

Abstract
Objective
Abdominal aortic aneurysm (AAA) is characterized as a progressive dilation and degradation of the aortic wall, associated with activation of matrix metalloproteinases (MMPs) and inflammation. Emerging evidence indicates a role for microRNAs (miRNAs) in AAA pathogenesis, but it is unclear whether abdominal aortic endothelial miRNAs play a role in the disease process. We aimed to identify miRNAs in the abdominal aortic endothelium that play a critical role in AAA development.
Approach and Results
The mouse model of AAA induced by Angiotensin II infusion was used in this study.Through a miRNA array and validation study, we initially identified the murine-specific miR-712 and subsequently its human/murine homolog miR-205 as Angiotensin II (AngII)-induced miRNAs in the abdominal aortic endothelium in vivo and in vitro. Mechanistically, miR-712 stimulated MMP activity in the aortic wall by directly targeting two MMP inhibitors:tissue inhibitor of metalloproteinase 3 (TIMP3) and reversion inducing cysteine-rich protein with kazal motifs (RECK). Silencing of miR-712 and miR-205 by using anti-miR-712 and anti-miR-205, respectively, significantly decreased the aortic MMP activity and inflammation, preventing AAA development in AngII-infused ApoE-/- mice. Further, upregulation of four AngII-sensitive miRNAs, miR-205, -21, -133b and -378, identified in this murine study were confirmed in human AAA samples compared to non-diseased control.
Conclusions
Our results demonstrate that AngII-sensitive miR-712 and its human homolog miR-205, downregulate TIMP3 and RECK, which in turn stimulate aortic MMP activity and inflammation, leading to AAA development.Targeting these miRNAs may be a novel therapeutic strategy to prevent AAA.
Keywords: microRNA, abdominal aortic aneurysm, angiotensin II, TIMP3, RECK
Introduction
Abdominal aortic aneurysm (AAA) is defined as a permanent dilation of the abdominal aorta due to a loss of the structural integrity of the vascular wall. AAA is a fairly common disease, especially in men age 65 and older and continues to have high mortality.1, 2 The most significant cause of mortality from AAA is acute rupture,with ~80% mortality, accounting for ~15,000 deaths a year in the United States2. The progressive weakening and dilation of the aorta observed in AAA is due to degradation and remodeling of the extracellular matrix (ECM) of the aortic wall.3, 4 Although several types of elastinolytic proteases are elevated in AAA tissues, matrix metalloproteinases (MMPs), particularly MMP2 and MMP9, are known to play critical roles in the pathological remodeling of vascular wall in this disease.2, 5-9. Currently surgical interventions are the only known effective treatments to prevent the aortic rupture10, there is no consensus regarding effective pharmacological treatments as the underlying disease mechanisms are still poorly understood. It is however, interesting to note that some of the ongoing clinical trials involve the use of the angiotensin receptor blockers (telmisartan and valsartan), angiotensin converting enzyme (ACE) inhibitor (perindopril), and MMP inhibitor (doxycycline), suggesting the importance of AngII and MMPs both as the critical mechanisms of the AAA development and therapeutic targets.10, 11
The microRNAs (miRNAs) are non-coding, small RNAs (21-23 nucleotides) that bind to 3’ untranslated region (UTR) of specific mRNA targets, inducing the target degradation or translational repression.12 Moreover, a single miRNA is capable of targeting multiple mRNAs, and a single mRNA may bind to multiple miRNAs.13 They are potent regulators of diverse biological and pathophysiological processes, and are considered as potential diagnostic and prognostic markers for cardiovascular diseases.14 Recent studies have shown the important roles of miRNAs in AAA. The miR-21 was shown to prevent AAA development by targeting PTEN in two different murine models of AAA - the porcine pancreatic elastase (PPE) infusion model in C57BL/6 and AngII infusion model in ApoE-/- mice.15 While miR-29 has been shown to play a role in various AAA models, its expression level was shown to decrease in the murine PPE and AngII infusion models16, but to increase in AngII-infused aged mouse model and fibulin-4R/R genetic model of AAA.17 Interestingly, however, silencing of miR-29 in both studies reduced aneurysmal pathology by targeting ECM components, collagens and elastin.16, 17 These studies not only demonstrate a role for miRNAs in AAA, but also illustrate a complex and unresolved role of miRNAs in AAA.
AngII infusion model of murine AAA, developed by Daugherty and colleagues18, has become an important experimental platform to study pathophysiology of AAA. While various aortic wall cell types are implicated in AAA development, the role of endothelial cells has been controversial.19-22 In addition, whether endothelial miRNAs play a critical role in AAA development has not been addressed. Therefore, here we searched for miRNAs that are regulated in abdominal aortic endothelium in the AngII infused mouse model and the role of one such miRNA, miR-712 and its human homolog miR-205, in AAA development. Our study shows that miR-712 and miR-205 directly target two key upstream inhibitors of MMPs, tissue inhibitor of metalloproteinases 3 (TIMP3) and reversion inducing cysteine-rich protein with kazal motifs (RECK), that in turn led to ECM degradation and AAA development. We also show therapeutic potentials of anti-miR-712 and anti-miR-205 in prevention of AAA development.
Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Identification of miRNAs regulated by AngII in mouse abdominal aortic endothelium
To identify which miRNAs were regulated by AngII in abdominal aortic endothelium in vivo, we carried out miRNA microarray analyses (Exiqon miRNA Array chip) using endothelial-enriched RNAs23, 24 obtained from the abdominal aortas at 12 or 36 h post-AngII or control pump implantation. As shown in the heat map analysis, we found that 21 (18 up- and 3 down-regulated) out of 401 miRNAs were significantly altered at 36 h by more than 50% and 35% respectively in the AngII-infused endothelium compared to the vehicle control (Supplement Figure I, Supplemental Table I). Next we validated the miRNA microarray results for the 12 and 36 h time points for 18 up-regulated and 3-down regulated miRNAs (Figure1A) by miScript qPCR assay. Of those, the qPCR results confirmed that 7 miRNAs (miRNA-21, -1, -133b, -207, -378, -1957, -712) were significantly up-regulated, while 2 miRNAs (miR-1952, -1249) were down-regulated by AngII at 36 h time-point. These results showed that miR-21 and miR-712 are two of the most robustly upregulated by AngII treatment in vivo. Since miR-21 was already shown to play an important role in AAA15, we decided to focus on studying the novel AngII-sensitive miRNA further.
Figure 1. Identification and validation of AngII-sensitive miRNA, miR-712 in in vivo and in vitro.
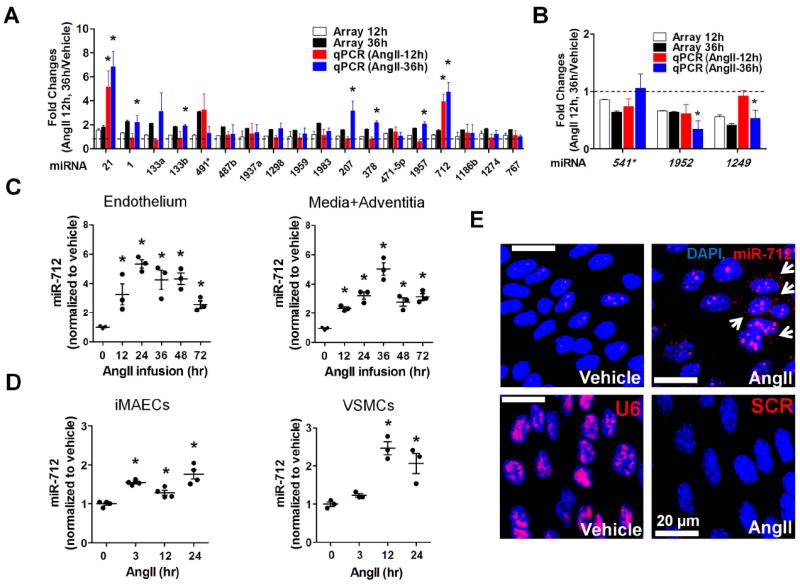
(A, B) Endothelial-enriched total RNAs, obtained from the abdominal aorta (C57BL/6 mice) at 12 and 36hr post-AngII infusion (1 μg/kg/min), were analyzed by miRNA array and quantitative PCR (qPCR). Twenty one miRNAs (18 up-, 3 down-regulated miRNAs at 36 hr post-AngII implantation compared to the vehicle control) were selected based on fold-change (18 up-regulated miRNAs : >1.5 fold and 3 down-regulated miRNAs : <0.65 fold) and statistical significance (p<0.05) in the miRNA array study. These miRNAs were further validated by qPCR and their fold-change over the vehicle control are shown along with the microarray results (compared to vehicle control; mean±S.E.; *p<0.05). Data were analyzed by Student’s t-test. (C) Expression of miR-712 was determined by qPCR using endothelial-enriched RNA from AngII-infused abdominal aorta and leftover RNA (media/adventitia) obtained from AngII-infused C57BL/6 mouse (0-72hr). (D) miR-712 expression was tested in immortalized mouse aortic endothelial cells (iMAECs) and vascular smooth muscle cells (VSMC) in vitro (0-24hr). (C, D : Data were analyzed using ANOVA followed by Tukey’s post hoc test, mean±S.E. *p<0.05; n=4). (E) Abdominal aortas of C57BL/6 mice obtained at 2-days post AngII-infusion were subjected to fluorescence in situ hybridization using digoxigenin-labeled miR-712 probe (red) and confocal microscopy, (n=4). Blue: DAPI nuclear stain; Arrows indicate cytosolic miR-712.
Ang II induced miR-712 expression in vivo and in vitro
To confirm the array and qPCR data for miR-712, we tested a time-dependent miR-712 expression in AngII infused abdominal aortic endothelium as well as the leftover sample (containing RNAs of medial smooth muscle cells and adventitial cells). AngII infusion increased miR-712 expression between 12-72 h time-points in both the endothelial and media/adventitia samples (Figure 1C). We further confirmed that AngII treatment induced miR-712 expression in both iMAECs and VSMCs in vitro (Figure 1D). Next, we performed in situ hybridization to further validate the AngII-sensitivity of miR-712 expression in the abdominal aortic endothelium. Studies using in situ hybridization with a miR-712 probe (Exiqon) showed a robust expression of miR-712 in the cytoplasm (arrows) and nuclei of the abdominal aortic endothelium, compared to the vehicle (Figure 1E). These results suggest that AngII treatment increases miR-712 expression both in endothelial cells and smooth muscle cells in the mouse abdominal aorta in vivo as well as in vitro.
TIMP3 and RECK are direct targets of miR-712
We recently showed that miR-712 is a mechanosensitive miRNA, upregulated by disturbed flow and oscillatory shear stress in endothelial cells and that it targets TIMP3.24 Here, through our in silico analysis using TargetScan,we identified an additional potential target of miR-712, RECK in response to the humoral AngII stimulation. Since TIMP3 and RECK are well-known negative regulators of MMP activity, a critical player in AAA development and progression2, we examined whether miR-712 indeed targeted TIMP3 and RECK expression using gain-of-function (premiR-712) and loss-of-function (anti-miR-712) approaches in the AngII-dependent manner. Treatment with premiR-712 and AngII significantly decreased TIMP3 and RECK mRNA expression, both of which were blocked by anti-miR-712 treatment in both iMAEC (Figure 2A and 2B) and VSMCs (Supplement Figure III-A and III-B) in vitro. Further in vivo study using mouse abdominal aorta endothelial-enriched RNA showed that AngII infusion decreased TIMP3 and RECK expression after 36h and 48h time-point, respectively (Supplement Figure III-C and III-D). In addition, AngII-stimulated miR-712 induction as well as downregulation of TIMP3 and RECK were significantly reversed in mice treated with anti-miR-712 (Figure 2D and 2E and Supplement Figure III-F and III-G). For this study, anti-miR-712 was subcutaneously injected twice (1 and 2 days prior to AngII implantation) at 5 mg/kg/day dose, and effectively silenced AngII-induced miR-712 expression (Figure 2C and Supplement Figure III-E).
Figure 2. Identification of TIMP3 and RECK as direct targets of miR-712.
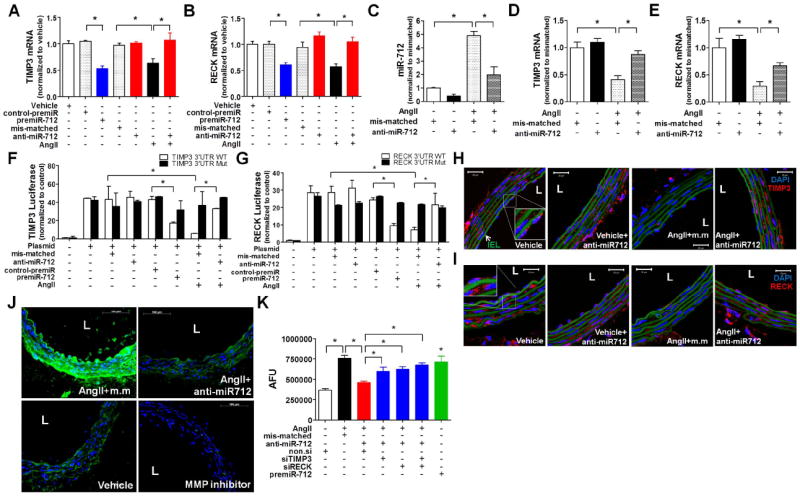
(A,B) TIMP3 and RECK expression were determined by qPCR in iMAECs treated with AngII (100 ng/ml) and pre-miR-712 (20 nM) with or without anti-miR-712 (400 nM) (n=4; p<0.05). (C-E) Endothelial-enriched RNAs were prepared from suprarenal artery of AngII (1 μg/kg/min)-infused mice, which were injected with mis-matched control or anti-miR-712 (5 mg/kg; injected subcutaneously, s.c.) twice (one and two days before AngII infusion). miR-712, TIMP3 and RECK expression was determined by qPCR . (n=4; p<0.05). (F,G) iMAECs transfected with dual-luciferase reporter plasmids containing wild-type (WT) or mutant (Mut) of TIMP3-3’UTR (F) and RECK-3’UTR (G), were treated with AngII (100 ng/ml), pre-miR-712 (20 nM) or control pre-miR (20 nM) and anti-miR-712 (400 nM). Firefly luciferase activity (normalized to control Renilla luciferase) indicating TIMP3 and RECK expression was determined using Luc-Pair miR Luciferase Assay Kit. (H-J) Frozen sections of abdominal aortas obtained from AngII-infused C57BL/6 mice were used for immunofluorescence staining with antibody specific to TIMP3 (H) and RECK (I) shown in red (scale bar =20 μm) and in situ zymography (J) using DQ-gelatin (green) to determine MMP activity. As a control, some abdominal aorta sections were incubated with the MMP inhibitor GM6001 (J, right bottom panel; scale bar =100 μm). (K) iMAECs, pretreated with AngII (100 ng/ml) and/or premiR-712 (20 nM) for 1 day, were further treated with anti-miR-712 or mismatched control at 400 nM each as well as siRNAs against TIMP3 and RECK (siRECK or siTIMP3), respectively, at 100 nM each for 1 day. MMP activity was determined by cell-based ELISA using DQ-gelatin. Data (Figure 2A-G and K) were analyzed using ANOVA followed by Tukey’s post hoc test, and values represent the mean±S.E.(*p<0.05;n=4; in triplicate). Blue: DAPI, and green: auto-fluorescent elastic lamina, IEL : internal elastic lamina, L : lumen, AFU : arbitrary fluorescence unit.
To further determine whether miR-712 bound to and inhibited TIMP3 and RECK expression directly in an AngII-dependent manner, we performed the luciferase assay, in which a construct containing the 3′-UTR region of TIMP3 or RECK mRNA containing the putative miR-712 binding sequence was used. Treatment of iMAECs with premiR-712 and AngII inhibited luciferase activity of TIMP3 and RECK, while their mutants or control-premiR showed no effect (Figure 2F and 2G). In addition, anti-miR-712 blocked the inhibitory effect of premiR-712 or AngII on TIMP3 and RECK luciferase activity (Figure 2F and 2G). Together, these data suggest that TIMP3 and RECK are direct targets of miR-712 in response to AngII. We next tested whether AngII downregulates TIMP3 and RECK expression by a miR-712-dependent mechanism in vivo by immunostaining. Expression of TIMP3 and RECK were evident in endothelial and smooth muscle cells in the vehicle control groups (Figure 2H and 2I). AngII infusion decreased the expression of TIMP3 and RECK compared to the vehicle, but anti-miR-712 treatment reversed it (Figure 2H and 2I). Since TIMP3 and RECK are well-known inhibitors of MMPs, we examined the effect of anti-miR-712 on MMP activity in vivo by using an in situ zymography assay using the fluorescent DQ-gelatin. As shown in Figure 2J, AngII infusion increased MMP activity as evidenced by the green fluorescent signal intensity, but was prevented by treating mice with anti-miR-712 in vivo or the MMP inhibitor GM6001 added during the zymography assay. This in situ zymography result was further confirmed in an in vitro cell-based assay using iMAEC (Figure 2K). The in vitro study showed that AngII induced MMP activity, which was prevented by anti-miR-712 treatment. Next we determined whether TIMP3 or RECK, or both were important player in regulation of the AngII-dependent MMP activity. For this study, cells pre-treated with AngII and anti-miR-712 were treated with siRNAs to knockdown TIMP3, RECK, or both. We found that the inhibitory anti-miR-712 effect on the MMP activity was partially blunted when cells were treated with TIMP3 siRNA or RECK siRNA (Figure 2K). Interestingly, knockdown of both TIMP3 and RECK together did not produce the additive effect, which may be due to an insensitive assay condition or an unknown cooperation between the two inhibitors. Together, these results demonstrate that AngII stimulates MMP activity by inducing expression of miR-712, which in turn downregulates TIMP3 and RECK, both of which seem to be equally important in MMP activity regulation.
Anti-miR-712 inhibits AAA induced by AngII infusion
Next, we tested whether anti-miR-712 can prevent AAA induced by AngII infusion in ApoE-/- mice using the Daugherty method.18 For this study, mice were treated with anti-miR-712 using the same dosage and protocol used above in Figure 2C-E, except for additional anti-miR-712 injections every 4 days for 3 weeks following AngII implantation. As expected, AngII infusion (1μg/kg/min) for 3 weeks induced pronounced AAA phenotype in the suprarenal region of the abdominal aorta, which was significantly blunted in the anti-miR-712-treated mice compared to the mis-matched or saline-treated control groups (Supplement Figure IV). While 20% (2 out of 10 mice each) of AngII-infused mice died during the first week in the saline and mis-matched groups, none of the anti-miR-712 treated mice showed mortality for the duration of the study (Figure 3A). Also, treatment with anti-miR-712 dramatically reduced AAA incidence to 20% (2 of 10 mice) compared to the mis-matched (80%; 8/10) or saline controls (70%; 7/10) (Figure 3B). Similarly, the increase in AngII-induced abdominal aortic diameter was also significantly reduced by the anti-miR-712 treatment compared to the saline and mis-matched controls (Figure 3C).
Figure 3. Inhibition of miR-712 prevents AngII-induced AAA.
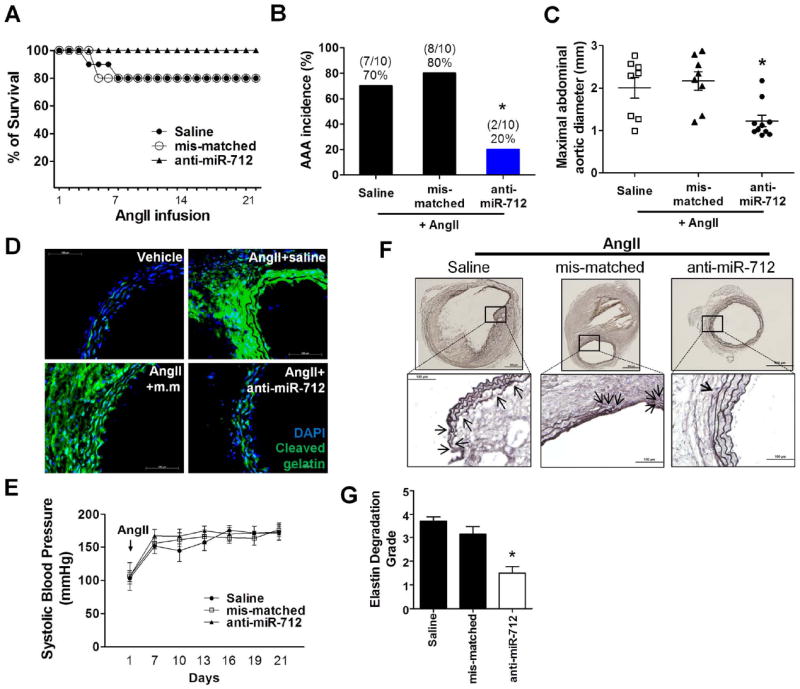
ApoE-/- mice (24-26 weeks) injected with mis-matched control or anti-miR-712 (5 mg/kg each) were infused with AngII (1 μg/kg/min) or vehicle for 3 weeks via osmotic minipumps. (A,B) The survival and incidence rate of AngII-induced AAA in anti-mR-712 treated group compared to saline or mis-matched anti-miR controls are shown. Survival rates were estimated with Kaplan-Meier analysis (p=0.34). The significance of the incidence were tested by Fisher’s exact test (p<0.05; n=10 each group). (C) Maximal abdominal aortic diameter quantitated using the H&E stained section; compared to saline or mis-match control group.The data was analyzed using the Kruskal-Wallis test followed by the Mann-Whitney U-test using Bonferroni correction to adjust the probability (*p<0.05; n=10 each group). (D) shows in situ zymography using DQ-gelatin (green) to determine MMP activity in AngII-infused abdominal aortic sections with saline, mis-matched control or anti-miR-712 injected group (scale bar =100 μm). (E) shows systolic blood pressure measured in AngII-infused mice treated with anti-miR-712 compared to saline or mis-matched controls (n=10 each group). (F) Elastin fragmentation was evaluated by histochemical staining with Orcein elastin stain kit using aorta sections of saline, mis-matched control and anti-miR-712 treated groups.Upper panels : low magnification (scale : 500 μm); lower panels : high magnification (scale bar : 100 μm; elastin : dark brown). (G) shows quantitation of elastin degradation. The data was analyzed using the Kruskal-Wallis test followed by the Mann-Whitney U-test using Bonferroni correction to adjust the probability (*p<0.05; n=6 each group).
We next tested whether anti-miR-712 could inhibit AngII-induced MMP activity by the in situ zymography using DQ-gelatin. As expected, the MMP activity (shown as the intense fluorescent gelatin signal) was dramatically higher in AngII-infused mice (AngII+saline group) compared to the vehicle control, but it was remarkably reduced in the anti-miR-712-treated mice compared to the mis-matched control (Figure 3D and Supplement Figure V). We next tested whether the anti-AAA effect of anti-miR-712 was mediated through normalizing blood pressure in the AngII-infused mice. AngII-infusion significantly increased blood pressure as expected within one week following the AngII infusion18, but anti-miR-712 treatment did not alter the blood pressure (Figure 3E). Aortic wall elastin fragmentation is an important feature of AAA in both humans and mouse models.18 We graded the elastic lamina degradation on a scale of 1 (least) to 4 (worst) in our mouse samples (Supplement Figure VI). As expected, the elastic laminas were frequently disrupted and fragmented in AngII-infused mice treated with saline or mis-matched controls, whereas anti-miR-712 treated mice showed little signs of elastin fragmentation (Figure 3F and 3G). These results suggest that the anti-AAA effect of anti-miR-712 is mediated in an MMP-dependent manner, but is independent of the pressor response.
Anti-miR-712 inhibits both endothelial and circulating leukocyte inflammation
Given the importance of inflammation in AAA development25, we examined whether the effect of anti-miR-712 (delivered systemically via s.c. injection) was mediated through the aortic endothelial cells, circulating leukocytes or both. To test this hypothesis, we performed an ex vivo leukocyte adhesion assay using abdominal aorta explants and peripheral blood monocytic cells (PBMCs) obtained from the mice treated with anti-miR-712 or mis-matched control along with vehicle or AngII-infusion. Three groups of aortas were obtained from the mice that were treated with 1) vehicle, 2) AngII + mis-matched control, or 3) AngII + anti-miR-712. In addition, PBMCs were also obtained from the same three groups of mice as above. These 3 groups of aortic explants and 3 groups of PBMCs were then used in a 3 × 3 combination study (Figure 4A). Here, PBMCs were added to an aortic explant with its endothelial surface up in a dish, and the number of PBMCs adhering to the endothelial surface after a 30 min incubation time was microscopically quantitated. First, adhesion of vehicle-control PBMCs to the vehicle-control endothelial surface was very low (Figure 4A, panel 1). Second, both PBMCs and aortic explants obtained from AngII-infused mice showed significantly increased adhesion as compared to the vehicle control (as indicated by the increased number of PBMCs shown as green dots) (Figure 4A: panel 1 vs. 2; panel 1 vs. 4).Third, both the aorta explants and PBMCs obtained from anti-miR-712 treated mice showed a significant reduction in PBMC adhesion to endothelium compared to the mis-matched controls (Figure 4A: panel 3 vs. 2; panel 7 vs. 4), suggesting the anti-inflammatory effect of anti-miR-712 treatment. The quantitative results shown in Figure 4B further supported these points. Consistent with these ex vivo results, we also found a robust F4/80+ monocyte/macrophage staining in AngII-infused mice with saline or mis-matched controls, but it became nearly undetectable in the anti-miR-712 treated mice (Figure 4C and supplement Figure VII). These findings suggest that the anti-AAA effect of anti-miR-712 is mediated, at least in part, by inhibiting inflammation of both aortic endothelial cells and circulating leukocytes.
Figure 4. Treatment with anti-miR-712 inhibits AngII-induced inflammation of endothelial cells and peripheral leukocytes (PBMCs).
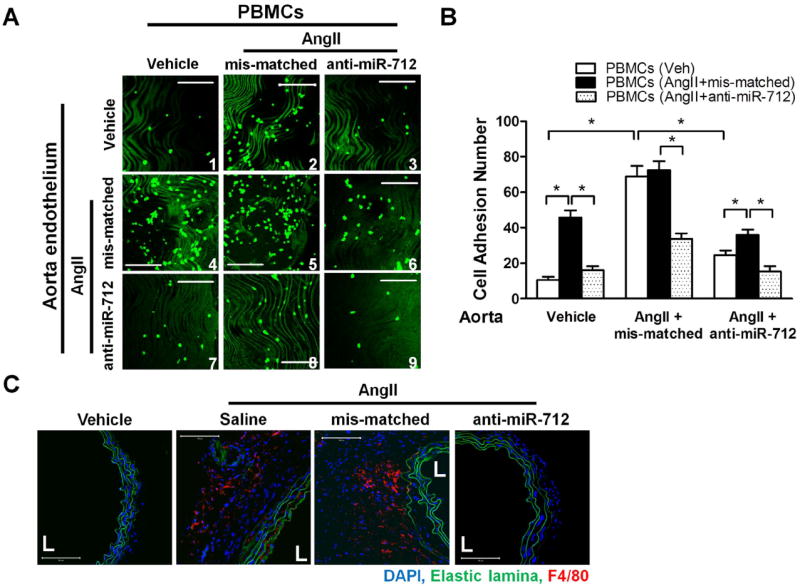
(A,B) Abdominal aortic explants as well as PBMCs were obtained from vehicle or AngII-infused (1 μg/kg/min; 2 days) C57BL/6 mice that were also treated with mis-matched or anti-miR-712 (5 mg/kg, s.c; 2 daily injections prior to AngII implantation). PBMCs (labeled with fluorescent Calcein) were then incubated for 30 min with the abdominal aortic explants with their endothelial side up, and the PBMCs adhered to the endothelial surface was counted by confocal microscopy. Data were analyzed using ANOVA followed by Tukey’s post hoc test, and values represent the mean±S.E. *p<0.05; n=4; green indicates PBMCs; scale bar = 200 μm). (C) Macrophage (F4/80+) infiltration was examined by the immunostaining the abdominal aorta section obtained in the frozen sections described in Figure 3D (scale bar, 100 μm; n=5 each group).
The miR-205, a human homolog of miR-712,directly targets TIMP3 and RECK
Since miR-712 is murine-specific24, its clinical implication for human disease could be limited. To address this potential concern, we searched and found its potential human homolog, miR-205, which shares the 7-mer “seed sequence” with miR-712 and is highly conserved in most mammalian species (TargetScan) including mouse and human (Supplement Figure VIII-A).24 We tested whether miR-205 also targets TIMP3 and RECK in endothelial cells in an AngII-dependent manner by using a gain-of-function (pre-miR-205) and loss-of-function (anti-miR-205) approaches. Like miR-712, AngII treatment increased expression of miR-205 in iMAEC in vitro (Supplement Figure VIII-B). Treatment of iMAEC with premiR-205 or AngII decreased TIMP3 and RECK mRNA expression, which was prevented by anti-miR-205 treatment (Figure 5A and 5B). Similarly, AngII treatment increased expression of miR-205 in mouse aortic endothelium and the media+adventitial cells in vivo (Figure5C and Supplement Figure VIII-C). AngII-induced miR-205 expression was effectively silenced by anti-miR-205 treatment in mice (Figure 5C), using the same dosage and injection protocol used for anti-miR-712 (Figure 2C-E). Next, we found that AngII treatment also resulted in a decreased TIMP3 and RECK expression, which was reversed by anti-miR-205 treatment in mouse aortic endothelium (Figure 5D and 5E). Taken together, these results suggest that miR-205 expression is AngII-sensitive and it targets TIMP3 and RECK, like miR-712. We also examined whether expression of LRP-1, a previously identified target of miR-20526 was regulated by AngII-induced miR-712 and miR-205 in our experimental model. As shown in Supplement Figure IX, LRP-1 was downregulated by AngII-infusion in the abdominal aortic endothelial RNA, however, its expression was not rescued by treatment with anti-miR-712 or anti-miR-205. These results demonstrate that although LRP-1 expression is downregulated by AngII infusion, it is not regulated by miR-712 or miR-205.
Figure 5. TIMP3 and RECK are direct targets of miR-205, a human homolog of miR-712.
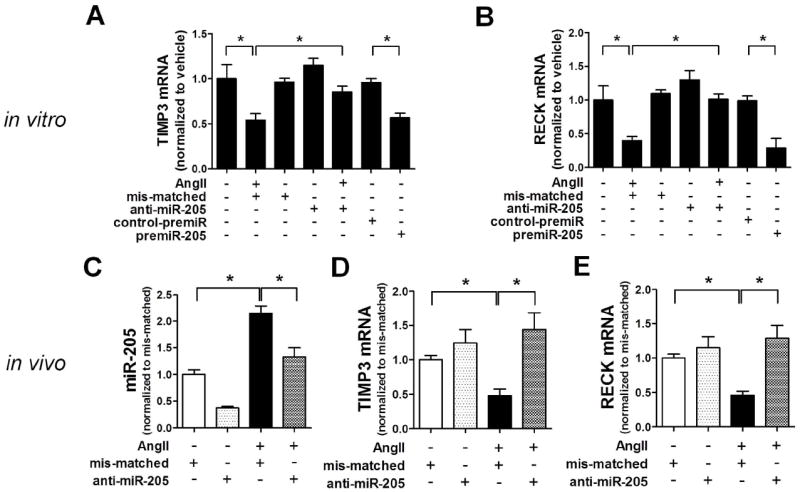
(A) TIMP3 and (B) RECK expression was determined by qPCR in iMAECs pre-treated with premiR-205 (20 nM), anti-miR-205 (400 nM) or mis-matched control (400 nM) for 2 days, followed by AngII (100 ng/ml) treatment for another 1 day (n=4; *p<0.05; in triplicate). (C-E) Endothelial-enriched RNAs were prepared from the abdominal aortas of vehicle or AngII-infused mice, which were pre-treated with mis-matched control or anti-miR-205 (5 mg/kg; injected subcutaneously, s.c.) twice (one and two days before AngII infusion), and miR-205, TIMP3 and RECK expression was determined by qPCR (n=4; *p<0.05; in triplicate). All data were analyzed using ANOVA followed by Tukey’s post hoc test, and values represent the mean±S.E.
Treatment with anti-miR-205 prevents Ang II-induced AAA
To determine whether miR-205 plays an important role in AAA, AngII-infused ApoE-/- mice were treated with anti-miR-205 or mis-matched control using the same protocol as anti-miR-712. As shown in Figure 6A-C and Supplement Figure X, anti-miR-205 treatment significantly decreased AAA incidence, mortality, and abdominal aorta dilation compared to the mis-matched control. While the survival rate of AngII-infused and mis-matched group was 56% (5/9), the anti-miR-205-treated group showed 88.8% survival rate at the 3 week time-point (Figure 6A). Anti-miR-205 treatment reduced AAA incidence (2/9; 22.2 %) compared to the mis-matched group (7/9; 77.7 %) (Figure 6B, p=0.056). Next, we tested the effect of anti-miR-205 treatment on MMP activity as well in the same groups of mice by in situ zymography. AngII-induced MMP activity was nearly blocked by treating the aorta section of the AngII-treated mice with the MMP inhibitor GM6001 during the in situ zymography assay (Figure 6D; upper right panel and Supplement Figure X). Anti-miR-205 treatment dramatically reduced the MMP activity compared to the mis-matched control (Figure 6D and Supplement Figure XI).These results suggest that the AngII- and miR-205-sensitive metalloproteinase activity is mostly accounted for by the MMP activity. Like anti-miR-712, anti-miR-205 also did not affect AngII-induced systemic hypertension in these mice (Figure 6E). Taken together, these results demonstrate that, like anti-miR-712, anti-miR-205 treatment has a potent preventive effect on AAA induced by AngII infusion.
Figure 6. anti-miR-205 prevents AAA development in AngII-infused mice and some of AngII-sensitive miRNAs that are upregulated in human AAA tissues.
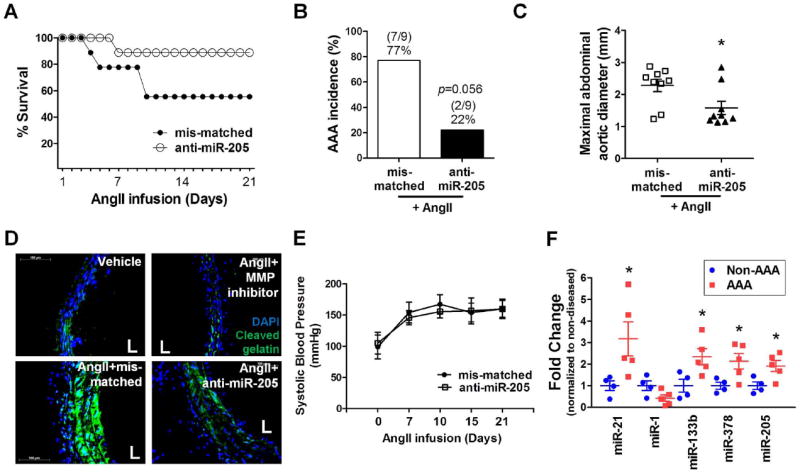
ApoE-/- mice treated with anti-miR-205 or mis-matched control (5 mg/kg) were infused with AngII for 3 weeks. (A, B) The incidence and survival rate of AngII-induced AAA in anti-mR-205 treated group compared to mis-matched controls are shown. Survival rates were estimated with Kaplan-Meier analysis (p=0.13). The significance of the incidence were tested by Fisher’s exact test (p=0.056; n=9 each group). (C) Maximal abdominal aortic diameter was quantitated using the H&E stained section.The data was analyzed using the Kruskal-Wallis test followed by the Mann-Whitney U-test using Bonferroni correction to adjust the probability (*p<0.05 compared to mis-matched control, n=9 each group). (D) shows in situ zymography using DQ-gelatin (green) to determine MMP activity in vehicle or AngII-infused abdominal aortic sections with mis-matched control or anti-miR-205 treated group (n=4, scale bar =100 μm). Some sections from the AngII + mis-matched control treated mice were incubated with GM6001 (upper right panel). (E) shows systolic blood pressure measured in AngII-infused mice treated with anti-miR-205 compared to mis-matched controls (n=9). (F) Expression of AngII-sensitive miRNAs, miR-21, miR-1, miR-133b, miR-378 and miR-205 was determined by qPCR using total RNAs obtained from paraffin sections of human AAA tissues (n=5) and non-diseased (non-AAA) tissues (n=4) using RNU6B as an internal control (*p<0.05 as determined by student’s t-test compared to non-AAA; value represent the mean±S.E.).
AngII-sensitive miRNAs in the murine AAA are also upregulated in human AAA tissues
We tested whether some of the AngII-sensitive miRNAs identified in this mouse study, including miR-205, -21, -1, 133b and 378 were also upregulated in human AAA tissues. For this study, total RNAs prepared from de-identified human AAA paraffin sections (n=5) were compared to those without the disease (non-AAA, n=4) from Origene. The detailed patient and donor clinical information is described in Supplement Table III. miR-205 expression was ;2-fold higher in AAA samples compared to the non-AAA (Figure 6F). Also, expression of miR-21, miR-133b, and miR-378, but not miR-1,was significantly higher in human AAA samples compared to the non-AAA. This result suggests that miRNAs, especially the human homolog of miR-712, miR-205, identified in our AngII-induced murine AAA model appears to be relevant in human AAA as well.
Discussion
While searching for miRNAs regulated in abdominal aortic endothelium in the AngII infused mouse model of AAA, we initially found miR-712 as an AngII-sensitive miRNA both in vivo and in vitro. We showed that miR-712 directly targets the two key inhibitors of MMPs, TIMP3 and RECK, that lead to activation of the metalloproteinases in the aorta wall and AAA development in the mouse model. Moreover, we demonstrated the potent anti-AAA effect of the anti-miR-712 in the AngII infused mice. Since miR-712 is a murine-specific miRNA24, however, its therapeutic potential in human AAA may be limited. This led us to the identification of miR-205, a human homolog of the murine miR-712. Like miR-712, expression of miR-205 was also increased by AngII, it regulated MMP activity by targeting TIMP3 and RECK, and that anti-miR-205 treatment effectively prevented AAA development in the AngII infusion model. Moreover, we found that expression of at least four miRNAs including miR-205 identified in our AngII infusion mouse study was significantly higher in the human AAA samples compared to the non-AAA samples. These results suggest a potential clinical relevance of these AngII-sensitive miRNAs identified in this mouse model in human AAA.
One of the exciting findings of this study was to observe that some of the miRNAs identified from our AAA mouse model was also upregulated in human AAA tissues compared to that of non-AAA. These miRNAs are miR-205, -21, -133b, and -378. Other investigators have shown previously that miR-21 is upregulated in human AAA tissues as well as in the abdominal aortas of AngII-infused mice15, consistent to our data. The miR-133b expression was shown to be decreased in human AAA and thoracic aortic dissection tissues27-29. These results are different from our current data and it may be due to the difference in the aortic wall tissue RNA vs. endothelial RNA. While miR-29 was shown to be either increased or decreased in the whole abdominal aortic wall tissue with AAA in mice16, 17, 30, our array result using endothelial RNA did not show a significant change and therefore we did not pursue it further. It is important to point out there are important limitations of our miRNA expression results in human AAA samples due to the small number of samples used. Additional studies involving much larger number of human AAA tissues and blood samples with proper controls are warranted.
In this study, we used in vivo endothelial miRNA array to identify AngII-sensitive miRNAs using the AngII-infused mice. The reason that we used endothelial-enriched RNAs rather than the whole aortic wall samples containing RNA mixtures coming from multiple cell types including endothelial cells, smooth muscle cells and immune cells that could vary greatly in different samples, which could largely dictate the outcomes of the array results depending on the various cell mixtures in each sample. We have successfully used the same approaches using endothelial-enriched RNAs obtained from the arteries exposed to disturbed blood flow or stable flow to identify the mechanosensitive mRNA transcripts and miRNAs in mice.23, 24
Recently, we showed that miR-712 is a mechanosensitive miRNA, upregulated by disturbed blood flow in in mouse arterial endothelial cells. We also showed that miR-712 is derived from an unexpected source, pre-ribosomal RNA, RN45S gene, in the exoribonuclease XRN1-dependent, but DiGeorge Syndrome Critical Region-8 (DGCR8)-independent manner, suggesting that it is an atypical miRNA.24 In addition, we showed that miR-205 is also mechanosensitive,human and mouse homolog of miR-712.24 Here, we now show that a non-mechanical, humoral stimulus, AngII can also stimulate miR-712 and -205 expression in endothelial cells in the mouse aorta in vivo and in vitro. In addition, AngII also stimulated miR-712 and -205 expression in vascular smooth muscle cells as well. These results suggest that AngII increases miR-712 and -205 production in multiple cell types of the aortic wall, stimulating MMP activities in the overall aortic wall, resulting in AAA development.
We previously showed that the mechanosensitive miR-712 and -205 target TIMP324, but here we identified another key endogenous MMP inhibitor, RECK, in response to AngII. TIMP3 is a well-known endogenous inhibitor of MMPs and ADAM family members and plays a key role in regulating ECM remodeling, endothelial permeability, and inflammation, atherosclerosis and AAA.24, 31-33. TIMP3 expression is decreased in Marfan syndrome patients with increased rate of aortic rupture.33 Also, the loss of TIMP3 has been shown to induce AAA in AngII infused mice34. RECK is a membrane-bound protein and is known to suppress MMP activity and to induce angiogenesis in the metastatic cascade.35, 36 However, its role in AAA has not been studied. Our current study clearly demonstrates the novel role of RECK, as a downstream target of miR-712/205 and a key regulator of MMP activity, in the murine AAA development. Also, an imbalance between MMPs and TIMPs/RECK has been reported in the aortas of AAA patients.32, 37 In our study, the relative importance of miR-712 vs.miR-205 and TIMP3 vs. RECK remains unresolved. Our result showed that AngII upregulates both miR-712 and -205, which target both TIMP3 and RECK as well, all of which lead to activation of the downstream MMP activity to a similar degree. These results suggest that the two miRNAs as well as the two endogenous MMP inhibitors seem to have similar contribution in regulation of MMP activity and/or that these miRNAs and TIMP3/RECK may coordinate each other by an unknown mechanism.
AAA is a progressive and deadly disease, especially when the aorta ruptures, but the effective treatments are limited to surgical interventions. Unfortunately, there are no proven effective pharmacological treatments that can regress, halt, or slow down the progressive enlargement of the AAA, leading to its rupture, due in large part to a lack of mechanistic insight into the pathogenesis of the disease.10 Currently, multiple clinical trials are underway, testing the safety and efficacy of pharmacological treatments. These include drugs that target AngII related pathways such as the angiotensin receptor blockers (telmisartan and valsartan) and the ACE inhibitor; perindopril, and the broad MMP inhibitor doxycycline.10, 11 It is interesting that the miRNAs, miR-712/205 identified in this study, are AngII-sensitive and are potent and direct regulators of TIMP3 and RECK, the two key endogenous upstream regulators of MMP activity. In addition, our study showed that miR-205 is upregulated in the human AAA tissues, further supporting the hypothesis that targeting this AngII-sensitive miRNA by anti-miR-205 would be a potential anti-AAA therapy in human patients. Our mouse studies using anti-miR-712 or anti-miR-205, which showed the potent preventive effect on AAA development by inhibiting MMP activity by protecting TIMP3 and RECK expression in the AngII infused mouse model provides additional support for the hypothesis.
In summary, we demonstrate that miR-712 and its human/murine homolog miR-205 are induced by AngII in the abdominal aorta cells and these miRNAs downregulate TIMP3 and RECK expression, which in turn activates MMP activity, leading to AAA development. Our study demonstrates a potent preventive effect of anti-miR-712 and anti-miR-205 in murine models of AngII-infused AAA. Since miR-205 is also increased in human AAA tissues, use of specific miR-205 inhibitors may be considered as a potential anti-AAA therapy.
Significance
AAA is a progressive and deadly disease with surgical intervention as the only treatment option. Unfortunately, lack of effective pharmacological treatments result in progressive enlargement of AAA, leading to its rupture, in large part due to incomplete mechanistic insight of the disease pathophysiology. Here, using aortic endothelial miRNA microarray from AngII-infused mice and subsequent validation studies in vivo, in vitro and human tissues, we demonstrate that expression of miR-712 and its human homolog miR-205 is increased in the abdominal aortic wall cells both in murine and human AAA development. Mechanistically, miR-712/205 family directly downregulate the two master controllers of MMPs, TIMP3 and RECK, leading to activation of MMPs and eventual AAA development. We also demonstrate a potent preventive effect of anti-miR-712/205 in the mouse model of AngII-infused AAA. Since miR-205 is also increased in human AAA tissues, use of specific miR-205 inhibitors may be considered as a potential anti-AAA therapy.
Supplementary Material
Acknowledgments
This work was supported by funding from NIH grants HL095070, HL114772, HL113451 to HJ. This work was also supported by the National Heart Lung and Blood Institute of the NIH as a Program of Excellence in Nanotechnology award HHSN268201000043C to HJ. SK is an American Heart Association Postdoctoral fellow. HJ thanks Ada Lee and Pete Correll and John and Jan Portman for the Professorships. We thank Dr. Oskar Laur at the Emory Custom Cloning Core Facility for generating TIMP3 and RECK mutants.
Non-standard Abbreviations and Acronyms
- miRNA
microRNA
- AngII
Angiotensin II
- LNA
locked nucleic acid
- MMP
matrix metalloproteinases
- TIMP
tissue inhibitor of metalloproteinase 3
- RECK
reversion inducing cysteine-rich protein with kazal motifs
- qPCR
quantitative polymerase chain reaction
Footnotes
DISCLOSURES
None.
References
- 1.Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: A best-evidence systematic review for the u.S.Preventive services task force. Annals of internal medicine. 2005;142:203–211. doi: 10.7326/0003-4819-142-3-200502010-00012. [DOI] [PubMed] [Google Scholar]
- 2.Maegdefessel L, Dalman RL, Tsao PS. Pathogenesis of abdominal aortic aneurysms: Micrornas, proteases, genetic associations. Annual review of medicine. 2013 doi: 10.1146/annurev-med-101712-174206. [DOI] [PubMed] [Google Scholar]
- 3.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 4.Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2605–2613. doi: 10.1161/01.ATV.0000245819.32762.cb. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Sukhova GK, Yang JT, Sun J, Ma L, Ren A, Xu WH, Fu H, Dolganov GM, Hu C, Libby P, Shi GP. Cathepsin l expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis. 2006;184:302–311. doi: 10.1016/j.atherosclerosis.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. The Journal of clinical investigation. 1998;102:1900–1910. doi: 10.1172/JCI2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. The Journal of clinical investigation. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase b) suppresses development of experimental abdominal aortic aneurysms. The Journal of clinical investigation. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:1625–1633. doi: 10.1161/01.atv.18.10.1625. [DOI] [PubMed] [Google Scholar]
- 10.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyake T, Morishita R. Pharmacological treatment of abdominal aortic aneurysm. Cardiovascular research. 2009;3:436–443. doi: 10.1093/cvr/cvp155. [DOI] [PubMed] [Google Scholar]
- 12.Bartel DP. Micrornas: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 13.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E, Olson EN. Microrna therapeutics for cardiovascular disease: Opportunities and obstacles. Nature reviews Drug discovery. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, Leeper NJ, Raaz U, Schoelmerich AM, McConnell MV, Dalman RL, Spin JM, Tsao PS. Microrna-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Science translational medicine. 2012;4:122ra122. doi: 10.1126/scitranslmed.3003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS. Inhibition of microrna-29b reduces murine abdominal aortic aneurysm development. The Journal of clinical investigation. 2012;122:497–506. doi: 10.1172/JCI61598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boon RA, Seeger T, Heydt S, et al. Microrna-29 in aortic dilation: Implications for aneurysm formation. Circulation research. 2011;109:1115–1119. doi: 10.1161/CIRCRESAHA.111.255737. [DOI] [PubMed] [Google Scholar]
- 18.Daugherty A, Manning MW, Cassis LA. Angiotensin ii promotes atherosclerotic lesions and aneurysms in apolipoprotein e-deficient mice. The Journal of clinical investigation. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito T, Hasegawa Y, Ishigaki Y, Yamada T, Gao J, Imai J, Uno K, Kaneko K, Ogihara T, Shimosawa T, Asano T, Fujita T, Oka Y, Katagiri H. Importance of endothelial nf-kappab signalling in vascular remodelling and aortic aneurysm formation. Cardiovascular research. 2013;97:106–114. doi: 10.1093/cvr/cvs298. [DOI] [PubMed] [Google Scholar]
- 20.Gao L, Siu KL, Chalupsky K, Nguyen A, Chen P, Weintraub NL, Galis Z, Cai H. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: Treatment with folic acid. Hypertension. 2012;59:158–166. doi: 10.1161/HYPERTENSIONAHA.111.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP, 3rd, Howatt DA, Subramanian V, Poduri A, Charnigo R, Cassis LA, Daugherty A. Endothelial cell-specific deficiency of ang ii type 1a receptors attenuates ang ii-induced ascending aortic aneurysms in ldl receptor-/- mice. Circulation research. 2011;108:574–581. doi: 10.1161/CIRCRESAHA.110.222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rateri DL, Moorleghen JJ, Knight V, Balakrishnan A, Howatt DA, Cassis LA, Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin ii type 1a receptors does not influence aortic aneurysms or atherosclerosis in ldl receptor deficient mice. PloS one. 2012;7:e51483. doi: 10.1371/journal.pone.0051483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66–73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, Jang IH, Kim S, Kim W, Won Kang S, Baker AH, Woong Seo J, Ferrara KW, Jo H. The atypical mechanosensitive microrna-712 derived from pre-ribosomal rna induces endothelial inflammation and atherosclerosis. Nature communications. 2013;4:3000. doi: 10.1038/ncomms4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of aaa progression.Part 1: Extracellular matrix degeneration. Nature reviews Cardiology. 2009;6:464–474. doi: 10.1038/nrcardio.2009.80. [DOI] [PubMed] [Google Scholar]
- 26.Song H, Bu G. Microrna-205 inhibits tumor cell migration through down-regulating the expression of the ldl receptor-related protein 1. Biochemical and biophysical research communications. 2009;388:400–405. doi: 10.1016/j.bbrc.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pahl MC, Derr K, Gabel G, Hinterseher I, Elmore JR, Schworer CM, Peeler TC, Franklin DP, Gray JL, Carey DJ, Tromp G, Kuivaniemi H. Microrna expression signature in human abdominal aortic aneurysms. BMC medical genomics. 2012;5:25. doi: 10.1186/1755-8794-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao M, Zou S, Weng J, Hou L, Yang L, Zhao Z, Bao J, Jing Z. A microrna profile comparison between thoracic aortic dissection and normal thoracic aorta indicates the potential role of micrornas in contributing to thoracic aortic dissection pathogenesis. Journal of vascular surgery. 2011;53:1341–1349 e1343. doi: 10.1016/j.jvs.2010.11.113. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Zhang M, He H, Chen J, Zeng H, Li J, Duan R. Microrna/mrna profiling and regulatory network of intracranial aneurysm. BMC medical genomics. 2013;6:36. doi: 10.1186/1755-8794-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. Mir-29b participates in early aneurysm development in marfan syndrome. Circulation research. 2012;110:312–324. doi: 10.1161/CIRCRESAHA.111.253740. [DOI] [PubMed] [Google Scholar]
- 31.Cardellini M, Menghini R, Martelli E, Casagrande V, Marino A, Rizza S, Porzio O, Mauriello A, Solini A, Ippoliti A, Lauro R, Folli F, Federici M. Timp3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by sirt1. Diabetes. 2009;58:2396–2401. doi: 10.2337/db09-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knox JB, Sukhova GK, Whittemore AD, Libby P. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation. 1997;95:205–212. doi: 10.1161/01.cir.95.1.205. [DOI] [PubMed] [Google Scholar]
- 33.Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS, Zeeshan A, Bavaria JE, Gorman JH, 3rd, Spinale FG, Gorman RC. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with bicuspid or tricuspid aortic valves. The Journal of thoracic and cardiovascular surgery. 2007;133:1028–1036. doi: 10.1016/j.jtcvs.2006.10.083. [DOI] [PubMed] [Google Scholar]
- 34.Basu R, Fan D, Kandalam V, Lee J, Das SK, Wang X, Baldwin TA, Oudit GY, Kassiri Z. Loss of timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin ii. The Journal of biological chemistry. 2012;287:44083–44096. doi: 10.1074/jbc.M112.425652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh J, Takahashi R, Kondo S, et al. The membrane-anchored mmp inhibitor reck is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 36.Meng N, Li Y, Zhang H, Sun XF. Reck, a novel matrix metalloproteinase regulator. Histology and histopathology. 2008;23:1003–1010. doi: 10.14670/HH-23.1003. [DOI] [PubMed] [Google Scholar]
- 37.Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS, Zeeshan A, Bavaria JE, Gorman JH, 3rd, Spinale FG, Gorman RC. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with marfan syndrome. Circulation. 2006;114:I365–370. doi: 10.1161/CIRCULATIONAHA.105.000810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.