

Abstract
The systematic exploration of a series of triazole-based agonists of the cation channel insect odorant receptor is reported. The structure-activity relationships of independent sections of the molecules are examined. Very small changes to the compound structure were found to exert a large impact on compound activity. Optimal substitutions were combined using a “mix-and-match” strategy to produce best-in-class compounds that are capable of potently agonizing odorant receptor activity and may form the basis for the identification of a new mode of insect behavior modification.
Keywords: Odorant Receptor, VUAA1, Structure Activity Relationship, Drosophila melanogaster
Insect chemosensory receptors from the odorant receptor (OR) superfamily are responsible for a large portion of the peripheral signal transduction that is required for the detection of a wide array of volatile odorant compounds that exist in an insect’s environment[1]. In contrast to their mammalian counterparts, which are G-protein coupled receptors, insect ORs act as heteromeric ligand (odorant)-gated cation channels consisting of two 7-transmembrane domain protein subunits: the obligate OR co-receptor (Orco) and an odorant-interacting “tuning” OR (ORx)[2-5]. While Orco is highly conserved across insect taxa, the tuning ORs tend to be highly divergent and, for the most part, specific to individual insect species[6]. Recognition of an odorant ligand by the tuning OR is thought to open the non-selective channel complex, which initiates action potentials in odorant receptor neurons (ORNs), leading to downstream neuronal activity that allows an insect to sample and respond to its chemical environment[4, 7]. As many aspects of insect behavior are directed on the basis of these olfactory processes, adversely affecting or otherwise modulating the ability of an insect to correctly sense and interpret environmental cues represents an established method of altering behavior to reduce the impact of a wide range of economically and medically important insect pests and disease vectors.
We have previously reported the identification and characterization of VUAA1 as an agonist of Orco action[8-10]. Similar independent efforts have identified additional active analogs in this compound class[13-14]. We have previously reported that more potent analogs from the same chemical class are capable of affecting the behavior of mosquito larvae in mobility assays at concentrations similar to, or lower than those required for VUAA1 activity [11]. Thus, we have continued to seek more potent analogs of VUAA1 that would be capable of agonizing the ion channel at more therapeutically useful doses. Here, we report the full details of the structure-activity relationships (SAR) in the VUAA1 series that led to the identification of more potent analogs. We have performed a systematic evaluation of each section of the chemical template and have noted that only an extremely narrow group of substitutions leads to agonist activity. Following this initial survey, a “mix and match” strategy was undertaken to produce potent compounds. The VUAA1-based agonists were assembled using a straightforward and flexible synthetic route (Scheme 1). The route begins with commercially available hydrazides (2), which were reacted with isothiocyanates and then subsequently cyclized to generate 3-thio-1,2,4-triazoles (3). If necessary, carboxylic acid esters (1) were first converted to the hydrazides. To construct the final analogs, the thiols (3) were alkylated in a two-stage process beginning with the condensation of anilines (4) with chloroacetyl chloride. The resulting crude intermediate was reacted with the thiol to generate the agonists (5).
Scheme 1.
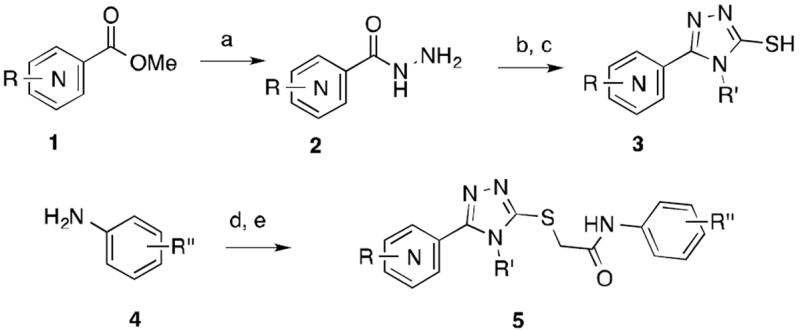
Synthetic route to VUAA1-based odorant agonists.
aReagents and Conditions: a) hydrazine, 150°C, μwave, 15min; b) isothiocyanates, 150°C, μwave, 15min; c) K2CO3, H2O, reflux, 16h; d) ClCH2COCl, triethyl amine, 2h; e) 3, CsCO3, acetonitrile, 16h
All analogs were evaluated for Orco agonist activity using a high-throughput calcium mobilization/imaging assay, as previously described[9, 10, 12]. For this SAR study, we used HEK293 cells stably expressing Orco derived from Drosophila melanogaster (DmOrco) on its own or, in some instances, together with additional tuning OR subunits. Using this assay across a range of compound concentrations allowed the determination of the half maximal effective concentration (EC50) of the compound, as well as the maximal agonist activity, which was expressed as a percentage of VUAA1 activity.
Initially, we examined the SAR of the aniline portion of the molecule. VUAA1 has a 4-ethyl aniline in this position. We observed an extremely narrow constraint for active compounds – only structures very close to the parent maintained activity (Table 1). VUAA1 (6a) activates DmOrco with an EC50 of 35 μM. Simply moving the ethyl substitution to the meta position (6b) completely abrogated activity (other substitutions in the 2- and 3-position of the phenyl ring also failed to demonstrate agonist activity, data not shown). While removing one carbon sharply reduced activity (6c), we found that the addition of one carbon to form the propyl analog (6d) was tolerated, while a butyl substitution (6e) was inactive. Although the binding site for these allosteric agonists is unknown, these results suggest a very narrow steric constraint at this end of the molecule. We next examined branching on the substituent at the 4-position of the aniline. The isopropyl analog (6f) was superior to VUAA1 in both EC50 and overall agonist activity. However, the tert-butyl moiety (6g) displayed reduced agonism.
Table 1.
Structure-activity relationships on the aniline ring.
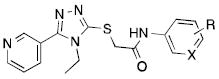
| ||||
|---|---|---|---|---|
| Cmpd | R | X | EC50 (μM)a | % VUAA1 efficacyb |
| 6a (VUAA1) | 4-ethyl | H | 35.1 | 100 % |
| 6b | 3-ethyl | H | - | No agonism |
| 6c | 4-methyl | H | - | No agonism |
| 6d | 4-propyl | H | 94.1 | 57 % |
| 6e | 4-butyl | H | - | No agonism |
| 6f | 4-isopropyl | H | 11.7 | 127 % |
| 6g | 4-tertbutyl | H | LA d | 42 % d |
| 6h | 4-acetyl | H | 84.7 | 96 % |
| 6i | 4-methoxy | H | - | No agonism |
| 6j | 4-bromo | H | - | No agonism |
| 6k | 4-vinyl | H | 102 | 57 % |
| 6l | 4-ethynyl | H | - | No agonism |
| 6m | 2-methyl-4-ethyl | H | - | No agonism |
| 6n | 2-bromo-4-ethyl | H | - | No agonism |
| 6o | cyclohexylc | - | - | No agonism |
| 6p | 4-ethyl | N | - | No agonism |
Mean result of 4 experiments.
Maximum agonism of the compound, normalized to the activity of VUAA1.
Entire ring replaced.
LA = Low agonism. Compound shows agonism only at the highest concentration tested, but no EC50 could be calculated. Maximum observed agonism, but from an incomplete curve.
Along with the steric constraints at the 4-position of the phenyl ring, we also explored changes to the nature of the substitution. Although a methyl ketone (6h) was somewhat tolerated, other changes (e.g. methoxy and halides) were not (6i-j). Unsaturated carbon chains were partially effective, with a vinyl group (6k) showing activity, although reduced in potency relative to VUAA1. With the addition of a further degree of unsaturation using an alkynyl group (6l), all activity was lost. In general, additional substitutions on the ethylaniline were not tolerated (e.g. 6m-n), and non-phenyl analogs were ineffective as agonists (6o-p).
We surveyed a small set of indoline-based amides (Table 2) with substitution at the 5-position, which corresponds to the aniline 4-position that generated positive results. Similar to the anilines, a very narrow SAR was observed, with only the bromo (7b) and ethyl (7e) derivatives maintaining agonist activity. In addition, we examined tetrahydroquinoline analogs, exemplified by 7k, which were also inactive. Surprisingly, however, the indole (7j) was noted as a moderate agonist, with activity inferior to VUAA1.
Table 2.
Indoline-based agonists.
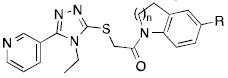
| ||||
|---|---|---|---|---|
| Cmpd | R | n | EC50 (μM)a | % VUAA1 efficacyb |
| 6a (VUAA1) | N/A | 35.1 | 100 % | |
| 7a | H | 1 | - | No agonism |
| 7b | Br | 1 | 21.5 | 82% |
| 7c | F | 1 | - | No agonism |
| 7d | Methyl | 1 | - | No agonism |
| 7e | Ethyl | 1 | 38.6 | 128% |
| 7f | Methoxy | 1 | - | No agonism |
| 7g | N,N-dimethylamino | 1 | - | No agonism |
| 7h | Isopropyl | 1 | - | No agonism |
| 7i | Tertbutyl | 1 | - | No agonism |
| 7j |
 c c
|
47.6 | 71% | |
| 7k | Br | 2 | - | No agonism |
Mean result of 4 experiments.
Maximum agonism of the compound, normalized to the activity of VUAA1.
Entire ring replaced.
Several changes to the sulfide-containing linker region were examined, including the addition of a methylene to the linker length, as well as substitution on the linker and replacement of the sulfur with a oxygen atom or a sulfonyl moiety. Although these were explored within the context of the optimal aniline of 6f, no activity was noted with any of these analogs (not shown). Even simple methylation of the amide nitrogen produced inactive analogs (not shown), reinforcing a preference for the unmodified linker.
We also examined the contribution of the aromatic ring at the 3-position of the triazole, using the 4-isopropyl aniline found for compound 6f. Once again, a narrow SAR space for active compounds was observed. Compounds without an aromatic ring (e.g. 8a, 8b) were inactive. While a 2-pyridyl was inactive, a 4-pyridyl analog showed improved potency and agonist efficacy relative to both VUAA1 and 6f, both of which contain a 3-pyridyl in this position. Analogs with various heterocyclic isosteres (8f-l) met with minimal success, although both a methylated pyrazole (8k) and a thiazole (8l) were noted as weak agonists. Interestingly, 8l displayed good potency although the overall agonist activity was weak.
As the final portion of the systematic evaluation of the SAR of the compound class, we examined the impact of the nitrogen substitution of the triazole. For this study, we employed the 4-pyridyl moiety and the 4-isopropyl aniline of 8d. Notably, an oxadiazole derivative with no substitution at this position was not active (not shown), nor was a methyl substitution (9a). Substitution on the ethyl moiety was not tolerated (9b,c), and lengthening the carbon chain was only marginally successful (9d,e). Interestingly, unsaturation was tolerated (9f), although it led to reduced activity. Although larger carbocycles were poorly tolerated (9h-9j), a cyclopropyl analog (9g) produced one of the most potent and efficacious analogs in the study.
Having thoroughly explored the contributions of the individual segments of the molecule, we engaged in an organized “mix and match” strategy to combine the best substituents at each position with each other to seek possible complementarity in the SAR. Selected successful examples are shown in Table 5. As expected, the combination of the 3- pyridyl and 4-pyridyl aryl groups with ethyl and cyclopropyl triazole substitutions produced consistently good results compared with VUAA1. In particular, the cyclopropyl triazole substitution allowed a range of anilines to have activity (10a-10d). Notably, some of these generated an EC50 below 5 μM and showed efficacy superior to VUAA1 (e.g. 10b). Surprisingly, however, the 4-pyridyl moiety did not appear to provide as large a boost in this context (compare 10d with 10a). Interesting results were also obtained with the methyl pyrazole substitution (10e,f). Strikingly, some of the compounds showed massive increases in agonist efficacy, but without corresponding potency improvements (e.g. 10e). The mix and match combination strategy revealed a surprising tolerance for the indoline-based compounds, with several examples possessing EC50 values superior to VUAA1, along with comparable efficacy (10g-k). Interestingly, a wider than anticipated range of triazole substitutions were allowed (10j, 10k) with some substitution patterns. Representative dose response curves (Figure 1) demonstrate the range of agonist activities observed with selected compounds from this combination strategy.
Table 5.
Selected results from the combination strategy.
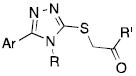
| |||||
|---|---|---|---|---|---|
| Cmpd | Ar | R | R’ | EC50 (μM)a | % VUAA1 efficacyb |
| 6a (VUAA1) | 3-pyridyl | Ethyl | 4-ethylaniline | 35.1 | 100 % |
| 10a | Cyclopropyl | 4-ethylaniline | 11.9 | 171 % | |
| 10b | 4-isopropyl aniline | 3.64 | 178 % | ||
| 10c | 4-vinylaniline | 34.4 | 126 % | ||
| 10d | 4-pyridyl | 4-ethylaniline | 37.0 | 150 % | |
| 10e | 1-methyl,4-pyrazolyl | Ethyl | 4-ethylaniline | LA | 122 % d |
| 10f | Cyclopropyl | 4-isopropyl aniline | 10.7 | 175 % | |
| 10g | 4-pyridyl | Ethyl | 5-ethylindoline | 22.0 | 130 % |
| 10h | Cyclopropyl | 5-ethylindoline | 15.0 | 113 % | |
| 10i | 3-pyridyl | 5-bromoindoline | 16.7 | 98 % | |
| 10j | n-propyl | 5-ethylindoline | 17.6 | 99 % | |
| 10k | 4-pyridyl | Allyl | 5-ethylindoline | 17.9 | 76 % |
Mean result of 4 experiments.
Maximum agonism of the compound, normalized to the activity of VUAA1.
LA = Low agonism. Compound shows agonism only at the highest concentration tested, but no EC50 could be calculated.
Maximun observed agonism, but from an incomplete curve.
Figure 1. VUAA analogs as agonists of DmOrco.
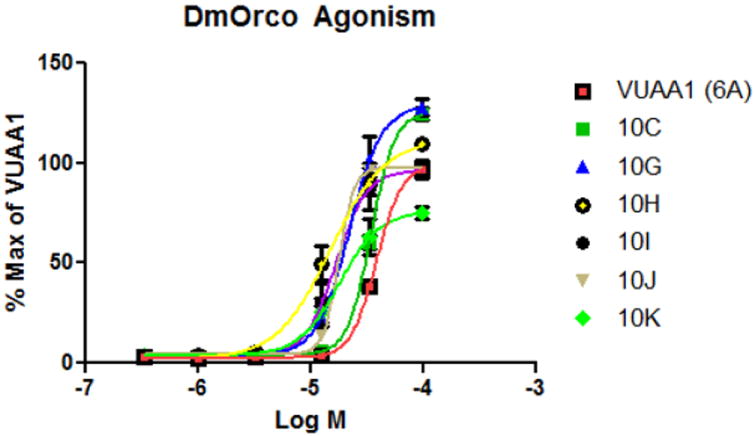
Concentration response curves of DmOrco expressing HEK cells in the presence of a series of increasing concentrations of test compound (Log M) vs. the % Max fluorescence using VUAA1.
In summary, we have thoroughly explored the SAR around a triazole-based series of DmOrco agonists. Significant activity was obtained within only a very narrow set of compound structures. The most successful analogs presented a five- or sixmembered heterocycle at the 3-position of a 1,2,4-triazole, and a small alkyl substituent on the nitrogen. A range of aniline-based end pieces were tolerated, but all active analogs possessed a small 4-position substitution (or the equivalent substitution on an indoline ring). This narrow, but definable, SAR may be indicative of a specific binding location with tight steric and electronic tolerances. However, the binding site and the precise kinetics of these allosteric ORco agonists remain unknown. Compounds in this series have been shown to affect the behavior of insect larvae [11] as well as adults (LJZ, unpublished observations). Thus, these compounds may have potential as excito-repellents that may be able to manipulate of the destructive insect behaviors and/or alter the behavior of economically and medically important insects. Further work to define the binding interactions of these compounds and to explore their use in the context of insect behavior modification is in progress and will be reported in due course.
Table 3.
Examination of the pyridyl ring SAR.
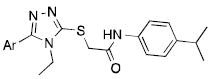
| |||
|---|---|---|---|
| Cmpd | Ar | EC50 (μM)a | % VUAA1 efficacyb |
| 6f | 3-pyridyl | 11.7 | 127 % |
| 8a | H | - | No agonism |
| 8b | Br | - | No agonism |
| 8c | 2-pyridyl | - | No agonism |
| 8d | 4-pyridyl | 6.8 | 150 % |
| 8e | 2-F, 4-Pyridyl | 60.1 | 148 % |
| 8f | 3-pyrrolyl | - | No agonism |
| 8g | 3-furyl | - | No agonism |
| 8h | 3-thiophenyl | - | No agonism |
| 8i | 1-methyl,3-pyrazolyl | - | No agonism |
| 8j | 4-pyrazolyl | - | No agonism |
| 8k | 1-methyl,4-pyrazolyl | 107 | 34 % |
| 8l | 5-thiazolyl | 10.7 | 36 % |
Mean result of 4 experiments.
Maximum agonism of the compound, normalized to the activity of VUAA1.
Table 4.
Examination of substitution on the triazole nitrogen.
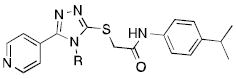
| |||
|---|---|---|---|
| Cmpd | R | EC50 (μM)a | % VUAA1 efficacyb |
| 8d | Ethyl | 6.8 | 150 % |
| 9a | Methyl | - | No agonism |
| 9b | i-Propyl | - | No agonism |
| 9c | t-Butyl | - | No agonism |
| 9d | n-Propyl | 84.0 | 28 % |
| 9e | n-Butyl | - | No agonism |
| 9f | Allyl | 35.9 | 111 % |
| 9g | Cyclopropyl | 3.9 | 162 % |
| 9h | Cyclopentyl | LAc | 13 % |
| 9i | Cyclohexyl | - | No agonism |
| 9j | Phenyl | - | No agonism |
Mean result of 4 experiments.
Maximum agonism of the compound, normalized to the activity of VUAA1.
LA = Low agonism. Compound shows agonism only at the highest concentration tested, but no EC50 could be calculated.
Acknowledgments
We would like to thank Paul Reid for his invaluable insight and contributions to this project. This work was supported by a grant from the Foundation for the National Institutes of Health through the Grand Challenges in Global Health Initiative (to L.J.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaupp UB. Nat Rev Neurosci. 2010;11(3):188–200. doi: 10.1038/nrn2789. [DOI] [PubMed] [Google Scholar]
- 2.Smart R, Kiely A, Beale M, Vargas E, Carraher C, Kralicek AV, Christie DL, Chen C, Newcomb RD, Warr CG. Insect Biochem Mol Biol. 2008;38(8):770–80. doi: 10.1016/j.ibmb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Benton R, Sachse S, Michnick SW, Vosshall LB. PLoS Biol. 2006;4(2):e20. doi: 10.1371/journal.pbio.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato K, Pellegrino M, Nakagawa T, Nakagawa T, Vosshall LB, Touhara K. Nature. 2008;452(7190):1002–6. doi: 10.1038/nature06850. [DOI] [PubMed] [Google Scholar]
- 5.Wicher D, Schäfer R, Bauernfeind R, Stensmyr MC, Heller R, Heinemann SH, Hansson BS. Nature. 2008;452(7190):1007–11. doi: 10.1038/nature06861. [DOI] [PubMed] [Google Scholar]
- 6.Jones WD, Nguyen T-AT, Kloss B, Lee KJ, Vosshall LB. Curr Biol. 2005;15(4):R119–21. doi: 10.1016/j.cub.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa T. Science. 2005;307(5715):1638–42. doi: 10.1126/science.1106267. [DOI] [PubMed] [Google Scholar]
- 8.Jones PL, Pask GM, Rinker DC, Zwiebel LJ. Proc Natl Acad Sci USA. 2011;108(21):8821–5. doi: 10.1073/pnas.1102425108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rinker DC, Jones PL, Pitts RJ, Rützler M, Camp G, Sun L, Xu P, Dorset DC, Weaver D, Zwiebel LJ. Physiological Entomology. 2012;37(1):33–41. doi: 10.1111/j.1365-3032.2011.00821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones P, Pask G, Romaine I, Taylor R. Plos One. 2012;7(1):e30304. doi: 10.1371/journal.pone.0030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor RW, Romaine IM, Liu C, Murthi P, Jones PL, Waterson AG, Sulikowski GA, Zwiebel LJ. ACS Chem Biol. 2012;7(10):1647–52. doi: 10.1021/cb300331z. [DOI] [PubMed] [Google Scholar]
- 12.Bohbot JD, Jones PL, Wang G, Pitts RJ, Pask GM, Zwiebel LJ. Chem Senses. 2011;36(2):149–60. doi: 10.1093/chemse/bjq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohbot JD, Dickens JC. Neuropharmacology. 2012;62(5-6):2086–95. doi: 10.1016/j.neuropharm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Sisi C, Luetje CW. PLoS One. 2012;7(5):e36784. doi: 10.1371/journal.pone.0036784. [DOI] [PMC free article] [PubMed] [Google Scholar]