/mc.2940130408. [DOI] [PubMed] [Google Scholar]
- Bentz S, Cee A, Endlicher E, Wojtal KA, Naami A, Pesch T, Lang S, Schubert P, Fried M, Weber A, Coy JF, Goelder S, Knuchel R, Hausmann M, Rogler G. Hypoxia Induces the Expression of Transketolase-Like 1 in Human Colorectal Cancer. Digestion. 2013;88(3):182–192. doi: 10.1159/000355015. [DOI] [PubMed] [Google Scholar]
- Key G, Becker MH, Baron B, Duchrow M, Schluter C, Flad HD, Gerdes J. New Ki-67-equivalent murine monoclonal antibodies (MIB 1–3) generated against bacterially expressed parts of the Ki-67 cDNA containing three 62 base pair repetitive elements encoding for the Ki-67 epitope. Lab Invest. 1993;68(6):629–636. [PubMed] [Google Scholar]
- Alexander D, Schafer F, Olbrich M, Friedrich B, Buhring HJ, Hoffmann J, Reinert S. MSCA-1/TNAP selection of human jaw periosteal cells improves their mineralization capacity. Cell Physiol Biochem. 2010;26(6):1073–1080. doi: 10.1159/000323985. [DOI] [PubMed] [Google Scholar]
- Grimm M, Lazariotou M, Kircher S, Stuermer L, Reiber C, Hofelmayr A, Gattenlohner S, Otto C, Germer CT, von Rahden BH. MMP-1 is a (pre-)invasive factor in Barrett-associated esophageal adenocarcinomas and is associated with positive lymph node status. J Transl Med. 2010;8:99. doi: 10.1186/1479-5876-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander D, Hoffmann J, Munz A, Friedrich B, Geis-Gerstorfer J, Reinert S. Analysis of OPLA scaffolds for bone engineering constructs using human jaw periosteal cells. J Mater Sci Mater Med. 2008;19(3):965–974. doi: 10.1007/s10856-007-3351-8. [DOI] [PubMed] [Google Scholar]
- von Rahden BH, Kircher S, Kafka M, Stuermer L, Reiber C, Gattenlohner S, Germer CT, Grimm M. Glucocorticoid-induced TNFR family-related receptor (GITR)-expression in tumor infiltrating leucocytes (TILs) is associated with the pathogenesis of esophageal adenocarcinomas with and without Barrett’s mucosa. Cancer Biomark. 2010;7(6):285–294. doi: 10.3233/CBM-2010-0192. [DOI] [PubMed] [Google Scholar]
- Fulda S. Exploiting mitochondrial apoptosis for the treatment of cancer. Mitochondrion. 2010;10(6):598–603. doi: 10.1016/j.mito.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP, Sotgia F. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10(23):4047–4064. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- Chen V, Staub RE, Fong S, Tagliaferri M, Cohen I, Shtivelman E. Bezielle selectively targets mitochondria of cancer cells to inhibit glycolysis and OXPHOS. PLoS One. 2012;7(2):e30300. doi: 10.1371/journal.pone.0030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475(7355):231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Lee P, Vousden KH, Cheung EC. TIGAR, TIGAR, burning bright. Cancer Metab. 2014;2(1):1. doi: 10.1186/2049-3002-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kioka H, Kato H, Fujikawa M, Tsukamoto O, Suzuki T, Imamura H, Nakano A, Higo S, Yamazaki S, Matsuzaki T, Takafuji K, Asanuma H, Asakura M, Minamino T, Shintani Y, Yoshida M, Noji H, Kitakaze M, Komuro I, Asano Y, Takashima S. Evaluation of intramitochondrial ATP levels identifies G0/G1 switch gene 2 as a positive regulator of oxidative phosphorylation. Proc Natl Acad Sci U S A. 2014;111(1):273–278. doi: 10.1073/pnas.1318547111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3(10):768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- Ding Y, Yao H, Yao Y, Fai LY, Zhang Z. Protection of dietary polyphenols against oral cancer. Nutr. 2013;5(6):2173–2191. doi: 10.3390/nu5062173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG, Newgard CB, Chi JT. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013;1(1):23. doi: 10.1186/2049-3002-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Phan L, Yeung S-C, Lee M-H. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014;11:1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR. Targeting apoptosis with dietary bioactive agents. Exp Biol Med. 2006;231(2):117–129. doi: 10.1177/153537020623100201. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Neuzil J. Mitochondria as targets for cancer therapy. Mol Nutr Food Res. 2009;53(1):9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Moreno-Sanchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol Aspects Med. 2010;31(1):29–59. doi: 10.1016/j.mam.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer & metabolism. 2013;1(1):5. doi: 10.1186/2049-3002-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, An JM, Chung WY, Park KK, Hwang JK, du Kim S, Seo SR, Seo JT. Xanthorrhizol induces apoptosis through ROS-mediated MAPK activation in human oral squamous cell carcinoma cells and inhibits DMBA-induced oral carcinogenesis in hamsters. Phytother Res. 2013;27(4):493–498. doi: 10.1002/ptr.4746. [DOI] [PubMed] [Google Scholar]
- Bhattarai G, Lee YH, Lee NH, Lee IK, Yun BS, Hwang PH, Yi HK. Fomitoside-K from Fomitopsis nigra induces apoptosis of human oral squamous cell carcinomas (YD-10B) via mitochondrial signaling pathway. Biol Pharm Bull. 2012;35(10):1711–1719. doi: 10.1248/bpb.b12-00297. [DOI] [PubMed] [Google Scholar]
- Yen CY, Chiu CC, Haung RW, Yeh CC, Huang KJ, Chang KF, Hseu YC, Chang FR, Chang HW, Wu YC. Antiproliferative effects of goniothalamin on Ca9-22 oral cancer cells through apoptosis, DNA damage and ROS induction. Mutat Res. 2012;747(2):253–258. doi: 10.1016/j.mrgentox.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Gonzales CB, Kirma NB, De La Chapa JJ, Chen R, Henry MA, Luo S, Hargreaves KM. Vanilloids induce oral cancer apoptosis independent of TRPV1. Oral Oncol. 2014;50:437–447. doi: 10.1016/j.oraloncology.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CC, Yang JI, Lee JC, Tseng CN, Chan YC, Hseu YC, Tang JY, Chuang LY, Huang HW, Chang FR, Chang HW. Anti-proliferative effect of methanolic extract of Gracilaria tenuistipitata on oral cancer cells involves apoptosis, DNA damage, and oxidative stress. BMC Complement Altern Med. 2012;12:142. doi: 10.1186/1472-6882-12-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN, Singh HB, Singh A, Naqvi AH, Singh BR. Dietary phytochemicals alter epigenetic events and signaling pathways for inhibition of metastasis cascade: Phytoblockers of metastasis cascade. Cancer Metastasis Rev. 2014. [DOI] [PubMed]
- Upadhyay J, Kesharwani RK, Misra K. Comparative study of antioxidants as cancer preventives through inhibition of HIF-1 alpha activity. Bioinformation. 2009;4(6):233–236. doi: 10.6026/97320630004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriti M, Varoni EM. Chemopreventive potential of flavonoids in oral squamous cell carcinoma in human studies. Nutr. 2013;5(7):2564–2576. doi: 10.3390/nu5072564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anti Cancer Agents Med Chem. 2010;10(7):571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Inhibition of PI3K/Akt/mTOR signaling by natural products. Anti Cancer Agents Med Chem. 2013;13(7):967–970. doi: 10.2174/1871520611313070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Montoya A, Lee WN, Bassilian S, Lim S, Trebukhina RV, Kazhyna MV, Ciudad CJ, Noe V, Centelles JJ, Cascante M. Pentose phosphate cycle oxidative and nonoxidative balance: a new vulnerable target for overcoming drug resistance in cancer. Int J Cancer. 2006;119(12):2733–2741. doi: 10.1002/ijc.22227. [DOI] [PubMed] [Google Scholar]
- Thomasset SC, Berry DP, Garcea G, Marczylo T, Steward WP, Gescher AJ. Dietary polyphenolic phytochemicals–promising cancer chemopreventive agents in humans? A review of their clinical properties. Int J Cancer. 2007;120(3):451–458. doi: 10.1002/ijc.22419. [DOI] [PubMed] [Google Scholar]
- Koeppen H, Yu W, Zha J, Pandita A, Penuel E, Rangell L, Raja R, Mohan S, Patel R, Desai R, Fu L, Do A, Parab V, Xia X, Januario T, Louie SG, Filvaroff E, Shames DS, Wistuba II, Lipkind M, Huang J, Lazarov M, Ramakrishnan V, Amler LC, Phan SC, Patel P, Peterson A, Yauch RL. Biomarker analyses from a placebo-controlled phase ii study evaluating erlotinib {+/-} onartuzumab in advanced non-small-cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res. 2014. [DOI] [PMC free article] [PubMed]
- Naksuriya O, Okonogi S, Schiffelers RM, Hennink WE. Curcumin nanoformulations: a review of pharmaceutical properties and preclinical studies and clinical data related to cancer treatment. Biomaterials. 2014;35(10):3365–3383. doi: 10.1016/j.biomaterials.2013.12.090. [DOI] [PubMed] [Google Scholar]
- Prasad S, Tyagi AK, Aggarwal BB. Recent Developments in delivery, bioavailability, absorption and metabolism of curcumin: the golden pigment from golden spice. Cancer Res Treat. 2014;46(1):2–18. doi: 10.4143/crt.2014.46.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogorski A, Dayan A, Dayan D, Chaushu G, Salo T, Vered M. Nutraceuticals as new treatment approaches for oral cancer–I: Curcumin. Oral Oncol. 2013;49(3):187–191. doi: 10.1016/j.oraloncology.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Kim JY, Cho TJ, Woo BH, Choi KU, Lee CH, Ryu MH, Park HR. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Arch Oral Biol. 2012;57(8):1018–1025. doi: 10.1016/j.archoralbio.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Clark CA, McEachern MD, Shah SH, Rong Y, Rong X, Smelley CL, Caldito GC, Abreo FW, Nathan CO. Curcumin inhibits carcinogen and nicotine-induced Mammalian target of rapamycin pathway activation in head and neck squamous cell carcinoma. Cancer Prev Res. 2010;3(12):1586–1595. doi: 10.1158/1940-6207.CAPR-09-0244. [DOI] [PubMed] [Google Scholar]
- Gao W, Chan JY, Wei WI, Wong TS. Anti-cancer effects of curcumin on head and neck cancers. Anti Cancer Agents Med Chem. 2012;12(9):1110–1116. doi: 10.2174/187152012803529736. [DOI] [PubMed] [Google Scholar]
- Chen J, Wang FL, Chen WD. Modulation of apoptosis-related cell signalling pathways by curcumin as a strategy to inhibit tumor progression. Mol Biol Rep. 2014;41:4583–4594. doi: 10.1007/s11033-014-3329-9. [DOI] [PubMed] [Google Scholar]
- Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, Ko JY, Lin JT, Lin BR, Ming-Shiang W, Yu HS, Jee SH, Chen GS, Chen TM, Chen CA, Lai MK, Pu YS, Pan MH, Wang YJ, Tsai CC, Hsieh CY. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21(4B):2895–2900. [PubMed] [Google Scholar]
- Goel A, Aggarwal BB. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer. 2010;62(7):919–930. doi: 10.1080/01635581.2010.509835. [DOI] [PubMed] [Google Scholar]
- Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, Kakarala M, Carpenter PM, McLaren C, Meyskens FL Jr, Brenner DE. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res. 2011;4(3):354–364. doi: 10.1158/1940-6207.CAPR-10-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Chun YS, Kim SW, Kim MS, Park JW. Curcumin inhibits hypoxia-inducible factor-1 by degrading aryl hydrocarbon receptor nuclear translocator: a mechanism of tumor growth inhibition. Mol Pharmacol. 2006;70(5):1664–1671. doi: 10.1124/mol.106.025817. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Makita H, Ohnishi M, Hirose Y, Wang A, Mori H, Satoh K, Hara A, Ogawa H. Chemoprevention of 4-nitroquinoline 1-oxide-induced oral carcinogenesis by dietary curcumin and hesperidin: comparison with the protective effect of beta-carotene. Cancer Res. 1994;54(17):4653–4659. [PubMed] [Google Scholar]
- Beevers CS, Zhou H, Huang S. Hitting the golden TORget: curcumin’s effects on mTOR signaling. Anti Cancer Agents Med Chem. 2013;13(7):988–994. doi: 10.2174/1871520611313070004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liu F. Targeting mTOR: evaluating the therapeutic potential of resveratrol for cancer treatment. Anti Cancer Agents Med Chem. 2013;13(7):1032–1038. doi: 10.2174/18715206113139990113. [DOI] [PubMed] [Google Scholar]
- Zlotogorski A, Dayan A, Dayan D, Chaushu G, Salo T, Vered M. Nutraceuticals as new treatment approaches for oral cancer: II. Green tea extracts and resveratrol. Oral Oncol. 2013;49(6):502–506. doi: 10.1016/j.oraloncology.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Gescher A, Steward WP, Brown K. Resveratrol in the management of human cancer: how strong is the clinical evidence? Ann N Y Acad Sci. 2013;1290:12–20. doi: 10.1111/nyas.12205. [DOI] [PubMed] [Google Scholar]
- Singh CK, George J, Ahmad N. Resveratrol-based combinatorial strategies for cancer management. Ann N Y Acad Sci. 2013;1290:113–121. doi: 10.1111/nyas.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YA, Lin CH, Chi WH, Wang CY, Hsieh YT, Wei YH, Chen YJ. Resveratrol impedes the stemness, epithelial-mesenchymal transition, and metabolic reprogramming of cancer stem cells in nasopharyngeal carcinoma through p53 activation. Evid Based Complement Altern Med. 2013;2013:590393. doi: 10.1155/2013/590393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24(5A):2783–2840. [PubMed] [Google Scholar]
- Jung KH, Lee JH, Thien Quach CH, Paik JY, Oh H, Park JW, Lee EJ, Moon SH, Lee KH. Resveratrol suppresses cancer cell glucose uptake by targeting reactive oxygen species-mediated hypoxia-inducible factor-1alpha activation. J Nucl Med. 2013;54(12):2161–2167. doi: 10.2967/jnumed.112.115436. [DOI] [PubMed] [Google Scholar]
- Widlund AL, Baur JA, Vang O. mTOR: more targets of resveratrol? Expert Rev Mol Med. 2013;15:e10. doi: 10.1017/erm.2013.11. [DOI] [PubMed] [Google Scholar]
- Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010;285(47):36387–36394. doi: 10.1074/jbc.M110.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta GN, Salamone P, Sprio AE, Di Scipio F, Marinos LM, Sapino S, Carlotti ME, Cavalli R, Di Carlo F. Chemoprevention of 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamster cheek pouch by topical application of resveratrol complexed with 2-hydroxypropyl-beta-cyclodextrin. Oral Oncol. 2010;46(1):42–48. doi: 10.1016/j.oraloncology.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Tome-Carneiro J, Larrosa M, Gonzalez-Sarrias A, Tomas-Barberan FA, Garcia-Conesa MT, Espin JC. Resveratrol and clinical trials: the crossroad from in vitro studies to human evidence. Curr Pharm Des. 2013;19(34):6064–6093. doi: 10.2174/13816128113199990407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Porte C, Voduc N, Zhang G, Seguin I, Tardiff D, Singhal N, Cameron DW. Steady-State pharmacokinetics and tolerability of trans-resveratrol 2000 mg twice daily with food, quercetin and alcohol (ethanol) in healthy human subjects. Clin Pharmacokinet. 2010;49(7):449–454. doi: 10.2165/11531820-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Ramshankar V, Krishnamurthy A. Chemoprevention of oral cancer: Green tea experience. J Nat Sci Biol Med. 2014;5(1):3–7. doi: 10.4103/0976-9668.127272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CM, Chang PY, Tu MG, Lu CC, Kuo SC, Amagaya S, Lee CY, Jao HY, Chen MY, Yang JS. Epigallocatechin gallate sensitizes CAL-27 human oral squamous cell carcinoma cells to the anti-metastatic effects of gefitinib (Iressa) via synergistic suppression of epidermal growth factor receptor and matrix metalloproteinase-2. Oncol Rep. 2012;28(5):1799–1807. doi: 10.3892/or.2012.1991. [DOI] [PubMed] [Google Scholar]
- Tao L, Forester SC, Lambert JD. The role of the mitochondrial oxidative stress in the cytotoxic effects of the green tea catechin, (-)-epigallocatechin-3-gallate, in oral cells. Mol Nutr Food Res. 2014;58(4):665–676. doi: 10.1002/mnfr.201300427. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Tang X, Lu Q, Zhang Z, Rao J, Le AD. Green tea extract and (-)-epigallocatechin-3-gallate inhibit hypoxia- and serum-induced HIF-1alpha protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Mol Cancer Ther. 2006;5(5):1227–1238. doi: 10.1158/1535-7163.MCT-05-0490. [DOI] [PubMed] [Google Scholar]
- Wang H, Bian S, Yang CS. Green tea polyphenol EGCG suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha. Carcinogenesis. 2011;32(12):1881–1889. doi: 10.1093/carcin/bgr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casto BC, Knobloch TJ, Galioto RL, Yu Z, Accurso BT, Warner BM. Chemoprevention of oral cancer by lyophilized strawberries. Anticancer Res. 2013;33(11):4757–4766. [PMC free article] [PubMed] [Google Scholar]
- Vidya Priyadarsini R, Kumar N, Khan I, Thiyagarajan P, Kondaiah P, Nagini S. Gene expression signature of DMBA-induced hamster buccal pouch carcinomas: modulation by chlorophyllin and ellagic acid. PLoS One. 2012;7(4):e34628. doi: 10.1371/journal.pone.0034628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou DX, Fujii M, Terahara N, Yoshimoto M. Molecular mechanisms behind the chemopreventive effects of anthocyanidins. J Biomed Biotechnol. 2004;2004(5):321–325. doi: 10.1155/S1110724304403040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvorovic J, Tramer F, Granzotto M, Candussio L, Decorti G, Passamonti S. Oxidative stress-based cytotoxicity of delphinidin and cyanidin in colon cancer cells. Arch Biochem Biophys. 2010;501(1):151–157. doi: 10.1016/j.abb.2010.05.019. [DOI] [PubMed] [Google Scholar]
- Vanella L, Di Giacomo C, Acquaviva R, Barbagallo I, Cardile V, Kim DH, Abraham NG, Sorrenti V. Apoptotic markers in a prostate cancer cell line: effect of ellagic acid. Oncol Rep. 2013;30(6):2804–2810. doi: 10.3892/or.2013.2757. [DOI] [PubMed] [Google Scholar]
- Lee YK, Lee WS, Kim GS, Park OJ. Anthocyanins are novel AMPKalpha1 stimulators that suppress tumor growth by inhibiting mTOR phosphorylation. Oncol Rep. 2010;24(6):1471–1477. doi: 10.3892/or_00001007. [DOI] [PubMed] [Google Scholar]
- Quintos L, Lee IA, Kim HJ, Lim JS, Park J, Sung MK, Seo YR, Kim JS. Significance of p27 as potential biomarker for intracellular oxidative status. Nut Res Pract. 2010;4(5):351–355. doi: 10.4162/nrp.2010.4.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavitha K, Thiyagarajan P, Rathna Nandhini J, Mishra R, Nagini S. Chemopreventive effects of diverse dietary phytochemicals against DMBA-induced hamster buccal pouch carcinogenesis via the induction of Nrf2-mediated cytoprotective antioxidant, detoxification, and DNA repair enzymes. Biochimie. 2013;95(8):1629–1639. doi: 10.1016/j.biochi.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Park SJ, Kim MJ, Kim YK, Kim SM, Park JY, Myoung H. Combined cetuximab and genistein treatment shows additive anti-cancer effect on oral squamous cell carcinoma. Cancer Lett. 2010;292(1):54–63. doi: 10.1016/j.canlet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Johnson TL, Lai MB, Lai JC, Bhushan A. Inhibition of Cell Proliferation and MAP Kinase and Akt Pathways in Oral Squamous cell Carcinoma by Genistein and Biochanin A. Evid Based Complement Altern Med. 2010;7(3):351–358. doi: 10.1093/ecam/nen011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JI, Shim JH, Kim KH, Choi HS, Kim JW, Lee HG, Kim BY, Park SN, Park OJ, Yoon DY. Sensitization of the apoptotic effect of gamma-irradiation in genistein-pretreated CaSki cervical cancer cells. J Microbiol Biotechnol. 2008;18(3):523–531. [PubMed] [Google Scholar]
- Sahin K, Tuzcu M, Basak N, Caglayan B, Kilic U, Sahin F, Kucuk O. Sensitization of cervical cancer cells to cisplatin by genistein: the role of NFkappaB and Akt/mTOR signaling pathways. J Oncol. 2012;2012:461562. doi: 10.1155/2012/461562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Gupta V, Zhang H, Banerjee S, Kong D, Raffoul JJ, Sarkar FH, Hillman GG. Radiation-induced HIF-1alpha cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer. 2009;124(7):1675–1684. doi: 10.1002/ijc.24015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman GG, Singh-Gupta V. Soy isoflavones sensitize cancer cells to radiotherapy. Free Radic Biol Med. 2011;51(2):289–298. doi: 10.1016/j.freeradbiomed.2011.04.039. [DOI] [PubMed] [Google Scholar]
- Maggioni D, Garavello W, Rigolio R, Pignataro L, Gaini R, Nicolini G. Apigenin impairs oral squamous cell carcinoma growth in vitro inducing cell cycle arrest and apoptosis. Int J Oncol. 2013;43(5):1675–1682. doi: 10.3892/ijo.2013.2072. [DOI] [PubMed] [Google Scholar]
- Chan LP, Chou TH, Ding HY, Chen PR, Chiang FY, Kuo PL, Liang CH. Apigenin induces apoptosis via tumor necrosis factor receptor- and Bcl-2-mediated pathway and enhances susceptibility of head and neck squamous cell carcinoma to 5-fluorouracil and cisplatin. Biochim Biophys Acta. 2012;1820(7):1081–1091. doi: 10.1016/j.bbagen.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Silvan S, Manoharan S. Apigenin prevents deregulation in the expression pattern of cell-proliferative, apoptotic, inflammatory and angiogenic markers during 7,12-dimethylbenz[a]anthracene-induced hamster buccal pouch carcinogenesis. Arch Oral Biol. 2013;58(1):94–101. doi: 10.1016/j.archoralbio.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Swanson HI, Choi EY, Helton WB, Gairola CG, Valentino J. Impact of apigenin and kaempferol on human head and neck squamous cell carcinoma. Oral Surg Oral Med Oral pathol Oral Radiol. 2014;117(2):214–220. doi: 10.1016/j.oooo.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Pelling JC. Targeting the PI3K/Akt/mTOR axis by apigenin for cancer prevention. Anti Cancer Agents Med Chem. 2013;13(7):971–978. doi: 10.2174/18715206113139990119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Gupta S. Apigenin: a promising molecule for cancer prevention. Pharm Res. 2010;27(6):962–978. doi: 10.1007/s11095-010-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melstrom LG, Salabat MR, Ding XZ, Strouch MJ, Grippo PJ, Mirzoeva S, Pelling JC, Bentrem DJ. Apigenin down-regulates the hypoxia response genes: HIF-1alpha, GLUT-1, and VEGF in human pancreatic cancer cells. J Surg Res. 2011;167(2):173–181. doi: 10.1016/j.jss.2010.10.041. [DOI] [PubMed] [Google Scholar]
- Gates MA, Tworoger SS, Hecht JL, De Vivo I, Rosner B, Hankinson SE. A prospective study of dietary flavonoid intake and incidence of epithelial ovarian cancer. Int J Cancer. 2007;121(10):2225–2232. doi: 10.1002/ijc.22790. [DOI] [PubMed] [Google Scholar]
- Makita H, Tanaka T, Fujitsuka H, Tatematsu N, Satoh K, Hara A, Mori H. Chemoprevention of 4-nitroquinoline 1-oxide-induced rat oral carcinogenesis by the dietary flavonoids chalcone, 2-hydroxychalcone, and quercetin. Cancer Res. 1996;56(21):4904–4909. [PubMed] [Google Scholar]
- Chen SF, Nieh S, Jao SW, Liu CL, Wu CH, Chang YC, Yang CY, Lin YS. Quercetin suppresses drug-resistant spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer cells. PLoS One. 2012;7(11):e49275. doi: 10.1371/journal.pone.0049275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma H, Sen S, Singh N. Molecular pathways in the chemosensitization of cisplatin by quercetin in human head and neck cancer. Cancer Biol Ther. 2005;4(9):949–955. doi: 10.4161/cbt.4.9.1908. [DOI] [PubMed] [Google Scholar]
- Kim GT, Lee SH, Kim JI, Kim YM. Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int J Mol Med. 2014;33(4):863–869. doi: 10.3892/ijmm.2014.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anti Cancer Agents Med Chem. 2013;13(7):1025–1031. doi: 10.2174/18715206113139990114. [DOI] [PubMed] [Google Scholar]
- Wang K, Liu R, Li J, Mao J, Lei Y, Wu J, Zeng J, Zhang T, Wu H, Chen L, Huang C, Wei Y. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1alpha-mediated signaling. Autophagy. 2011;7(9):966–978. doi: 10.4161/auto.7.9.15863. [DOI] [PubMed] [Google Scholar]
- Lee DH, Lee YJ. Quercetin suppresses hypoxia-induced accumulation of hypoxia-inducible factor-1alpha (HIF-1alpha) through inhibiting protein synthesis. J Cell Biochem. 2008;105(2):546–553. doi: 10.1002/jcb.21851. [DOI] [PubMed] [Google Scholar]
- Chang WW, Hu FW, Yu CC, Wang HH, Feng HP, Lan C, Tsai LL, Chang YC. Quercetin in elimination of tumor initiating stem-like and mesenchymal transformation property in head and neck cancer. Head Neck. 2013;35(3):413–419. doi: 10.1002/hed.22982. [DOI] [PubMed] [Google Scholar]
- Babaee N, Moslemi D, Khalilpour M, Vejdani F, Moghadamnia Y, Bijani A, Baradaran M, Kazemi MT, Khalilpour A, Pouramir M, Moghadamnia AA. Antioxidant capacity of calendula officinalis flowers extract and prevention of radiation induced oropharyngeal mucositis in patients with head and neck cancers: a randomized controlled clinical study. Daru: journal of Faculty of Pharmacy, Tehran University of Medical Sciences. 2013;21(1):18. doi: 10.1186/2008-2231-21-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JF, Tsai TF, Liao PC, Lin YH, Lin YC, Chen HE, Chou KY, Hwang TI. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis. 2013;34(2):406–414. doi: 10.1093/carcin/bgs359. [DOI] [PubMed] [Google Scholar]
- Shabany K, Chiu PC, Raghian A, Chang KW, Solt DB. Rapid in vivo assay for topical oral cancer chemopreventive agents. Int J Oncol. 2002;21(1):159–164. [PubMed] [Google Scholar]
- Cho NP, Han HS, Leem DH, Choi IS, Jung JY, Kim HJ, Moon KS, Choi KH, Soh Y, Kong G, Cho SD, Choi SH. Sulforaphane enhances caspase-dependent apoptosis through inhibition of cyclooxygenase-2 expression in human oral squamous carcinoma cells and nude mouse xenograft model. Oral Oncol. 2009;45(8):654–660. doi: 10.1016/j.oraloncology.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kojima T, Morishita Y, Mori H. Inhibitory effects of the natural products indole-3-carbinol and sinigrin during initiation and promotion phases of 4-nitroquinoline 1-oxide-induced rat tongue carcinogenesis. Jpn J Cancer Res. 1992;83(8):835–842. doi: 10.1111/j.1349-7006.1992.tb01988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Lew KL, Zeng Y, Xiao H, Marynowski SW, Dhir R, Singh SV. Phenethyl isothiocyanate-induced apoptosis in PC-3 human prostate cancer cells is mediated by reactive oxygen species-dependent disruption of the mitochondrial membrane potential. Carcinogenesis. 2006;27(11):2223–2234. doi: 10.1093/carcin/bgl087. [DOI] [PubMed] [Google Scholar]
- Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, Ye L, Talalay P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: a clinical phase I study. Nutr Cancer. 2006;55(1):53–62. doi: 10.1207/s15327914nc5501_7. [DOI] [PubMed] [Google Scholar]
- Yao H, Wang H, Zhang Z, Jiang BH, Luo J, Shi X. Sulforaphane inhibited expression of hypoxia-inducible factor-1alpha in human tongue squamous cancer cells and prostate cancer cells. Int J Cancer. 2008;123(6):1255–1261. doi: 10.1002/ijc.23647. [DOI] [PubMed] [Google Scholar]
- Camacho-Alonso F, Lopez-Jornet P, Tudela-Mulero MR. Synergic effect of curcumin or lycopene with irradiation upon oral squamous cell carcinoma cells. Oral Dis. 2013;19(5):465–472. doi: 10.1111/odi.12025. [DOI] [PubMed] [Google Scholar]
- Zu K, Mucci L, Rosner BA, Clinton SK, Loda M, Stampfer MJ, Giovannucci E. Dietary lycopene, angiogenesis, and prostate cancer: a prospective study in the prostate-specific antigen era. J Natl Cancer Inst. 2014;106(2):djt430. doi: 10.1093/jnci/djt430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozza P, Parrone N, Simone R, Catalano A. Role of lycopene in the control of ROS-mediated cell growth: implications in cancer prevention. Curr Med Chem. 2011;18(12):1846–1860. doi: 10.2174/092986711795496845. [DOI] [PubMed] [Google Scholar]
- Ettorre A, Frosali S, Andreassi M, Di Stefano A. Lycopene phytocomplex, but not pure lycopene, is able to trigger apoptosis and improve the efficacy of photodynamic therapy in HL60 human leukemia cells. Exp Biol Med. 2010;235(9):1114–1125. doi: 10.1258/ebm.2010.009386. [DOI] [PubMed] [Google Scholar]
- Palozza P, Colangelo M, Simone R, Catalano A, Boninsegna A, Lanza P, Monego G, Ranelletti FO. Lycopene induces cell growth inhibition by altering mevalonate pathway and Ras signaling in cancer cell lines. Carcinogenesis. 2010;31(10):1813–1821. doi: 10.1093/carcin/bgq157. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Shnimizu M, Moriwaki H. Cancer chemoprevention by carotenoids. Molecules. 2012;17(3):3202–3242. doi: 10.3390/molecules17033202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuvaneswari V, Velmurugan B, Balasenthil S, Ramachandran CR, Nagini S. Chemopreventive efficacy of lycopene on 7,12-dimethylbenz[a]anthracene-induced hamster buccal pouch carcinogenesis. Fitoterapia. 2001;72(8):865–874. doi: 10.1016/s0367-326x(01)00321-5. [DOI] [PubMed] [Google Scholar]
- Rahman MA, Amin AR, Shin DM. Chemopreventive potential of natural compounds in head and neck cancer. Nutr Cancer. 2010;62(7):973–987. doi: 10.1080/01635581.2010.509538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seren S, Lieberman R, Bayraktar UD, Heath E, Sahin K, Andic F, Kucuk O. Lycopene in cancer prevention and treatment. Am J Ther. 2008;15(1):66–81. doi: 10.1097/MJT.0b013e31804c7120. [DOI] [PubMed] [Google Scholar]
- Sahin K, Orhan C, Tuzcu M, Sahin N, Ali S, Bahcecioglu IH, Guler O, Ozercan I, Ilhan N, Kucuk O. Orally administered lycopene attenuates diethylnitrosamine-induced hepatocarcinogenesis in rats by modulating Nrf-2/HO-1 and Akt/mTOR pathways. Nutr Cancer. 2014;1:1. doi: 10.1080/01635581.2014.894092. [DOI] [PubMed] [Google Scholar]
- Takeshima M, Ono M, Higuchi T, Chen C, Hara T, Nakano S. Anti-proliferative and apoptosis-inducing activity of lycopene against three subtypes of human breast cancer cell lines. Cancer Sci. 2014;105(3):252–257. doi: 10.1111/cas.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Yuan Y, Vaishnav A, Tessel MA, Nonn L, van Breemen RB. Effects of lycopene on protein expression in human primary prostatic epithelial cells. Cancer Prev Res. 2013;6(5):419–427. doi: 10.1158/1940-6207.CAPR-12-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuri FR, Lee JJ, Lippman SM, Kim ES, Cooper JS, Benner SE, Winn R, Pajak TF, Williams B, Shenouda G, Hodson I, Fu K, Shin DM, Vokes EE, Feng L, Goepfert H, Hong WK. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. J Natl Cancer Inst. 2006;98(7):441–450. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- Perry CF, Stevens M, Rabie I, Yarker ME, Cochrane J, Perry E, Traficante R, Coman W. Chemoprevention of head and neck cancer with retinoids: a negative result. Arch Otolaryngol Head Neck Surg. 2005;131(3):198–203. doi: 10.1001/archotol.131.3.198. [DOI] [PubMed] [Google Scholar]
- Kadara H, Lacroix L, Lotan D, Lotan R. Induction of endoplasmic reticulum stress by the pro-apoptotic retinoid N-(4-hydroxyphenyl)retinamide via a reactive oxygen species-dependent mechanism in human head and neck cancer cells. Cancer Biol Ther. 2007;6(5):705–711. doi: 10.4161/cbt.6.5.3963. [DOI] [PubMed] [Google Scholar]
- Xie H, Zhu F, Huang Z, Lee MH, Kim DJ, Li X, Lim Do Y, Jung SK, Kang S, Li H, Reddy K, Wang L, Ma W, Lubet RA, Bode AM, Dong Z. Identification of mammalian target of rapamycin as a direct target of fenretinide both in vitro and in vivo. Carcinogenesis. 2012;33(9):1814–1821. doi: 10.1093/carcin/bgs234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi G, Sardella A, Bez C, Demarosi F, Carrassi A. Interventions for treating oral leukoplakia. Cochrane Database Syst Rev. 2006;4:CD001829. doi: 10.1002/14651858.CD001829.pub3. [DOI] [PubMed] [Google Scholar]
- Poveda-Roda R, Bagan JV, Jimenez-Soriano Y, Diaz-Fernandez JM, Gavalda-Esteve C. Retinoids and proliferative verrucous leukoplakia (PVL). A preliminary study. Medicina oral, patologia oral y cirugia bucal. 2010;15(1):e3–e9. doi: 10.4317/medoral.15.e3. [DOI] [PubMed] [Google Scholar]
- Haussler MR, Haussler CA, Bartik L, Whitfield GK, Hsieh JC, Slater S, Jurutka PW. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66(10 Suppl 2):S98–S112. doi: 10.1111/j.1753-4887.2008.00093.x. [DOI] [PubMed] [Google Scholar]
- Meier JD, Enepekides DJ, Poirier B, Bradley CA, Albala JS, Farwell DG. Treatment with 1-alpha,25-dihydroxyvitamin D3 (vitamin D3) to inhibit carcinogenesis in the hamster buccal pouch model. Arch Otolaryngol Head Neck Surg. 2007;133(11):1149–1152. doi: 10.1001/archotol.133.11.1149. [DOI] [PubMed] [Google Scholar]
- Abe M, Akeno N, Ohida S, Horiuchi N. Inhibitory effects of 1,25-dihydroxyvitamin D3 and 9-cis-retinoic acid on parathyroid hormone-related protein expression by oral cancer cells (HSC-3) J Endocrinol. 1998;156(2):349–357. doi: 10.1677/joe.0.1560349. [DOI] [PubMed] [Google Scholar]
- Yang J, Ikezoe T, Nishioka C, Ni L, Koeffler HP, Yokoyama A. Inhibition of mTORC1 by RAD001 (everolimus) potentiates the effects of 1,25-dihydroxyvitamin D(3) to induce growth arrest and differentiation of AML cells in vitro and in vivo. Exp Hematol. 2010;38(8):666–676. doi: 10.1016/j.exphem.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Ben-Shoshan M, Amir S, Dang DT, Dang LH, Weisman Y, Mabjeesh NJ. 1alpha,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol Cancer Ther. 2007;6(4):1433–1439. doi: 10.1158/1535-7163.MCT-06-0677. [DOI] [PubMed] [Google Scholar]
- Weitsman GE, Koren R, Zuck E, Rotem C, Liberman UA, Ravid A. Vitamin D sensitizes breast cancer cells to the action of H2O2: mitochondria as a convergence point in the death pathway. Free Radic Biol Med. 2005;39(2):266–278. doi: 10.1016/j.freeradbiomed.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Ling MT, Luk SU, Al-Ejeh F, Khanna KK. Tocotrienol as a potential anticancer agent. Carcinogenesis. 2012;33(2):233–239. doi: 10.1093/carcin/bgr261. [DOI] [PubMed] [Google Scholar]
- Kannappan R, Ravindran J, Prasad S, Sung B, Yadav VR, Reuter S, Chaturvedi MM, Aggarwal BB. Gamma-tocotrienol promotes TRAIL-induced apoptosis through reactive oxygen species/extracellular signal-regulated kinase/p53-mediated upregulation of death receptors. Mol Cancer Ther. 2010;9(8):2196–2207. doi: 10.1158/1535-7163.MCT-10-0277. [DOI] [PubMed] [Google Scholar]
- Tiwari RV, Parajuli P, Sylvester PW. Gamma-Tocotrienol-induced autophagy in malignant mammary cancer cells. Exp Biol Med (Maywood) 2014;239(1):33–44. doi: 10.1177/1535370213511022. [DOI] [PubMed] [Google Scholar]
- Bi S, Liu JR, Li Y, Wang Q, Liu HK, Yan YG, Chen BQ, Sun WG. Gamma-Tocotrienol modulates the paracrine secretion of VEGF induced by cobalt(II) chloride via ERK signaling pathway in gastric adenocarcinoma SGC-7901 cell line. Toxicology. 2010;274(1–3):27–33. doi: 10.1016/j.tox.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Zhang W, Negoro T, Satoh K, Jiang Y, Hashimoto K, Kikuchi H, Nishikawa H, Miyata T, Yamamoto Y, Nakano K, Yasumoto E, Nakayachi T, Mineno K, Satoh T, Sakagami H. Synergistic cytotoxic action of vitamin C and vitamin K3. Anticancer Res. 2001;21(5):3439–3444. [PubMed] [Google Scholar]
- Na YR, Han KC, Park H, Yang EG. Menadione and ethacrynic acid inhibit the hypoxia-inducible factor (HIF) pathway by disrupting HIF-1alpha interaction with p300. Biochem Biophys Res Commun. 2013;434(4):879–884. doi: 10.1016/j.bbrc.2013.04.044. [DOI] [PubMed] [Google Scholar]
- Taper HS. Altered deoxyribonuclease activity in cancer cells and its role in non toxic adjuvant cancer therapy with mixed vitamins C and K3. Anticancer Res. 2008;28(5A):2727–2732. [PubMed] [Google Scholar]
- Lamson DW, Gu YH, Plaza SM, Brignall MS, Brinton CA, Sadlon AE. The vitamin C:vitamin K3 system - enhancers and inhibitors of the anticancer effect. Altern Med Rev. 2010;15(4):345–351. [PubMed] [Google Scholar]
- Lamson DW, Plaza SM. The anticancer effects of vitamin K. Altern Med Rev. 2003;8(3):303–318. [PubMed] [Google Scholar]
- Beck R, Pedrosa RC, Dejeans N, Glorieux C, Leveque P, Gallez B, Taper H, Eeckhoudt S, Knoops L, Calderon PB, Verrax J. Ascorbate/menadione-induced oxidative stress kills cancer cells that express normal or mutated forms of the oncogenic protein Bcr-Abl. An in vitro and in vivo mechanistic study. Invest New Drugs. 2011;29(5):891–900. doi: 10.1007/s10637-010-9441-3. [DOI] [PubMed] [Google Scholar]
- Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6(222):222ra218. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- Kawada H, Kaneko M, Sawanobori M, Uno T, Matsuzawa H, Nakamura Y, Matsushita H, Ando K. High concentrations of L-ascorbic acid specifically inhibit the growth of human leukemic cells via downregulation of HIF-1alpha transcription. PLoS One. 2013;8(4):e62717. doi: 10.1371/journal.pone.0062717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen B, Summers JL, Jamison JM, Neal DR, McGuire K, Gerson L, Diokno A. A 12 week, open label, phase I/IIa study using apatone for the treatment of prostate cancer patients who have failed standard therapy. Int J Med Sci. 2008;5(2):62–67. doi: 10.7150/ijms.5.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy JF. The Usage Of Oxybenfotiamine As A Thiamin Analog In Cancer Therapy. 2014. (Personal Communication).
- Demidov LV, Manziuk LV, Kharkevitch GY, Pirogova NA, Artamonova EV. Adjuvant fermented wheat germ extract (Avemar) nutraceutical improves survival of high-risk skin melanoma patients: a randomized, pilot, phase II clinical study with a 7-year follow-up. Cancer Biother Radiopharm. 2008;23(4):477–482. doi: 10.1089/cbr.2008.0486. [DOI] [PubMed] [Google Scholar]
- Mueller T, Voigt W. Fermented wheat germ extract–nutritional supplement or anticancer drug? Nutr J. 2011;10:89. doi: 10.1186/1475-2891-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comin-Anduix B, Boros LG, Marin S, Boren J, Callol-Massot C, Centelles JJ, Torres JL, Agell N, Bassilian S, Cascante M. Fermented wheat germ extract inhibits glycolysis/pentose cycle enzymes and induces apoptosis through poly(ADP-ribose) polymerase activation in Jurkat T-cell leukemia tumor cells. J Biol Chem. 2002;277(48):46408–46414. doi: 10.1074/jbc.M206150200. [DOI] [PubMed] [Google Scholar]
- Mouradian M, Kikawa KD, Dranka BP, Komas SM, Kalyanaraman B, Pardini RS. Docosahexaenoic acid attenuates breast cancer cell metabolism and the Warburg phenotype by targeting bioenergetic function. Mol Carcinog. 2014. [DOI] [PubMed]
- Nikolakopoulou Z, Nteliopoulos G, Michael-Titus AT, Parkinson EK. Omega-3 polyunsaturated fatty acids selectively inhibit growth in neoplastic oral keratinocytes by differentially activating ERK1/2. Carcinogenesis. 2013;34(12):2716–2725. doi: 10.1093/carcin/bgt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Luis DA, Izaola O, Cuellar L, Terroba MC, de la Fuente B, Cabezas G. A randomized clinical trial with two doses of a omega 3 fatty acids oral and arginine enhanced formula in clinical and biochemical parameters of head and neck cancer ambulatory patients. Eur Rev Med Pharmacol Sci. 2013;17(8):1090–1094. [PubMed] [Google Scholar]
- Shin S, Jing K, Jeong S, Kim N, Song KS, Heo JY, Park JH, Seo KS, Han J, Park JI, Kweon GR, Park SK, Wu T, Hwang BD, Lim K. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. BioMed Res Int. 2013;2013:568671. doi: 10.1155/2013/568671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviello G, Di Nicuolo F, Gragnoli S, Piccioni E, Serini S, Maggiano N, Tringali G, Navarra P, Ranelletti FO, Palozza P. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and -2 and HIF-1alpha induction pathway. Carcinogenesis. 2004;25(12):2303–2310. doi: 10.1093/carcin/bgh265. [DOI] [PubMed] [Google Scholar]
- Abdelwahab MG, Fenton KE, Preul MC, Rho JM, Lynch A, Stafford P, Scheck AC. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS One. 2012;7(5):e36197. doi: 10.1371/journal.pone.0036197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BG, Bhatia SK, Buatti JM, Brandt KE, Lindholm KE, Button AM, Szweda LI, Smith BJ, Spitz DR, Fath MA. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin Cancer Res. 2013;19(14):3905–3913. doi: 10.1158/1078-0432.CCR-12-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement RJ. Calorie or carbohydrate restriction? The ketogenic diet as another option for supportive cancer treatment. Oncologist. 2013;18(9):1056. doi: 10.1634/theoncologist.2013-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer GD, Brucker DP, Bahr O, Harter PN, Hattingen E, Walenta S, Mueller-Klieser W, Steinbach JP, Rieger J. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer. 2011;11:315. doi: 10.1186/1471-2407-11-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavropoulos JC, Isaacs WB, Pizzo SV, Freedland SJ. Is there a role for a low-carbohydrate ketogenic diet in the management of prostate cancer? Urology. 2006;68(1):15–18. doi: 10.1016/j.urology.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Otto C, Kaemmerer U, Illert B, Muehling B, Pfetzer N, Wittig R, Voelker HU, Thiede A, Coy JF. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer. 2008;8:122. doi: 10.1186/1471-2407-8-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli A. Ketogenic diet for obesity: friend or foe? Int J Environ Res Public Health. 2014;11(2):2092–2107. doi: 10.3390/ijerph110202092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poff AM, Ari C, Seyfried TN, D’Agostino DP. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS One. 2013;8(6):e65522. doi: 10.1371/journal.pone.0065522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Pfetzer N, Schwab M, Strauss I, Kammerer U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: a pilot trial. Nutr Metab. 2011;8(1):54. doi: 10.1186/1743-7075-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder U, Himpe B, Pries R, Vonthein R, Nitsch S, Wollenberg B. Decline of lactate in tumor tissue after ketogenic diet: in vivo microdialysis study in patients with head and neck cancer. Nutr Cancer. 2013;65(6):843–849. doi: 10.1080/01635581.2013.804579. [DOI] [PubMed] [Google Scholar]
- Seyfried TN, Marsh J, Shelton LM, Huysentruyt LC, Mukherjee P. Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer? Epilepsy Res. 2012;100(3):310–326. doi: 10.1016/j.eplepsyres.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Klement RJ, Kammerer U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr Metab. 2011;8:75. doi: 10.1186/1743-7075-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, Kratzat S, Purfurst B, Walenta S, Mueller-Klieser W, Graler M, Hummel M, Keller U, Buck AK, Dorken B, Willmitzer L, Reimann M, Kempa S, Lee S, Schmitt CA. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–425. doi: 10.1038/nature12437. [DOI] [PubMed] [Google Scholar]
- Onodera Y, Nam JM, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Invest. 2014;124(1):367–384. doi: 10.1172/JCI63146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Hartman AL, Stafstrom CE, Thio LL. How does the ketogenic diet work? Four potential mechanisms. J Child Neurol. 2013;28(8):1027–1033. doi: 10.1177/0883073813487598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziech D, Anestopoulos I, Hanafi R, Voulgaridou GP, Franco R, Georgakilas AG, Pappa A, Panayiotidis MI. Pleiotrophic effects of natural products in ROS-induced carcinogenesis: the role of plant-derived natural products in oral cancer chemoprevention. Cancer Lett. 2012;327(1–2):16–25. doi: 10.1016/j.canlet.2012.02.025. [DOI] [PubMed] [Google Scholar]
- Lee KW, Bode AM, Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat Rev Cancer. 2011;11(3):211–218. doi: 10.1038/nrc3017. [DOI] [PubMed] [Google Scholar]
- Tan AC, Konczak I, Sze DM, Ramzan I. Molecular pathways for cancer chemoprevention by dietary phytochemicals. Nutr Cancer. 2011;63(4):495–505. doi: 10.1080/01635581.2011.538953. [DOI] [PubMed] [Google Scholar]
- Pratheeshkumar P, Sreekala C, Zhang Z, Budhraja A, Ding S, Son YO, Wang X, Hitron A, Hyun-Jung K, Wang L, Lee JC, Shi X. Cancer prevention with promising natural products: mechanisms of action and molecular targets. Anti Cancer Agents Med Chem. 2012;12(10):1159–1184. doi: 10.2174/187152012803833035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maund SL, Cramer SD. The tissue-specific stem cell as a target for chemoprevention. Stem Cell Rev. 2011;7(2):307–314. doi: 10.1007/s12015-010-9205-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos S. Cancer chemoprevention and chemotherapy: dietary polyphenols and signalling pathways. Mol Nutr Food Res. 2008;52(5):507–526. doi: 10.1002/mnfr.200700326. [DOI] [PubMed] [Google Scholar]
- Li Y, Wicha MS, Schwartz SJ, Sun D. Implications of cancer stem cell theory for cancer chemoprevention by natural dietary compounds. J Nutr Biochem. 2011;22(9):799–806. doi: 10.1016/j.jnutbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JS, Hong MZ, Ren JL. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol. 2009;15(14):1702–1707. doi: 10.3748/wjg.15.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Lai CH, Huang SF, Liao CT, Chen IH, Wang HM, Hsieh LL. Clinical significance in oral cavity squamous cell carcinoma of pathogenic somatic mitochondrial mutations. PLoS One. 2013;8(6):e65578. doi: 10.1371/journal.pone.0065578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, Zobalova R, Turanek J, Spitz DR, Domann FE, Scheffler IE, Ralph SJ, Neuzil J. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008;27(31):4324–4335. doi: 10.1038/onc.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci U S A. 2007;104(34):13632–13637. doi: 10.1073/pnas.0706290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalving M, Gietema JA, Lefrandt JD, de Jong S, Reyners AK, Gans RO, de Vries EG. Metformin: taking away the candy for cancer? Eur J Cancer. 2010;46(13):2369–2380. doi: 10.1016/j.ejca.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Vitale-Cross L, Molinolo AA, Martin D, Younis RH, Maruyama T, Patel V, Chen W, Schneider A, Gutkind JS. Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev Res. 2012;5(4):562–573. doi: 10.1158/1940-6207.CAPR-11-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoni EM, Lodi G, Sardella A, Carrassi A, Iriti M. Plant polyphenols and oral health: old phytochemicals for new fields. Curr Med Chem. 2012;19(11):1706–1720. doi: 10.2174/092986712799945012. [DOI] [PubMed] [Google Scholar]
- Dionne KR, Warnakulasuriya S, Binti Zain R, Cheong SC. Potentially malignant disorders of the oral cavity: Current practice and future directions in the clinic and laboratory. Int J Cancer. 2014. [DOI] [PubMed]
- Li N, Sun Z, Han C, Chen J. The chemopreventive effects of tea on human oral precancerous mucosa lesions. Proc Soc Exp Biol Med. 1999;220(4):218–224. doi: 10.1046/j.1525-1373.1999.d01-37.x. [DOI] [PubMed] [Google Scholar]
- Avril NE, Weber WA. Monitoring response to treatment in patients utilizing PET. Radiol Clin North Am. 2005;43(1):189–204. doi: 10.1016/j.rcl.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Abdelsalam M, Bazarbashi S, Abouzied M, Amin T, Soudy H, Rahal M, Darwish A. Whole body 18 F-FDG PET predicts progression free and overall survival in squamous cell carcinoma of the esophagus: results of a prospective trial. Hematol Oncol Stem Cell Ther. 2010;3(4):179–184. doi: 10.5144/1658-3876.2010.179. [DOI] [PubMed] [Google Scholar]
- Brucher BL, Swisher SG, Konigsrainer A, Zieker D, Hartmann J, Stein H, Kitagawa Y, Law S, Ajani JA. Response to preoperative therapy in upper gastrointestinal cancers. Ann Surg Oncol. 2009;16(4):878–886. doi: 10.1245/s10434-009-0315-x. [DOI] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337(6097):975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol. 2011;76:211–216. doi: 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, Priebe W, Myers JN. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117(13):2926–2938. doi: 10.1002/cncr.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens-Talcott SU, Percival SS. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett. 2005;218(2):141–151. doi: 10.1016/j.canlet.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Vander Broek R, Snow GE, Chen Z, Van Waes C. Chemoprevention of head and neck squamous cell carcinoma through inhibition of NF-kappaB signaling. Oral Oncol. 2013. [DOI] [PMC free article] [PubMed]
- Wirth LJ. Chemoprevention of squamous cell carcinoma of the head and neck: no time to lose momentum. Cancer Prev Res (Phila) 2014;7(3):279–282. doi: 10.1158/1940-6207.CAPR-13-0437. [DOI] [PubMed] [Google Scholar]
- Jameson MJ, Taniguchi LE, VanKoevering KK, Stuart MM, Francom CR, Mendez RE, Beckler AD, Carlson HT, Thomas CY, Khalil AA. Activation of the insulin-like growth factor-1 receptor alters p27 regulation by the epidermal growth factor receptor in oral squamous carcinoma cells. J Oral Pathol Med. 2013;42(4):332–338. doi: 10.1111/jop.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poff AM, Ari C, Arnold P, Seyfried TN, D’Agostino DP. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int J Cancer. 2014;1:1. doi: 10.1002/ijc.28809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granchi C, Paterni I, Rani R, Minutolo F. Small-molecule inhibitors of human LDH5. Future Med Chem. 2013;5(16):1967–1991. doi: 10.4155/fmc.13.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyedotun KS, Lemire BD. The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase. Homology modeling, cofactor docking, and molecular dynamics simulation studies. J Biol Chem. 2004;279(10):9424–9431. doi: 10.1074/jbc.M311876200. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–1715. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hematoxylin-Eosin (H&E) staining. H&E staining shows representative images of normal tissue (A), squamous intraepithelial neoplasia SIN I (B), SIN II (C), SIN III carcinoma in situ(D) and invasive OSCC (E). Original magnification: x100-fold. N.T., normal tissue.
Clonality, host species, dilution, and company of antibodies used for immunohistochemistry and western blot analysis [249-252].