

Abstract
The production of β-lactamase is one of the primary resistance mechanisms used by Gram-negative bacterial pathogens to counter β-lactam antibiotics, such as penicillins, cephalosporins and carbapenems. There is an urgent need to develop novel β-lactamase inhibitors in response to ever evolving β-lactamases possessing an expanded spectrum of β-lactam hydrolyzing activity. Whereas traditional high-throughput screening has proven ineffective against serine β-lactamases, fragment-based approaches have been successfully employed to identify novel chemical matter, which in turn has revealed much about the specific molecular interactions possible in the active site of serine and metallo β-lactamases. In this review, we summarize recent progress in the field, particularly: the identification of novel inhibitor chemotypes through fragment-based screening; the use of fragment-protein structures to understand key features of binding hot spots and inform the design of improved leads; lessons learned and new prospects for β-lactamase inhibitor development using fragment-based approaches.
The β-lactams are the most widely used antibiotics due to their effective inhibition of the penicillin binding proteins, which are essential enzymes in bacterial cell wall synthesis (Figure 1) [1,2]. The primary mediators of bacterial resistance to these antibiotics are β-lactamases, an evolutionarily ancient family of hydrolases that catalyze β-lactam hydrolysis and inactivation [3,4]. β-lactamases can be classified under the Ambler system by sequence similarity and catalytic mechanism, or under the Bush–Jacoby system by their substrate preferences [5,6]. The Ambler system recognizes three families of serine β-lactamases (Class A, C and D) which employ a catalytic serine to mediate the hydrolysis reaction, and a fourth family (Class B) of metallo enzymes.
Figure 1. β-lactam antibiotics and β-lactamase inhibitors.

Compounds represent the main classes of β-lactam antibiotics in clinical use such as penicillins, cephalosporins, including third-generation such as ceftazidime, and carbapenems. Clavulanate is a Class A β-lactamase inhibitor while avibactam and RPX7009 are broad-spectrum covalent inhibitors in clinical trials.
Since the discovery of penicillins, many new β-lactam drugs, such as cephalosporins and carbapenems, have been developed that can evade and/or inhibit β-lactamase hydrolysis, with many acting as mechanism-based suicide substrates for penicillin binding proteins and/or β-lactamases (Figure 1) [1]. Almost invariably however, mutations or new enzymes have emerged that enable the recognition and hydrolysis of the latest generation of β-lactam antibiotics. The CTX-M Class A β-lactamases, the most common of the extended-spectrum β-lactamases (ESBLs), continue to evolve enhanced activity against the third generation cephalosporins [7–11]. Other Class A ESBLs are also of increasing concern clinically [11,12]. In addition, in recent years, carbapenemase activity has been observed in many Class A, B and D enzymes, rendering even the venerable carbapenems ineffective for treating bacterial infections in life threatening situations [13–15].
The use of a β-lactamase inhibitor in combination with a β-lactam antibiotic is a well-established strategy to counter resistance [16]. Classical β-lactamase inhibitors such as clavulanate (Figure 1), tazobactam and sulbactam have been of significant clinical value in this regard, but these inhibitors are primarily effective against Class A enzymes and therefore are not broadly useful for countering the diversity of β-lactamase enzymes present today. These classical inhibitors also contain a β-lactam ring and, as a result, they are susceptible to resistance resulting from upregulation of β-lactamase expression, new β-lactamase acquisition and other mechanisms that have evolved over millions of years of competition between bacteria and β-lactam-producing microorganisms [17–20]. These limitations, as well as the widespread presence of ESBLs and carbapenemases in multidrug-resistant Gram-negative pathogens, have motivated the search for new classes of β-lactamase inhibitors, as reviewed recently by Drawz and Bonomo [16]. Two notable examples include avibactam (previously known as NXL104) [21–23], and RPX7009 [24,25] (Figure 1). These new β-lactamase inhibitors can form a stable covalent bond with the catalytic serine in a wide range of β-lactamases, and thus possess broad-spectrum activity. Avibactam is currently in late-stage clinical trials as cephalosporin or carbapenem combinations. RPX7009 is undergoing Phase I trials in combination with a carbapenem, biapenem. These recent successes demonstrate that novel, non-β-lactam compounds can be employed clinically to inhibit β-lactamases in resistant bacteria [26–29].
Developing novel antimicrobial chemotypes is crucial for addressing bacterial resistance, yet traditional high-throughput screening (HTS) against isolated drug targets has proven largely ineffective [30,31]. For example, 51 out of 67 HTS campaigns on antibacterial targets at GlaxoSmithKline (GSK) failed to yield any hits, and only five from the remaining 16 generated leads suitable for further optimization [30]. HTS against whole bacteria, however, has produced at least one new class of drugs, the oxazolidinones [32,33]. A problem with existing HTS screening libraries is their limited size and biased chemical composition favoring G-protein coupled receptor (GPCR)- and kinase-type ligands [34–37]. Fragment-based approaches instead focus on lower molecular weight (MW <250 Da) compounds that target sub-pockets of a binding site [38], usually with higher ‘ligand efficiency’ (binding energy per heavy atom) than typical HTS hits. Fragments, because of their smaller size, provide a more complete representation of chemical space [39–45]. This drug-discovery paradigm often offers an efficient lead optimization strategy through growing or merging of structurally characterized fragments into high-affinity compounds [46], and has led to several drug candidates in Phase II/III trials and one US FDA approved drug [47].
HTS versus fragment-based methods in inhibitor design against β-lactamases
HTS has produced mixed results against serine and metallo β-lactamases. For serine β-lactamases, HTS has consistently failed to yield meaningful lead compounds. The HTS campaign by GSK against P99 Class C β-lactamase did not uncover any hits out of half a million compounds screened [30]. Another screen of ~70,000 compounds from the NIH Chemical Genomics Center against AmpC Class C β-lactamase led to 12 promiscuous covalent inhibitors but no specific reversible inhibitors [31]. Computational docking methods were slightly more successful against AmpC, producing two inhibitors from 16 compounds chosen from the same NIH Chemical Genomics Center library [31], and three inhibitors from an earlier effort using 56 molecules selected from 200,000 compounds of Available Chemical Database [48]. Similar efforts using the Available Chemical Database against TEM Class A β-lactamases led to two high μM allosteric inhibitors [49].
In contrast to this poor track record against serine β-lactamases, HTS has been applied successfully to identify novel inhibitors against metallo β-lactamases. Although screening of the GSK library against CfiA metallo β-lactamase did not lead to any hits, HTS at Merck [50,51], Meiji Seika Kaisha Ltd [52], and academic groups [53,54] all identified novel chemotypes using large libraries containing hundreds of thousands of compounds. One reason for this success may be the presence in the screening compounds of functional groups such as thiol, carboxylate and tetrazole that can form strong interactions with the zinc center in these metallo-enzymes. This has led to the screening of more focused libraries containing some of these chemotypes [55–57]. The relatively shallow and open active sites of metallo β-lactamases also reduce the chances of steric clashes with potential inhibitors [58–60]. By comparison, there is no single feature in serine β-lactamases that can by itself attribute binding affinity comparable to the metal–ligand interaction in Class B enzymes. It thus becomes less likely that one will find HTS hits of reasonable affinity for serine β-lactamases, since multiple binding interactions would need to be satisfied. However, these very features of serine β-lactamases (multiple binding sub-sites) make them especially attractive targets for fragment-based approaches.
Fragment-based lead discovery (FBLD) has proven in the past several years to be a very promising method to develop new inhibitors against both serine and metallo β-lactamases, including CTX-M Class A [61,62], AmpC Class C [28,63] and IMP-1 Class B enzymes [64,65]. It has been successfully applied in both lead identification to uncover novel chemotypes for non-covalent inhibitors, and in lead optimization to probe additional binding hot spots for covalent inhibitors. Several trends have emerged from these studies.
First, the availability of high quality structural data has played a crucial role in this area [66]. The majority of β-lactamase crystals can diffract to very high resolutions in x-ray crystallographic analysis. Some extraordinary examples include Class A enzymes TEM-1 (0.85 Å) [67], CTX-M-9 (0.88 Å) [68], SHV-2 (0.9 Å) [69], KPC-2 (1.23 Å) [70], Class C enzymes AmpC (1.07 Å) [71], Class D enzymes OXA-10 (1.35 Å, PDB 2X02), and Class B enzymes NDM-1 (1.05 Å, PDB 4HL2). Accordingly, this group of enzymes is ideally suited to the FBLD approach, with the high resolution structures not only providing better templates for computational modeling, but also allowing more accurate determination of fragment binding modes. The low binding affinity of fragment inhibitors often results in their low occupancy in complex crystals and weak signals in electron density maps. For lower resolution crystal structures including those approximately 2.0 Å, it usually means significant ambiguities in the binding conformations of fragment inhibitors. The high quality of β-lactamase crystal structures overcomes this difficulty and the improved accuracy is particularly useful in elaborating fragments into leads via growing or merging fragments [28,61–63].
Second, computational docking technology has matured to the point that it is now very useful for prioritizing fragments for experimental testing [72,73]. The low binding affinity of fragment inhibitors means that they have to be tested at very high concentrations, usually in the high μM or low mM range. Traditional biochemical assays are often plagued by artifacts when performed in these concentration ranges and so biophysical methods such as NMR, x-ray crystallography, and surface plasmon resonance have more often been deployed to screen fragment compounds in a low-throughput fashion, usually testing up to only a few thousand molecules in each screening [66,74–76]. Computational docking allows a rapid initial prioritization of compounds from the approximately half a million commercially available fragment compounds [77], significantly expanding the overall chemical space being sampled in a fragment screening campaign. Despite initial skepticism, computational screens have uncovered novel fragment inhibitors against both Class A and Class C β-lactamases at a hit rate higher than the average of experimental fragment screening, demonstrating the potential of this technique in FBLD against β-lactamases. In addition, molecular docking performed well in predicting fragment binding poses when compared with subsequently determined crystal structures.
Finally, in vitro biochemical assays are available for β-lactamases that allow for the biochemical characterization (Ki) of fragments with very weak affinities. This is in contrast to many other classes of targets, where functional biochemical activity is not easily measured for weak affinity fragments. Many β-lactam substrates have been developed over the years to study β-lactamase activities in the context of bacterial resistance, and these reagents now provide convenient UV absorption-based assays for testing fragments [78]. More substrates are being developed, such as those for fluorescence-based activity assays [79], and this allows the selection of a suitable substrate for a particular β-lactamase and/or the identification of false positive hits by using substrates with different fluorophores. The use of molecular docking also reduces by orders of magnitude the number of compounds that need to be experimentally tested. This enables biochemical assays to be used for primary experimental testing alongside, or in place of, other techniques such as surface plasmon resonance [63].
Class A β-lactamases
Clinically, the Class A β-lactamase CTX-M is the most commonly encountered ESBLs [8]. This very diverse group of enzymes has evolved enhanced activities against third generation cephalosporins such as cefotaxime and ceftazidime. There are five subgroups of CTX-M β-lactamases, CTX-1, CTX-M-2, CTX-M-8, CTX-M-9 and CTX-M-25, based on sequence similarities. The active sites of all these subgroups share high sequence and structural conservation, with a slightly enlarged binding site (e.g., Asp240 vs Glu240 in TEM-1) to accommodate the bulky side chains of extended spectrum β-lactam antibiotics, as well as particular protein residues to enhance substrate binding (e.g., Ser237). Given the importance of this family of enzymes, it is not surprising that CTX-M has been the focus of the most significant FBLD efforts to date on β-lactamase.
Using CTX-M-9 as a model system, a series of fragment screening and optimization experiments have led to the first nM-affinity noncovalent inhibitor against any serine β-lactamase (Figures 2 &3) [61,62]. The program DOCK was employed to screen both the fragment and lead-like subset of the ZINC small molecule database [77,80,81]. Out of 70,000 fragment compounds, 69 were selected for experimental testing and ten were shown to be inhibitors (Figure 2A). In contrast, no inhibitors were found among the 34 candidates chosen from 1,000,000 lead-like compounds. The failure with screening lead-like compounds is likely due to a combination of the inaccuracy of docking methods in general and the more sporadic coverage of chemical space by the larger lead-like compounds. Inadequate sampling of conformational space and errors in scoring functions may pose a larger challenge for docking lead-like compounds than fragments because such problems can be amplified due to the larger number of conformations and interactions that need to be calculated for lead-like compounds. Also, as discussed below in studies using AmpC as a model system, many inhibitor chemotypes may be available at much lower percentages in commercial lead-like databases in comparison to fragment databases, increasing the difficulty of identifying these inhibitors in screening lead-like compounds.
Figure 2. Fragments targeting Class A β-lactamase.

(A) Anionic fragments identified by virtual screening against CTX-M Class A β-lactamase [61]. Many of these fragments also demonstrated activity against the Class C enzyme AmpC. (B) x-ray crystallographic structures of fragments in complex with CTX-M β-lactamase.
Figure 3. Fragment-based drug design against Class A β-lactamase.

Fragment-based approach to develop a lead-like inhibitor against CTX-M-9 β-lactamase.
Five crystal structures of CTX-M-9 were determined in complex with fragment inhibitors to resolutions of 1.5 Å or better. Several inhibitors only appeared in the active site with partial occupancy, and they overlapped with a phosphate and water molecules usually observed in the apo crystal. The value of high-resolution structures for FBLD against β-lactamases is demonstrated by details of the ligand electron density permitting the differentiation of the apo and complex states of the active site. These structures also demonstrated the ability of molecular docking to successfully predict ligand binding conformations, which played a significant role in subsequent lead optimization efforts.
The crystal structures with fragment inhibitors revealed several key binding hot spots (Figure 2B):
The subpocket (Ser130, Thr235) that normally harbors the C3(4)′-carboxylate group of the β-lactam substrates and that is well complemented by the tetrazole ring present in several fragments;
The polar side chain of Asn132, which also hydrogen bonds with the amide bond in β-lactam compounds;
An aromatic/hydrophobic site at the base of the active site formed by Tyr105;
The residues Gly236, Ser237 and Gly238 in the β3 strand.
A subsequent search for commercial tetrazole analogs uncovered a 21 μM inhibitor that served as the template for ensuing fragment-based lead optimization efforts (Figure 3). Interestingly, the 21 μM inhibitor was present in the lead-like subset of the ZINC database but did not rank especially high in the original docking screen, and therefore was not among the 34 docking hits initially analyzed. At this stage, chemical synthesis was employed to explore additional functional groups targeting either of two binding hot spots; a hydrophobic site formed by Pro167, and the side chain of Asp240. Compounds targeting each site were identified, and these exhibited binding affinities of 1–2 μM [62]. Combining these features in new molecules targeting both sites then yielded compounds with sub-μM affinities, including an 89 nM inhibitor that reduced the MIC of cefotaxime by 32–64-fold in clinical Escherichia coli isolates expressing CTX-M-9 (Figure 3). This early effort with CTX-M suggests FBLD can be a powerful platform for probing hot spots in the active sites of other Class A β-lactamases.
Due to the high structural conservation among CTX-M enzymes, the new tetrazole-based scaffolds are expected to inhibit other CTX-M β-lactamases, and some recent unpublished results have confirmed this hypothesis. The tetrazole functional group may also be useful in targeting other serine β-lactamases because the corresponding protein subpocket is mostly conserved in these enzymes. Interestingly, as discussed below, a tetrazole functional group has also been shown to be an effective chelator of the zinc ion in metallo β-lactamases.
Class C β-lactamases
For many years, AmpC Class C β-lactamase has been the subject of novel drug discovery efforts and a model system for investigating fragment-based methods. Overproduction of AmpC confers resistance to cefotaxime and other broad-spectrum β-lactam antibiotics [82], yet unlike CTX-M and Class A ESBLs, AmpC is resistant to classical β-lactamase inhibitors such as clavulanate [83].
A recent virtual screening campaign docked a library of 137,639 fragment compounds against E. coli AmpC [63]. A total of 48 molecules were selected for experimental testing from the top-ranking list and 23 were shown to be inhibitors (Figure 4A). The complex structures of these fragments, together with those from a previous study, highlighted several binding hot spots similar to those revealed by the fragments identified for CTX-M (Figure 4B):
Figure 4. Representative fragments identified for the Class C β-lactamase AmpC.

(A) Many fragments possess similar features, including anionic groups and pendant hydrophobic functionality. (B) x-ray crystallographic structures of fragments in complex with AmpC β-lactamase.
The subpocket (Tyr150, Thr316) for the C3(4)′-carboxylate group of the β-lactam substrates;
The polar side chain of Asn152, which serves as a hydrogen bond donor;
A hydrophobic shelf formed by Tyr150, Leu119, or in some cases Ile291;
The residues Gly317, Ala318 and Thr319 in the β3 strand, including the oxyanion hole formed by the backbone amide groups of the catalytic Ser64 and Ala318.
The fragment inhibitors also revealed some novel binding hot spots such as a distal sub-pocket surrounding Ser212 and Gly320, and suggested the merging of these fragment scaffolds with earlier boronic-acid based covalent inhibitors (Figure 5) [84–87]. These fragment-derived modifications were found to increase binding affinity of the covalent inhibitor by more than 20-fold [28]. Subsequent optimization efforts improved the Ki by an additional 25-fold to 50 pM. The development of this sub nanomolar inhibitor demonstrates the value of FBLD in modifying and improving existing lead compounds, including those identified through traditional screening and medicinal chemistry. The virtual screening study also highlighted molecular docking’s ability to accurately predict ligand binding poses. Four out of the eight structures determined showed close resemblance to the docking predictions, with root-mean-square deviations (RMSDs) ranging from 1.2 to 1.6 Å. Two additional complex structures showed RMSDs of approximately 2.0 Å. The remaining two fragments bound to the protein in conformations dramatically different from the docking predictions, due to protein flexibility not captured by the computational method.
Figure 5. Fragment-based drug design against Class C β-lactamase.

Fragment-merging approach to develop a lead-like inhibitor against AmpC β-lactamase [28,63].
Studies using AmpC as a model system have also produced important insights into the challenges and pitfalls of FBLD methods in general. Babaoglu et al. deconstructed a known lead-like AmpC inhibitor into three fragments and found that the fragments did not adopt the same binding conformation as they did as part of the lead compound [88]. In fact, the fragments interacted with two new sites in the active site. However, as the fragments were combined and elaborated into larger molecules, the new molecules did assume the same binding mode as the original lead-like molecule. These studies underscore the ability of fragments, particularly those with very few functional groups, to assume multiple binding poses. This work thus highlights the value of fragment-based approaches to define multiple binding sub-sites, while pointing to potential pitfalls for fragment optimization when structural information about binding poses is not available.
Several fragments identified from docking to AmpC were also among the high-scoring fragment hits in the screen against CTX-M. Moreover, biochemical testing of fragment hits from the CTX-M studies revealed that they also inhibit AmpC (Figure 2). These results demonstrate that the high hit rate associated with fragment screening is attributable to the ability of fragments to form a diversity of binding orientations and interactions. Despite some similarities between the active sites of the two enzymes, the cross-inhibiting fragments did not necessarily bind to the two proteins in the same orientation. Crystal structures of one compound bound by CTX-M and AmpC showed that the ligand occupies entirely different subpockets in the two enzymes. However, as the crystal structures in both cases capture only the most favorable binding conformation, it is possible that there are other alternative conformations that could be similar between the two proteins. Accordingly, it is likely that a shared fragment binding orientation could be exploited in the design of elaborated molecules that bind both sites with improved affinities.
The virtual screening experiments against AmpC were also used to investigate the chemical space covered by fragments in comparison to lead-like molecules [63]. The commercially available fragment databases have approximately 1% of all possible chemical scaffolds (<17 non-hydrogen atoms) containing validated fragment inhibitors as substructures. In comparison, the available lead-like databases have just 0.0000001% of all compounds (<25 non-hydrogen atoms) containing these fragments. Thus, the significantly better chemical space coverage by fragments is due both to the far fewer number of chemically possible compounds at the fragment level, and to the low specificity of fragment inhibitors as illustrated in these studies.
Class D β-lactamases
Although Class D enzymes include several problematic carbapenemases, FBLD against this group of enzymes has been scarcely investigated [89]. However, some fragment-size molecules, particularly polycarboxylic acids, have been found to bind these proteins and may serve as starting points for future development of novel inhibitors. Interestingly, similar fragments that have previously bound to the Class A enzyme BS3 were used to develop lead-like inhibitors against the Class D enzyme OXA-10. Specifically, citrate, isocitrate and aminocitrate had been previously discovered with modest affinity against BS3, with Ki values of 490 μM, 2200 μM and 250 μM, respectively [90]. Using these fragments as starting points, Beck et al. were able to develop a series of lead-like derivatives (Figure 6A). When tested against a wide range of β-lactamases, several of these derivatives showed binding affinity against OXA-10 in the low μM range. The close similarities between the various polycarboxylate chemotypes suggest their ability to target both Class A and D enzymes (Figure 6B) [91,92].
Figure 6. Fragment-based drug design against classes D β-lactamase.

(A) Fragment-based approach to develop a lead-like inhibitor against OXA-10, a Class D β-lactamase. Initially, the fragments were identified inhibiting the Class A β-lactamase BS3. Lead-like optimization of these fragments allowed for the identification of compounds inhibiting OXA-10 [90]. (B) x-ray crystal structures of Class D, OXA-46 in complex with tartaric acid (PDB ID 3IF6), and Class A BS3 in complex with aminocitrate (PDB ID 3B3X), showing polycarboxylic acid molecules binding to the active sites of both proteins.
Class B β-lactamases
Metallo β-lactamases have recently received much attention due to the emergence of NDM-1 and several other enzymes in Gram-negative pathogens [93]. The broad substrate spectrum of these β-lactamases is of great concern in the medical community, and consequently metallo β-lactamases have been the subject of many drug discovery efforts in recent years [78,94]. Although HTS has proved more effective for metallo β-lactamases than serine β-lactamases, FBLD remains a viable and underexploited alternative approach to uncover novel chemotypes for metallo-enzymes.
Using a chromogenic substrate CENTA, a 500-compound fragment library from May-bridge was screened against Pseudomonas aeruginosa IMP-1 β-lactamase [64]. Ten inhibitors were identified (Figure 7A). Most of these compounds exhibited mixed competitive and uncompetitive inhibition, and molecular docking was used to understand both inhibition mechanisms. One of these compounds functioned mainly as a competitive inhibitor and served as the starting point of a lead-optimization effort (Figure 8) [65]. This resulted in a modest improvement of approximately tenfold in the IC50 of inhibitors of the same chemotype. Interestingly, many precursors for chemical synthesis also showed activity against IMP-1 and they were subjected to further derivatization. These experiments led to an inhibitor with a Ki of 11 μM.
Figure 7. Fragments targeting Class B β-lactamase.

(A) Fragment inhibitors discovered using a fragment-based experimental screening approach against IMP-1 β-lactamase. (B) Fragment inhibitors discovered by virtual screening against a metallo β-lactamase from Bacteroides fragilis. (C) Various fragment inhibitors including thiol derivatives and dicarboxylates.
Figure 8. Fragment-based lead discovery for a Class B β-lactamase.
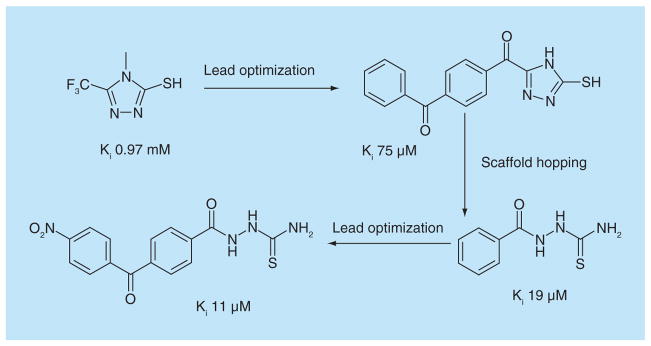
Fragment-based approach to develop a lead-like inhibitor against IMP-1, a metallo β-Lactamase [64,65].
A docking screen of the fragment set in ZINC against a metallo β-lactamase from Bacteroides fragilis uncovered five validated inhibitors among the 15 compounds selected for testing from the 50 highest-scoring hits (Figure 7B) [95]. In addition to the very impressive enrichment rate of true inhibitors in the high-ranking molecules, these inhibitors displayed Ki - values ranging from 120 to 2 μM, significantly better than the more typical high μM to low mM affinities observed for the fragment inhibitors against serine β-lactamases. This result highlights the general ability of metal centers in these enzymes to form strong interactions with specific functional groups (e.g., sulfhydryl) as well as the relative abundance of such moieties in existing small molecule databases. The selectivity exhibited by such inhibitors for metallo β-lactamases over other metallo-enzymes is likely to be an important factor in the further development of such inhibitors if previous experience with other drug targets (e.g., histone deacetylases, matrix metalloproteinases) is any guide.
Many additional fragment-sized inhibitors have been identified from other screening assays not necessarily involving fragment-based approaches (Figure 7C, compounds: 1 [96], 2 [97], 3 [98], 4 [99,100], 5 [101], 6 [65], 7 [102], 8 [103], 9 [104], 10 [52], 11 [105,106], 12 [107], 13 [108,109], 14 [110], 15 [64], 16 [104], 17 [111], 18 [112]). They cover a diverse range of chemotypes such as compounds containing thiol [99] (including mercaptoacetic acids [96,98,113] and mercaptophosphonates [103]) and dicarboxylate groups (including those with heterocyclic rings [104]). Many of these small compounds were identified by traditional screening at much lower concentrations than normally employed for fragment testing. This again reflects the intrinsic affinity of metal centers for metal-chelating functionality. In addition to the zinc ions, these fragments and previously-identified larger inhibitors have revealed other binding hot spots such as the hydrophobic residues on a flexible loop that closes down upon substrate binding as well as a lysine/serine residue that anchors carboxylate groups from substrates/inhibitors. Some of the fragment inhibitors have already been incorporated into larger lead-like molecules, providing good starting scaffolds for future optimization efforts. The different binding modes of D-captopril (Figure 7C, 2) in complex with BlaB subclass B1 [97], CphA subclass B2 [99], and FEZ-1 subclass B3 metallo β-lactamase [114] again demonstrate the ability of fragment inhibitors to adapt to the particular binding environment of a target protein (Figure 9A & B) [97,115]. Comparing the D-captopril-BlaB structure with another complex structure of IMP-1 also suggests that functional groups from D-captopril can be fused with the mercaptocarboxylate inhibitor of IMP-1, underscoring the utility of fragment compounds in probing binding hot spots (Figure 9C) [58,97].
Figure 9. Fragment binding hot spots for Class B β-lactamases.
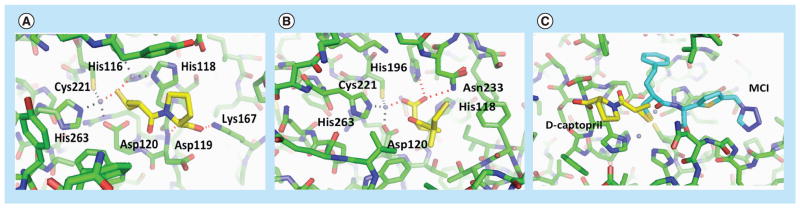
(A) x-ray crystallographic structure of the inhibitor D-captopril in complex with BlaB metallo β-lactamase (PDB ID 1M2X). (B) x-ray crystallographic structure of D-captopril in complex with CphA metallo β-lactamase (PDB ID 2QDS). (C) The binding pose of D-captopril (yellow) from the BlaB complex structure (enzyme structure not shown, PDB ID 1M2X) is superimposed on the complex structure of IMP-1 with MCI (cyan), a mercaptocarboxylate inhibitor (PDB ID 1DD6). The IMP-1 residues are in green. The zinc ions are in grey.
Perspective
In the past several years fragment-based approaches have been successfully applied to identify novel inhibitors for both serine and metallo β-lactamases. The strength of FBLD lies in its ability to effectively probe protein binding hot spots and efficiently identify fragment compounds complementing these subpockets. When structural information is also obtained, the elaboration of weak binding fragments into more potent lead compounds can occur rapidly, with modest synthetic chemistry resources. As still more β-lactamase fragments are identified and characterized, the specific molecular recognition properties and ‘druggability’ of the various sub-sites will become better understood. New fragments for β-lactamases are likely to originate both from denovo screening as detailed herein, and from the deconstruction of existing inhibitors identified through traditional non-fragment based approaches. Most importantly, testing these various fragments and leads across classes of β-lactamases and their mutants will reveal which hot spots are best conserved across enzymes, and might therefore be employed in developing expanded spectrum inhibitors. From the early efforts detailed herein, it is already clear that viable inhibitors of β-lactamases can be drawn from a much wider swath of chemical space than has been previously appreciated. Of course, much more work will be required to advance these new leads towards molecules that can be deployed clinically to counter bacterial resistance in Gram-negative pathogens.
Executive summary.
Introduction
β-lactamases hydrolyze β-lactam antibiotics including penicillins, cephalosporins and carbapenems, representing one of the primary resistance mechanisms used by Gram-negative bacterial pathogens against these antibiotics.
Four classes of β-lactamases are commonly found in resistant bacteria. Class A, C and D use a catalytic serine to catalyze the reaction, whereas Class B are metallo enzymes.
High-throughput screening versus fragment-based lead discovery against β-lactamases
High-throughput screening has been ineffective in identifying novel inhibitor chemotypes against serine β-lactamases, but has been used with some success in inhibitor discovery against metallo β-lactamases.
Fragment-based approaches use low-molecular-weight compounds (<250 Da) to probe small molecule binding sites in proteins and enzymes. Their small size allows for better coverage of chemical space than larger drug-like molecules. Fragments can be converted into more potent lead compounds by employing fragment linking or growing strategies, which are facilitated when fragments are structurally characterized.
Fragment-based lead discovery (FBLD) against β-lactamases have benefitted from the high quality crystal structures of these enzymes, many of which can be determined in the range of ~1–1.5 Å.
Molecular docking has played an important role in prioritizing fragments for experimental testing.
Biochemical inhibition assays have been used as primary assays for fragment testing in inhibitor discovery against β-lactamases.
FBLD case studies against four classes of β-lactamases
FBLD has been applied to identify a novel tetrazole-based inhibitor scaffold against CTX-M Class A β-lactamase and to develop the first nM-affinity non-covalent inhibitor of a serine β-lactamase.
Fragment inhibitors were used to probe protein binding hot spots of AmpC Class C β-lactamase, and to uncover new functional groups that were merged with an existing boronic acid covalent inhibitor scaffold. This led to a sub-nM inhibitor.
Fragment binders were also identified against Class B and D β-lactamases. Lead optimization resulted in μM-affinity inhibitors.
Acknowledgments
The authors thank E Lewandowski and O Pemberton for reading the manuscript.
Key Terms
- Extended-spectrumβ-lactamases
Group of enzymes that are able to hydrolyze not only penicillins and early-generation cephalosporins, but also third-generation extended-spectrum cephalosporins (e.g., ceftazidime) and the monobactam aztreonam
- High-throughput screening
Experimental procedure that uses robotic systems and software to quickly test and analyze the effects of individual compounds from a large library on target proteins or organisms. These experiments usually focus on lead-like and drug-like molecules and can screen millions of compounds
- Fragment-based lead discovery
Experimental approach to lead discovery that seeks to first identify small organic fragments (typically <250 Da) that bind to a drug target of interest. These fragments can then be linked or grown into larger inhibitors with higher affinity and specificity
- ZINC
Free online database containing >20 million commercially available compounds grouped into various subsets, including fragment-like and lead-like databases
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
This work was supported by the NIH (grant AI103158). The authors have a US patent on the tetrazole-based inhibitors against CTX-M Class A β-lactamase. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Testero SA, Fisher JF, Mobashery S. Beta-lactam antibiotics. In: Abraham DJ, Rotella DP, editors. Burger’s Medicinal Chemistry, Drug Discovery and Development. Wiley and Sons; NJ, USA: 2010. pp. 259–404. [Google Scholar]
- 2.Llarrull LI, Testero SA, Fisher JF, Mobashery S. The future of the beta-lactams. Curr Opin Microbiol. 2010;13(5):551–557. doi: 10.1016/j.mib.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frere JM. Beta-lactamases and bacterial resistance to antibiotics. Mol Microbiol. 1995;16(3):385–395. doi: 10.1111/j.1365-2958.1995.tb02404.x. [DOI] [PubMed] [Google Scholar]
- 4.Taubes G. The bacteria fight back. Science. 2008;321(5887):356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- 5.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39(6):1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livermore DM. Beta-lactamases in laboratory and clinical resistance. Clin Microbiol Rev. 1995;8(4):557–584. doi: 10.1128/cmr.8.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev. 2005;105(2):395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet R. Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother. 2004;48(1):1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Delmas J, Sirot J, Shoichet B, Bonnet R. Atomic resolution structures of CTX-M beta-lactamases: extended spectrum activities from increased mobility and decreased stability. J Mol Biol. 2005;348(2):349–362. doi: 10.1016/j.jmb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Shoichet B, Bonnet R. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J Am Chem Soc. 2005;127(15):5423–5434. doi: 10.1021/ja042850a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradford PA. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev. 2001;14(4):933–951. doi: 10.1128/CMR.14.4.933-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez F, Endimiani A, Hujer KM, Bonomo RA. The continuing challenge of ESBLs. Curr Opin Pharmacol. 2007;7(5):459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Queenan AM, Shang W, Flamm R, Bush K. Hydrolysis and inhibition profiles of beta-lactamases from molecular classes A to D with doripenem, imipenem, and meropenem. Antimicrob Agents Chemother. 2010;54(1):565–569. doi: 10.1128/AAC.01004-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel G, Bonomo RA. Status report on carbapenemases: challenges and prospects. Expert Rev Anti Infect Ther. 2012;9(5):555–570. doi: 10.1586/eri.11.28. [DOI] [PubMed] [Google Scholar]
- 15.Babic M, Hujer AM, Bonomo RA. What’s new in antibiotic resistance? Focus on beta-lactamases. Drug Resist Updat. 2006;9(3):142–156. doi: 10.1016/j.drup.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 16▪▪.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23(1):160–201. doi: 10.1128/CMR.00037-09. Comprehensive review of different classes of β-lactamase inhibitors in clinical use or under development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett PM, Chopra I. Molecular basis of beta-lactamase induction in bacteria. Antimicrob Agents Chemother. 1993;37(2):153–158. doi: 10.1128/aac.37.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobs C, Frere JM, Normark S. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in gram-negative bacteria. Cell. 1997;88:823–832. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 19.Petrosino J, Cantu C, 3rd, Palzkill T. Beta-lactamases: protein evolution in real time. Trends Microbiol. 1998;6(8):323–327. doi: 10.1016/s0966-842x(98)01317-1. [DOI] [PubMed] [Google Scholar]
- 20.Pages JM, Lavigne JP, Leflon-Guibout V, et al. Efflux pump, the masked side of beta-lactam resistance in Klebsiella pneumoniae clinical isolates. PloS ONE. 2009;4(3):e4817. doi: 10.1371/journal.pone.0004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehmann DE, Jahic H, Ross PL, et al. Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor. Proc Natl Acad Sci USA. 2012;109(29):11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lahiri SD, Mangani S, Durand-Reville T, et al. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC beta-lactamases. Antimicrob Agents Chemother. 2013;57(6):2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehmann DE, Jahic H, Ross PL, et al. Kinetics of avibactam inhibition against class A, C, and D beta-lactamases. J Biol Chem. 2013;288(39):27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein EJ, Citron DM, Tyrrell KL, Merriam CV. In vitro activity of Biapenem plus RPX7009, a carbapenem combined with a serine beta-lactamase inhibitor, against anaerobic bacteria. Antimicrob Agents Chemother. 2013;57(6):2620–2630. doi: 10.1128/AAC.02418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livermore DM, Mushtaq S. Activity of biapenem (RPX2003) combined with the boronate beta-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J Antimicrob Chemother. 2013;68(8):1825–1831. doi: 10.1093/jac/dkt118. [DOI] [PubMed] [Google Scholar]
- 26.Rahil J, Pratt RF. Phosphonate monoester inhibitors of class A beta-lactamases. Biochem J. 1991;275(Pt 3):793–795. doi: 10.1042/bj2750793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drawz SM, Babic M, Bethel CR, et al. Inhibition of the class C beta-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry. 2010;49(2):329–340. doi: 10.1021/bi9015988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28▪.Eidam O, Romagnoli C, Dalmasso G, et al. Fragment-guided design of subnanomolar beta-lactamase inhibitors active in vivo. Proc Natl Acad Sci USA. 2012;109(43):17448–17453. doi: 10.1073/pnas.1208337109. An interesting example of how fragment binders offer insights for optimization of existing inhibitors against Class C β-lactamases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyrembak PN, Babaoglu K, Pelto RB, Shoichet BK, Pratt RF. O-aryloxycarbonyl hydroxamates: new beta-lactamase inhibitors that cross-link the active site. J Am Chem Soc. 2007;129(31):9548–9549. doi: 10.1021/ja072370u. [DOI] [PubMed] [Google Scholar]
- 30.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 31.Babaoglu K, Simeonov A, Irwin JJ, et al. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51(8):2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renslo AR. Antibacterial oxazolidinones: emerging structure-toxicity relationships. Expert Rev Anti Infect Ther. 2010;8(5):565–574. doi: 10.1586/eri.10.26. [DOI] [PubMed] [Google Scholar]
- 33.Brickner SJ, Barbachyn MR, Hutchinson DK, Manninen PR. Linezolid (ZYVOX), the first member of a completely new class of antibacterial agents for treatment of serious gram-positive infections. J Med Chem. 2008;51(7):1981–1990. doi: 10.1021/jm800038g. [DOI] [PubMed] [Google Scholar]
- 34.O’shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem. 2008;51(10):2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 35.Hert J, Irwin JJ, Laggner C, Keiser MJ, Shoichet BK. Quantifying biogenic bias in screening libraries. Nat Chem Biol. 2009;5(7):479–483. doi: 10.1038/nchembio.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fink T, Reymond JL. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J Chem Inf Model. 2007;47(2):342–353. doi: 10.1021/ci600423u. [DOI] [PubMed] [Google Scholar]
- 37.Bohacek RS, Mcmartin C, Guida WC. The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev. 1996;16(1):3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Jhoti H, Williams G, Rees DC, Murray CW. The ‘rule of three’ for fragment-based drug discovery: where are we now? Nat Rev Drug Discov. 2013;12(8):644. doi: 10.1038/nrd3926-c1. [DOI] [PubMed] [Google Scholar]
- 39▪.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3(8):660–672. doi: 10.1038/nrd1467. Introduces key concepts of fragment-based lead discovery (FBLD) including comparison with high-throughput screening (HTS) [DOI] [PubMed] [Google Scholar]
- 40.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6(3):211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 41.Schulz MN, Hubbard RE. Recent progress in fragment-based lead discovery. Curr Opin Pharmacol. 2009;9(5):615–621. doi: 10.1016/j.coph.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 42.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 43.Pellecchia M, Bertini I, Cowburn D, et al. Perspectives on NMR in drug discovery: a technique comes of age. Nat Rev Drug Discov. 2008;7(9):738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartshorn MJ, Murray CW, Cleasby A, Frederickson M, Tickle IJ, Jhoti H. Fragment-based lead discovery using x-ray crystallography. J Med Chem. 2005;48(2):403–413. doi: 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney ZK, Acharya S, Briggs A, et al. Discovery of triazolinone non-nucleoside inhibitors of HIV reverse transcriptase. Bioorg Med Chem Lett. 2008;18(15):4348–4351. doi: 10.1016/j.bmcl.2008.06.080. [DOI] [PubMed] [Google Scholar]
- 46.Scott DE, Coyne AG, Hudson SA, Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry. 2012;51(25):4990–5003. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- 47▪.Baker M. Fragment-based lead discovery grows up. Nat Rev Drug Discov. 2013;12(1):5–7. doi: 10.1038/nrd3926. General summary of recent successes using FBLD including those in clinical trials. [DOI] [PubMed] [Google Scholar]
- 48.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure. 2002;10(7):1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 49.Horn JR, Shoichet BK. Allosteric inhibition through core disruption. J Mol Biol. 2004;336(5):1283–1291. doi: 10.1016/j.jmb.2003.12.068. [DOI] [PubMed] [Google Scholar]
- 50.Toney JH, Hammond GG, Fitzgerald PM, et al. Succinic acids as potent inhibitors of plasmid-borne IMP-1 metallo-beta-lactamase. J Biol Chem. 2001;276(34):31913–31918. doi: 10.1074/jbc.M104742200. [DOI] [PubMed] [Google Scholar]
- 51.Toney JH, Fitzgerald PM, Grover-Sharma N, et al. Antibiotic sensitization using biphenyl tetrazoles as potent inhibitors of Bacteroides fragilis metallo-beta-lactamase. Chem Biol. 1998;5(4):185–196. doi: 10.1016/s1074-5521(98)90632-9. [DOI] [PubMed] [Google Scholar]
- 52.Hiraiwa Y, Morinaka A, Fukushima T, Kudo T. Metallo-beta-lactamase inhibitory activity of phthalic acid derivatives. Bioorg Med Chem Lett. 2009;19(17):5162–5165. doi: 10.1016/j.bmcl.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 53.Moloughney JGD, Thomas J, Toney JH. Novel IMP-1 metallo-beta-lactamase inhibitors can reverse meropenem resistance in Escherichia coli expressing IMP-1. FEMS Microbiol Lett. 2005;243(1):65–71. doi: 10.1016/j.femsle.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 54.Minond D, Saldanha SA, Spicer T, et al. Probe Reports from the NIH Molecular Libraries Program [Internet] 2010. HTS assay for discovery of novel metallo-beta-lactamase (MBL) inhibitors. [PubMed] [Google Scholar]
- 55.Sun Q, Law A, Crowder MW, Geysen HM. Homo-cysteinyl peptide inhibitors of the L1 metallo-beta-lactamase, and SAR as determined by combinatorial library synthesis. Bioorg Med Chem Lett. 2006;16(19):5169–5175. doi: 10.1016/j.bmcl.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 56.Lienard BM, Huting R, Lassaux P, Galleni M, Frere JM, Schofield CJ. Dynamic combinatorial mass spectrometry leads to metallo-beta-lactamase inhibitors. J Med Chem. 2008;51(3):684–688. doi: 10.1021/jm070866g. [DOI] [PubMed] [Google Scholar]
- 57.Minond D, Saldanha SA, Subramaniam P, et al. Inhibitors of VIM-2 by screening pharmacologically active and click-chemistry compound libraries. Bioorg Med Chem. 2009;17(14):5027–5037. doi: 10.1016/j.bmc.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Concha NO, Janson CA, Rowling P, et al. Crystal structure of the IMP-1 metallo beta-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry. 2000;39(15):4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 59.Zhang H, Hao Q. Crystal structure of NDM-1 reveals a common beta-lactam hydrolysis mechanism. FASEB J. 2011;25(8):2574–2582. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 60.King DT, Worrall LJ, Gruninger R, Strynadka NC. New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition. J Am Chem Soc. 2012;134(28):11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 61▪.Chen Y, Shoichet BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat Chem Biol. 2009;5(5):358–364. doi: 10.1038/nchembio.155. Insightful case of how FBLD can identify novel inhibitor chemotypes against challenging targets such as Class A β-lactamases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nichols DA, Jaishankar P, Larson W, et al. Structure-based design of potent and ligand-efficient inhibitors of CTX-M Class A beta-lactamase. J Med Chem. 2012;55(5):2163–2172. doi: 10.1021/jm2014138. [DOI] [PubMed] [Google Scholar]
- 63.Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for fragment inhibitors of AmpC beta-lactamase. Proc Natl Acad Sci USA. 2009;106(18):7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vella P, Hussein WM, Leung EW, et al. The identification of new metallo-beta-lactamase inhibitor leads from fragment-based screening. Bioorg Med Chem Lett. 2011;21(11):3282–3285. doi: 10.1016/j.bmcl.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 65.Faridoon, Hussein WM, Vella P, et al. 3-mercapto-1,2,4-triazoles and N-acylated thiosemicarbazides as metallo-beta-lactamase inhibitors. Bioorg Med Chem Lett. 2012;22(1):380–386. doi: 10.1016/j.bmcl.2011.10.116. [DOI] [PubMed] [Google Scholar]
- 66.Murray CW, Blundell TL. Structural biology in fragment-based drug design. Curr Opin Struct Biol. 2010;20(4):497–507. doi: 10.1016/j.sbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Minasov G, Wang X, Shoichet BK. An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc. 2002;124(19):5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Bonnet R, Shoichet BK. The acylation mechanism of CTX-M beta-lactamase at 0.88 Angstrom resolution. J Am Chem Soc. 2007;129(17):5378–5380. doi: 10.1021/ja0712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. Ultrahigh resolution structure of a class A beta-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J Mol Biol. 2003;328(1):289–301. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 70.Petrella S, Ziental-Gelus N, Mayer C, Renard M, Jarlier V, Sougakoff W. Genetic and structural insights into the dissemination potential of the extremely broad-spectrum class A beta-lactamase KPC-2 identified in an Escherichia coli strain and an Enterobacter cloacae strain isolated from the same patient in France. Antimicrob Agents Chemother. 2008;52(10):3725–3736. doi: 10.1128/AAC.00163-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Y, Minasov G, Roth TA, Prati F, Shoichet BK. The deacylation mechanism of AmpC beta-lactamase at ultrahigh resolution. J Am Chem Soc. 2006;128(9):2970–2976. doi: 10.1021/ja056806m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Y, Pohlhaus DT. In silico docking and scoring of fragments. Drug Discov Today Technol. 2010;7(3):149–156. doi: 10.1016/j.ddtec.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 73.Desjarlais RL. Using computational techniques in fragment-based drug discovery. Methods Enzymol. 2011;493:137–155. doi: 10.1016/B978-0-12-381274-2.00006-6. [DOI] [PubMed] [Google Scholar]
- 74▪.Murray CW, Verdonk ML, Rees DC. Experiences in fragment-based drug discovery. Trends Pharmacol Sci. 2012;33(5):224–232. doi: 10.1016/j.tips.2012.02.006. Includes in-depth and updated discussion of key concepts and challenges of FBLD. [DOI] [PubMed] [Google Scholar]
- 75.Giannetti AM. From experimental design to validated hits a comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods Enzymol. 2011;493:169–218. doi: 10.1016/B978-0-12-381274-2.00008-X. [DOI] [PubMed] [Google Scholar]
- 76.Lepre CA. Practical aspects of NMR-based fragment screening. Methods Enzymol. 2011;493:219–239. doi: 10.1016/B978-0-12-381274-2.00009-1. [DOI] [PubMed] [Google Scholar]
- 77.Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78▪.Fast W, Sutton LD. Metallo-beta-lactamase: inhibitors and reporter substrates. Biochim Biophys Acta. 2013;1834(8):1648–1659. doi: 10.1016/j.bbapap.2013.04.024. Summarizes recent development of both inhibitors against metallo enzymes and reporter substrates for β-lactamases in general. [DOI] [PubMed] [Google Scholar]
- 79.Van Berkel SS, Brem J, Rydzik AM, et al. Assay platform for clinically relevant metallo-beta-lactamases. J Med Chem. 2013;56(17):6945–6953. doi: 10.1021/jm400769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meng E, Shoichet Bk, Kuntz ID. Automated docking with grid-based energy evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 81.Lorber DM, Udo MK, Shoichet BK. Protein–protein docking with multiple ligand residue conformations and multiple residue identities. Protein Sci. 2002;11:1393–1408. doi: 10.1110/ps.2830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009;22(1):161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pai H, Kang CI, Byeon JH, et al. Epidemiology and clinical features of bloodstream infections caused by AmpC-type-beta-lactamase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 2004;48(10):3720–3728. doi: 10.1128/AAC.48.10.3720-3728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caselli E, Powers RA, Blasczcak LC, Wu CY, Prati F, Shoichet BK. Energetic, structural, and antimicrobial analyses of beta-lactam side chain recognition by beta-lactamases. Chem Biol. 2001;8(1):17–31. doi: 10.1016/s1074-5521(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 85.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Structures of ceftazidime and its transition-state analogue in complex with AmpC beta-lactamase: implications for resistance mutations and inhibitor design. Biochemistry. 2001;40(31):9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 86.Morandi F, Caselli E, Morandi S, et al. Nanomolar inhibitors of AmpC beta-lactamase. J Am Chem Soc. 2003;125(3):685–695. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 87.Morandi S, Morandi F, Caselli E, Shoichet BK, Prati F. Structure-based optimization of cephalothin-analogue boronic acids as beta-lactamase inhibitors. Bioorg Med Chem. 2008;16(3):1195–1205. doi: 10.1016/j.bmc.2007.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Babaoglu K, Shoichet BK. Deconstructing fragment-based inhibitor discovery. Nat Chem Biol. 2006;2(12):720–723. doi: 10.1038/nchembio831. [DOI] [PubMed] [Google Scholar]
- 89.Leonard DA, Bonomo RA, Powers RA. Class D beta-lactamases: a reappraisal after five decades. Acc Chem Res. 2013;46(11):2407–2415. doi: 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beck J, Vercheval L, Bebrone C, Herteg-Fernea A, Lassaux P, Marchand-Brynaert J. Discovery of novel lipophilic inhibitors of OXA-10 enzyme (class D beta-lactamase) by screening amino analogs and homologs of citrate and isocitrate. Bioorg Med Chem Lett. 2009;19(13):3593–3597. doi: 10.1016/j.bmcl.2009.04.149. [DOI] [PubMed] [Google Scholar]
- 91.Docquier JD, Benvenuti M, Calderone V, et al. Crystal structure of the narrow-spectrum OXA-46 class D beta-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob Agents Chemother. 2010;54(5):2167–2174. doi: 10.1128/AAC.01517-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beck J, Sauvage E, Charlier P, Marchand-Brynaert J. 2-aminopropane-1,2,3-tricarboxylic acid: synthesis and co-crystallization with the class A beta-lactamase BS3 of Bacillus licheniformis. Bioorg Med Chem Lett. 2008;18(13):3764–3768. doi: 10.1016/j.bmcl.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 93.Palzkill T. Metallo-beta-lactamase structure and function. Ann NY Acad Sci. 2013;1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94▪▪.King DT, Strynadka NC. Targeting metallo-beta-lactamase enzymes in antibiotic resistance. Future Med Chem. 2013;5(11):1243–1263. doi: 10.4155/fmc.13.55. Comprehensive review of the structure, function and inhibition of metallo-β-lactamases. [DOI] [PubMed] [Google Scholar]
- 95.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44(37):12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 96.Wachino J, Yamaguchi Y, Mori S, Kurosaki H, Arakawa Y, Shibayama K. Structural insights into the subclass B3 metallo-beta-lactamase SMB-1 and the mode of inhibition by the common metallo-beta-lactamase inhibitor mercaptoacetate. Antimicrob Agents Chemother. 2013;57(1):101–109. doi: 10.1128/AAC.01264-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garcia-Saez I, Hopkins J, Papamicael C, et al. The 1.5-A structure of Chryseobacterium meningosepticum zinc beta-lactamase in complex with the inhibitor, D-captopril. J Biol Chem. 2003;278(26):23868–23873. doi: 10.1074/jbc.M301062200. [DOI] [PubMed] [Google Scholar]
- 98.Mollard C, Moali C, Papamicael C, et al. Thiomandelic acid, a broad spectrum inhibitor of zinc beta-lactamases: kinetic and spectroscopic studies. J Biol Chem. 2001;276(48):45015–45023. doi: 10.1074/jbc.M107054200. [DOI] [PubMed] [Google Scholar]
- 99.Lienard BM, Garau G, Horsfall L, et al. Structural basis for the broad-spectrum inhibition of metallo-beta-lactamases by thiols. Org Biomol Chem. 2008;6(13):2282–2294. doi: 10.1039/b802311e. [DOI] [PubMed] [Google Scholar]
- 100.Kurosaki H, Yamaguchi Y, Yasuzawa H, Jin W, Yamagata Y, Arakawa Y. Probing, inhibition, and crystallographic characterization of metallo-beta-lactamase (IMP-1) with fluorescent agents containing dansyl and thiol groups. ChemMedChem. 2006;1(9):969–972. doi: 10.1002/cmdc.200600115. [DOI] [PubMed] [Google Scholar]
- 101.Hussein WM, Fatahala SS, Mohamed ZM, et al. Synthesis and kinetic testing of tetrahydropyrimidine-2-thione and pyrrole derivatives as inhibitors of the metallo-beta-lactamase from Klebsiella pneumonia and Pseudomonas aeruginosa. Chem Biol Drug Des. 2012;80(4):500–515. doi: 10.1111/j.1747-0285.2012.01440.x. [DOI] [PubMed] [Google Scholar]
- 102.Schlesinger SR, Bruner B, Farmer PJ, Kim SK. Kinetic characterization of a slow-binding inhibitor of Bla2: thiomaltol. J Enzyme Inhib Med Chem. 2013;28(1):137–142. doi: 10.3109/14756366.2011.640632. [DOI] [PubMed] [Google Scholar]
- 103.Lassaux P, Hamel M, Gulea M, et al. Mercaptophosphonate compounds as broad-spectrum inhibitors of the metallo-beta-lactamases. J Med Chem. 2010;53(13):4862–4876. doi: 10.1021/jm100213c. [DOI] [PubMed] [Google Scholar]
- 104.Horsfall LE, Garau G, Lienard BM, et al. Competitive inhibitors of the CphA metallo-beta-lactamase from Aeromonas hydrophila. Antimicrob Agents Chemother. 2007;51(6):2136–2142. doi: 10.1128/AAC.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ishii Y, Eto M, Mano Y, Tateda K, Yamaguchi K. In vitro potentiation of carbapenems with ME1071, a novel metallo-beta-lactamase inhibitor, against metallo-beta-lactamase- producing Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2010;54(9):3625–3629. doi: 10.1128/AAC.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Livermore DM, Mushtaq S, Morinaka A, Ida T, Maebashi K, Hope R. Activity of carbapenems with ME1071 (disodium 2,3-diethylmaleate) against Enterobacteriaceae and Acinetobacter spp. with carbapenemases, including NDM enzymes. J Antimicrob Chemother. 2013;68(1):153–158. doi: 10.1093/jac/dks350. [DOI] [PubMed] [Google Scholar]
- 107.Feng L, Yang KW, Zhou LS, et al. N-heterocyclic dicarboxylic acids: broad-spectrum inhibitors of metallo-beta-lactamases with co-antibacterial effect against antibiotic-resistant bacteria. Bioorg Med Chem Lett. 2012;22(16):5185–5189. doi: 10.1016/j.bmcl.2012.06.074. [DOI] [PubMed] [Google Scholar]
- 108.Walter MW, Felici A, Galleni M, et al. Trifluoromethyl alcohol and ketone inhibitors of metallo-beta-lactamases. Bioorg Med Chem Lett. 1996;6:2455–2458. [Google Scholar]
- 109.Walter MW, Adlington RM, Baldwin JE, Schofield CJ. Synthesis of metallo-beta-lactamase inhibitors. Tetrahedron. 1997;53:7275–7290. [Google Scholar]
- 110.Docquier JD, Benvenuti M, Calderone V, et al. High-resolution crystal structure of the subclass B3 metallo-beta-lactamase BJP-1: rational basis for substrate specificity and interaction with sulfonamides. Antimicrob Agents Chemother. 2010;54(10):4343–4351. doi: 10.1128/AAC.00409-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fitzgerald PM, Wu JK, Toney JH. Unanticipated inhibition of the metallo-beta-lactamase from Bacteroides fragilis by 4-morpholineethanesulfonic acid (MES): a crystallographic study at 1.85-A resolution. Biochemistry. 1998;37(19):6791–6800. doi: 10.1021/bi9730339. [DOI] [PubMed] [Google Scholar]
- 112.Chen P, Horton LB, Mikulski RL, et al. 2-substituted 4,5-dihydrothiazole-4-carboxylic acids are novel inhibitors of metallo-beta-lactamases. Bioorg Med Chem Lett. 2012;22(19):6229–6232. doi: 10.1016/j.bmcl.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hammond GG, Huber JL, Greenlee ML, et al. Inhibition of IMP-1 metallo-beta-lactamase and sensitization of IMP-1-producing bacteria by thioester derivatives. FEMS Microbiol Lett. 1999;179(2):289–296. doi: 10.1111/j.1574-6968.1999.tb08740.x. [DOI] [PubMed] [Google Scholar]
- 114.Garcia-Saez I, Mercuri PS, Papamicael C, et al. Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-beta-lactamase from Fluoribacter gormanii, in native form and in complex with D-captopril. J Mol Biol. 2003;325(4):651–660. doi: 10.1016/s0022-2836(02)01271-8. [DOI] [PubMed] [Google Scholar]
- 115.Heinz U, Bauer R, Wommer S, et al. Coordination geometries of metal ions in D- or L-captopril-inhibited metallo-beta-lactamases. J Biol Chem. 2003;278(23):20659–20666. doi: 10.1074/jbc.M212581200. [DOI] [PubMed] [Google Scholar]