

Summary
We have previously reported that LITAF is silenced by promoter hypermethylation in germinal center-derived B-cell lymphomas, but beyond these data the regulation and function of LITAF in B cells are unknown. Gene expression and immunohistochemical studies revealed that LITAF and BCL6 show opposite expression in tonsil B-cell subpopulations and B-cell lymphomas, suggesting that BCL6 may regulate LITAF expression. Accordingly, BCL6 silencing increased LITAF expression, and chromatin immunoprecipitation and luciferase reporter assays demonstrated a direct transcriptional repression of LITAF by BCL6. Gain- and loss-of-function experiments in different B-cell lymphoma cell lines revealed that, in contrast to its function in monocytes, LITAF does not induce LPS-mediated TNFα secretion in B cells. However, gene expression microarrays defined a LITAF-related transcriptional signature containing genes regulating autophagy, including MAP1LC3B (LC3B). In addition, immunofluorescence analysis co-localized LITAF with autophagosomes, further suggesting a possible role in autophagy modulation. Accordingly, ectopic LITAF expression in B-cell lymphoma cells enhanced autophagy responses to starvation, which were impaired upon LITAF silencing. Our results indicate that the BCL6-mediated transcriptional repression of LITAF may inhibit autophagy in B cells during the germinal center reaction, and suggest that constitutive repression of autophagy responses in BCL6-driven lymphomas may contribute to lymphomagenesis.
Keywords: Non-Hogdkin Lymphoma, B cells, Transcription Factors, Germinal Center, Autophagy
Introduction
LITAF was initially identified as the P53-inducible gene 7 (therefore termed PIG7) in the DLD-1 colon cancer cell line (Polyak, et al 1997). Subsequent studies have functionally characterized LITAF, for lipopolysaccharide (LPS)-induced TNF alpha (TNFα) factor, as an activator of the secretion of inflammatory cytokines such as TNFα upon LPS stimulation in monocytes, acting as a transcriptional activator of TNFA (Moriwaki, et al 2001, Myokai, et al 1999, Tang, et al 2005, Tang, et al 2006). In other cell types, however, LITAF can exert non-inflammatory functions. For instance, mutations in LITAF cause abnormalities in protein degradation in the demyelinating neuropathy termed Charcot-Marie-Tooth disease type 1C (Eaton, et al 2012, Somandin, et al 2012, Street, et al 2003). These mutations interfere in the association of the ESCRT machinery with the endosome membrane, a process regulated by LITAF, resulting in reduced endosome-to-lysosome trafficking in Schwann cells (Lee, et al 2012). Therefore, LITAF plays different functional roles that seem to be tissue-specific.
In addition, LITAF has been implicated as a possible tumor suppressor in different malignancies. For instance, in prostate cancer cells LITAF silencing induced cell proliferation and anchorage-independent growth in a xenograft model (Zhou, et al 2011), while in acute myeloid leukemia cells LITAF expression promoted apoptosis and cell differentiation (Liu, et al 2012). In this regard, we have previously shown that LITAF is inactivated by epigenetic mechanisms in mature B-cell lymphoma cells (Mestre-Escorihuela, et al 2007), but beyond these data the regulation and function of LITAF in these cells are presently unknown. LITAF RNA and protein expression was particularly decreased in germinal center (GC) B-cell-like diffuse large B-cell lymphoma (GCB-DLBCL), a tumor entity characterized by constitutively high BCL6 expression due to genetic alterations (Basso and Dalla-Favera 2010, Chen, et al 1998, Ci, et al 2008, Klein and Dalla-Favera 2008). BCL6 is a transcriptional repressor normally expressed in the GCs of secondary follicles, structures where antibodies with high affinity for the antigen are generated during T-cell mediated humoral immune responses, and acts as master regulator of the GC reaction (Basso and Dalla-Favera 2010, Klein and Dalla-Favera 2008). In fact, BCL6 expression promotes several cell functions essential for this process, such as cell proliferation (Parekh, et al 2007, Phan, et al 2005, Saito, et al 2009), attenuation of the DNA damage sensing and repair mechanisms (Phan and Dalla-Favera 2004, Ranuncolo, et al 2007, Ranuncolo, et al 2008) and blocking of terminal B-cell differentiation (Shaffer, et al 2002, Tunyaplin, et al 2004). The continuous activation of these functions upon genetic lesions that lead to constitutive expression of BCL6 is a key determinant of malignant transformation in GC-derived lymphomas (Ci, et al 2009, Ci, et al 2008, Klein and Dalla-Favera 2008).
In this article we show that LITAF is a transcriptional target of BCL6 in B cells. Subsequent experiments revealed that LITAF co-localized with autophagosomes and lysosomes, increasing autophagy responses in B-cell lymphoma cells.
Methods
Tissue samples and cell lines
Human mature B-cell lymphoma cell lines and biopsies were included in the study (full material and experimental procedures are provided as Supplemental Information). Isolation of B-cell subpopulations from human tonsil reactive lymphoid follicles were performed as previously reported (Vicente-Dueñas, et al 2012). Samples were obtained in accordance with the ethical guidelines and after approval of the corresponding Institutional Review Boards.
LITAF gain- and -loss-of-function experiments
LITAF and BCL6 were silenced with specific siRNAs in KARPAS-231 and VAL cells or in OCI-Ly1 cells, respectively. BCL6 function was also targeted with the BCL6 inhibitor peptide BPI (Bio-Synthesis, http://www.biosyn.com), as previously reported (Polo, et al 2004). To express LITAF in SC-1 and RL cells, LITAF cDNA was cloned in the tet-on RLT-GFP plasmid (Watsuji, et al 1997). Retroviral vectors were produced and stable transfectants were selected as previously reported (Richter-Larrea, et al 2010). LITAF expression was induced with 50 ng/mL doxycycline.
Western blot analysis
Western blotting was performed as previously described (Mestre-Escorihuela, et al 2007), using specific antibodies for LITAF (clone 30, BD Biosciences); BCL6 (clone PG-B6p, DakoCytomation), rabbit anti-LC3A/B (Cell Signaling) and Actin (JLA20, Calbiochem).
Chromatin immunoprecipitation (Q-ChIP) and ChIP sequencing (ChIP-Seq)
Q-ChIP and ChIP-Seq experiments were performed in OCI-Ly1, as previously reported (Duy, et al 2010). Primers were designed to amplify three regions containing a putative binding site each for BCL6 (Ci, et al 2009). ChIP-Seq data are published under GEO accession no. GSM763399.
Luciferase assays
The DNA sequence found enriched in the ChIP assays was cloned between the XhoI and KpnI restriction sites of the pGL3 Control vector (Promega). Six bases (TTCTTAAG to GGATGCTG) were mutated in the putative BCL6 binding site. Dual-Luciferase Reporter Assay System (Promega) experiments were performed as previously described (Malumbres, et al 2009).
Gene expression microarray analysis
Affymetrix gene expression microarray hybridization and data analyses were performed as previously reported (Vicente-Dueñas, et al 2012). Data are published under GEO Database accession numbers GSE25638, GSE25613 and GSE42204.
Evaluation of autophagy responses
To induce autophagy, DLBCL cells over-expressing LITAF, or cells in which its expression was silenced, were starved in Earle’s Balanced Salt Solution (Sigma-Aldrich) and stained with acridine orange (Invitrogen), followed by flow cytometry to measure the FL3/FL1 ratio, as previously reported (Takeuchi, et al 2005). Increases in the number of acidic vesicles augment the FL3/FL1 ratio after acridine orange staining and correlate with increased autophagy. LC3BI to LC3BII conversion, a required step for autophagosome membrane formation (Mizushima, et al 2010), was assessed by Western blot.
Immunohistochemical (IHC) and Immunofluorescence (IF) studies
IHC staining of tonsil lymphoid follicles was performed using the EnVision system (DakoCytomation) with antibodies for BCL6 (clone PG-B6p, DakoCytomation), LITAF (clone 30, BD Biosciences), CD3 (clone F7.2.38, DakoCytomation) and CD20 (clone L26, DakoCytomation). IF was performed as previously reported (Beltran, et al 2011), with the same antibodies as for Western blotting.
Q-RT-PCR
For RT reactions, 0.5 μg of total RNA were retro-transcribed using MMLV-RT (Invitrogen) following the manufacturer’s instructions. Q-PCRs were performed in an ABI PRISM 7500 device (Applied Biosystems) using Taqman Gene Expression Assays (Applied Biosystems) for LITAF (Hs00191583_m1) and BCL6 (Hs00277037_m1). Data were normalized to GAPDH expression (Hs.99999905_m1, Applied Biosystems) and to the corresponding controls using the ΔΔCt method for calculations.
Results
LITAF and BCL6 expression are inversely correlated in mature B-lymphocyte subpopulations and B-cell lymphomas
To elucidate the function of LITAF in B cells, we first assessed its expression in B-cell subpopulations isolated from tonsil reactive lymphoid follicles. LITAF mRNA and protein expression levels were higher in naïve and memory B lymphocytes than in GC B cells, exhibiting reciprocal expression to BCL6 (Fig. 1A). Accordingly, IHC analysis identified cytoplasmic expression of LITAF in naïve and memory B-cell areas surrounding GCs, which were devoid of BCL6 expression, whereas GC cells exhibited strong nuclear BCL6 expression without LITAF staining (Fig. 1B). Of note, a minor cell population in the GC displayed LITAF expression, but lacked BCL6 staining as revealed by double immunostaining (Fig. 1C), further suggesting a negative correlation between BLC6 and LITAF. These LITAF-expressing GC cells were identified as CD20+ B-cells and CD3+ T cells, both of which were also detected out of the GC (Figs 1D-E). In GC-derived lymphomas, BCL6 is targeted by chromosomal translocations or point mutations that induce its constitutive expression, leading to malignant transformation. We hypothesized that these lymphomas with high levels of BCL6 could show particularly low levels of LITAF. As expected, GCB-DLBCL cell lines displayed BCL6high/LITAFlow expression, while the non-GC-derived mantle cell lymphoma and splenic marginal-zone lymphoma cell lines displayed a BCL6low/LITAFhigh expression pattern, and activated B-cell like-DLBCL cells showed intermediate expression of both proteins (Fig. 1F). In addition, expression microarray data of biopsy specimens from mature B-cell lymphoma patients were analyzed for LITAF and BCL6 expression, and most of them also exhibited an inverse correlation in the expression of these genes: GC-derived lymphomas such as GCB-DLBCL and follicular lymphoma displayed a BCL6high/LITAFlow expression pattern, whereas splenic marginal-zone lymphoma and mucosa-associated lymphoid tissue lymphoma showed the opposite pattern (Fig. 1G). Accordingly, Q-RT-PCR analysis in an independent series of 119 DLBCL biopsies confirmed the inverse correlation of LITAF and BCL6 expression (Spearman correlation coefficient -0.326, p<0.001), being mean LITAF expression 2-fold higher in the subgroup of patients with BCL6 expression below the median of the cohort (Fig. 1H). Furthermore, we found that high LITAF expression correlated with a high mortality score based on the expression of 6 genes (Malumbres, et al 2008) in these patients (Fig. 1H and Suppl. Fig. 1A), probably by identifying patients of the bad prognosis ABC-DLBCL subgroup, that are characterized by low BCL6 expression. This association of high LITAF expression with worse overall survival was corroborated with data of a published series of 240 DLBCL patients (Rosenwald, et al 2002) when the ABC and GCB DLBCL subtypes were grouped, while high LITAF expression showed a tendency for better overall survival in the less frequent Type III subgroup (Suppl. Fig. S1B). We also found a tendency for worse overall survival in DLCBL patients with high LITAF expression measured by IHC in tissue microarrays (Suppl. Fig. S1C). In summary, our data suggest that LITAF may be transcriptionally repressed by BCL6 both in non-transformed and lymphoma B cells.
Figure 1. BCL6 represses LITAF transcription in B cells.
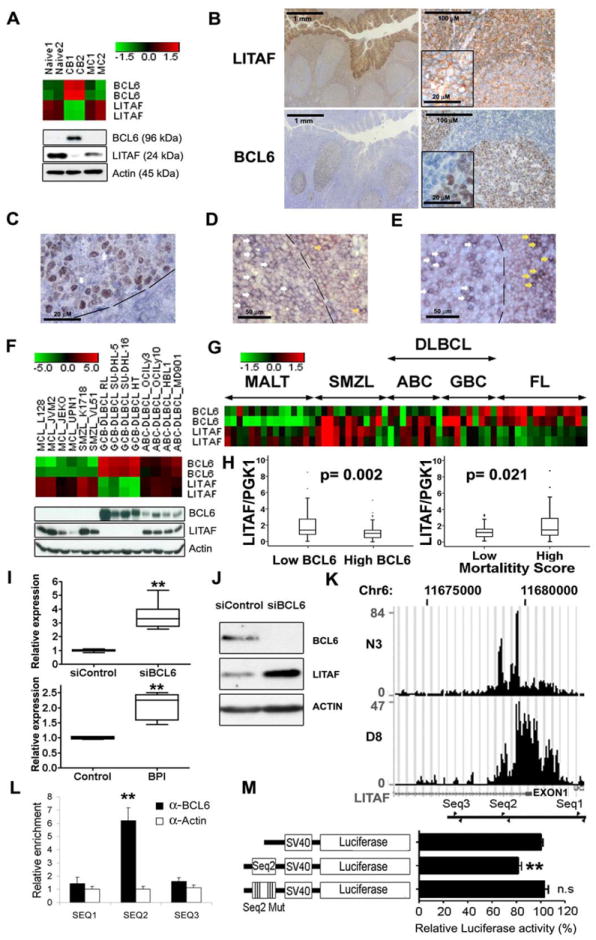
LITAF and BCL6 expression were assessed by analysis of gene expression microarrays (heatmap of RMA normalized log2 transformed values, two probe sets per gene) (A, upper panel) Western blotting (A, lower panel), immunohistochemistry (brown) (B) and double immunostaining (BCL6 brown and LITAF blue) (C) in reactive human tonsils. The white arrow in C points to a cell strongly expressing LITAF that is negative for BCL6 staining inside the GC, delimited by a discontinuous line. Details of higher magnification of the cells are embedded in two micrographs in B. Double staining for LITAF in red and CD20 (D) or CD3 (E) in blue showed that some B cells as well as T cells express LITAF inside (white arrows) and outside (yellow arrows) the GC. (F) Heatmap of microarray gene expression data (upper panel) and protein expression assessed by Western blot (lower panel) of LITAF and BLC6 in cell lines of mantle cell lymphoma (MCL), splenic marginal zone lymphoma (SMZL), diffuse large B-cell lymphoma of the GC subtype (GCB-DLBCL) and of the activated B-cell-like subtype (ABC-DLBCL). LITAF and BCL6 mRNA expression levels were also analyzed in microarray data of a series of B-cell lymphoma cases including 9 GCB-DLBCL, 9 ABC-DLBCL, 15 mucosa associated lymphoid tissue (MALT) lymphoma, 15 follicular lymphoma (FL) and 12 SMZL specimens (G), as well as analyzed by Q-RT-PCR in biopsies of 119 DLBCL cases and compared between patients with high (above cohort median) and low BCL6 expression or mortality score (H). LITAF mRNA expression assessed by Q-RT-PCR increased in the BCL6 expressing GCB-DLBCL cell line OCI-Ly1 after BCL6 silencing with a siRNA (I, upper panel) or BCL6 inhibition with the BPI peptide (I, lower panel). Q-RT-PCR data was normalized to the corresponding controls as 2-ΔΔCt. Boxplots of three independent experiments are shown. LITAF increase was also confirmed at protein level by Western Blot (J). (K) ChIP-Seq enrichment around LITAF exon 1 (small black square under the enrichment histograms) corresponding to two different anti-BCL6 antibodies (N3 and D8). The location of the primer pairs used for Q-ChIP experiments are depicted at the bottom. The enrichment of the sequence between the second primer pair was confirmed by Q-ChIP (L), using an anti-actin antibody as negative control. Data are displayed as mean±SD of 2 independent experiments. This sequence (Seq2) was cloned in a luciferase expression plasmid controlled by the SV40 promoter and showed a repressive effect abolished by the mutation of the putative binding site for BCL6 located in this sequence (Seq2 Mut) in Dual Luciferase assays (M). Mean±SEM data of 4 independent luciferase experiments in OCI-ly1 cells are shown. ** means statistical significance with p value below 0.01, n.s., non-significant differences (p> 0.05). CB, centroblasts; MC, memory B cells; Naïve, naïve B cells.
BCL6 directly binds to LITAF intron 1and represses LITAF transcription
Supporting the notion that BCL6 can regulate LITAF expression, peptide-mediated inhibition and siRNA-mediated silencing of BCL6 in OCI-Ly1 cells induced 2.1±0.2-fold and 3.4±0.4-fold increases, respectively, in LITAF mRNA levels (Fig. 1I). This increment resulted in an increased amount of LITAF protein measured by Western blot (Fig. 1J). To check whether BCL6 was able to bind to the LITAF promoter, we first analyzed data from high-throughput chromatin immunoprecipitation sequencing (ChIP-Seq) experiments performed in OCI-Ly1 cells using two different anti-BCL6 antibodies (Fig. 1K). This analysis showed a region of enrichment including the first exon of LITAF (as on RefSeq NM_004862.3, chr16: 11,680,229-11,680,003) and sequences nearby. Three sequences very similar to the consensus binding site for BCL6 ([A/T]TC[C/T][A/T][A/C]GA) (Ci, et al 2009) were located inside or in the vicinity of the ChIP-Seq enriched region. Quantitative real-time chromatin immunoprecipitation (Q-ChIP) experiments, using specific primers flanking each of these putative binding sites, showed a statistically significant 6-fold enrichment (p= 0.002) for the 98 bp sequence that included the second putative BCL6 binding sequence (TTCTTAAG) located at Chr.16:11,678,778-11,678,771, 1,224 bp downstream of the non-coding exon 1 in the LITAF gene (Genome Browser assembly GRCh37/hg19) (Fig. 1L). To confirm that this BCL6 binding site regulated LITAF expression, we cloned a region of 309 bp containing this motif upstream of the luciferase gene in a reporter vector. This resulted in a 20% decrease in luciferase activity in OCI-Ly1 cells, which was abolished when the BCL6 binding site was mutated (Fig. 1M). Taken together, these data indicate that LITAF is a novel direct transcriptional target of BCL6 in B cells.
LITAF is a positive regulator of autophagy in B cells
In monocytes, LITAF expression increases upon LPS exposure and induces TNFα secretion (Myokai, et al 1999). However, the function of LITAF in B cells is unknown. Unexpectedly, LITAF expression did not augment upon LPS exposure nor induced TNFα secretion in 8 out of 9 different B-cell lymphoma cell lines (Figs 2A-B), with the only exception of OCI-Ly10, which increased LITAF protein expression upon LPS exposure during 24h and showed a minor parallel increment of TNFα secretion from 0 to 16.1±14.7 pg/μl. To further confirm that the induction of TNF secretion is not the function of LITAF in B cells, TNFα was quantified in the supernatants of SC-1 and RL cell lines stably transfected with a tetracycline-inducible expression vector containing the full-length LITAF cDNA after doxycycline-induced LITAF expression, and no increment of this cytokine was found (Figs 2B-C). Moreover, TNFα secretion after LITAF silencing in KARPAS-231 and VAL cells was not decreased (Figs 2B, D). Interestingly these cell lines, as well as others studied by subcellular fractionation and Western blot, showed LITAF expression only in the cytoplasm and not in the nucleus (Fig. 2E), further supporting the hypothesis that LITAF is not acting as transcription factor in B cells. Likewise, LITAF expression had no effect on cell viability (Figs 2F-G). Interestingly, gene expression microarray analysis showed that the transcriptional signature driven by LITAF silencing in KARPAS-231 and VAL cells included genes involved in AMPK signaling, an important regulator of autophagy (Zhou, et al 2011) (Supplemental Fig. S2). In addition, MAP1LC3B (LC3B), an ortholog of the yeast autophagosome protein Atg8, was found 2- and 1.2-fold induced in gene microarray experiments in RL and SC-1 cells, respectively, upon LITAF over-expression. In addition, subsequent IF studies co-localized LITAF with autophagosomes and lysosomes in non-transformed and lymphoma B cells, further supporting the involvement of LITAF in autophagy regulation (Figs 3A-D). In agreement with this hypothesis, RL cells ectopically over-expressing LITAF displayed 1.6-fold increased FL3/FL1 ratio after acridine orange staining, which correlates with an increase in the number of acidic vesicles, and a 1.4-fold increase in LC3BII/LC3BI ratio, which implies an increment in the amount of autophagosomes, in comparison to control cells upon starvation (Figs 3E-F). This increased induction of autophagy was confirmed by the higher number of RL cells overexpressing LITAF that showed LC3B staining condensed in autophagy vacuoles, observed by confocal microscopy (Fig. 3I-L). Consistently, siRNA-mediated LITAF silencing in starved KARPAS-231 cells induced a 4.3-fold decrease in the FL3/FL1 ratio and a 2.5-fold reduction of the LC3BII/LC3BI ratio (Figs 3G-H), confirming that LITAF is involved in the regulation of autophagy responses. In addition, LITAF induction in OCI-LY1 cells upon BCL6 silencing resulted in an increase in LC3BI to LC3BII conversion, particularly in basal conditions but also after starvation, as assessed by Western Blot (Fig. 3M), further supporting the involvement of the BCL6/LITAF axis in the regulation of autophagy in B cells.
Figure 2. LITAF does not induce TNFα secretion upon LPS exposure in B cells.
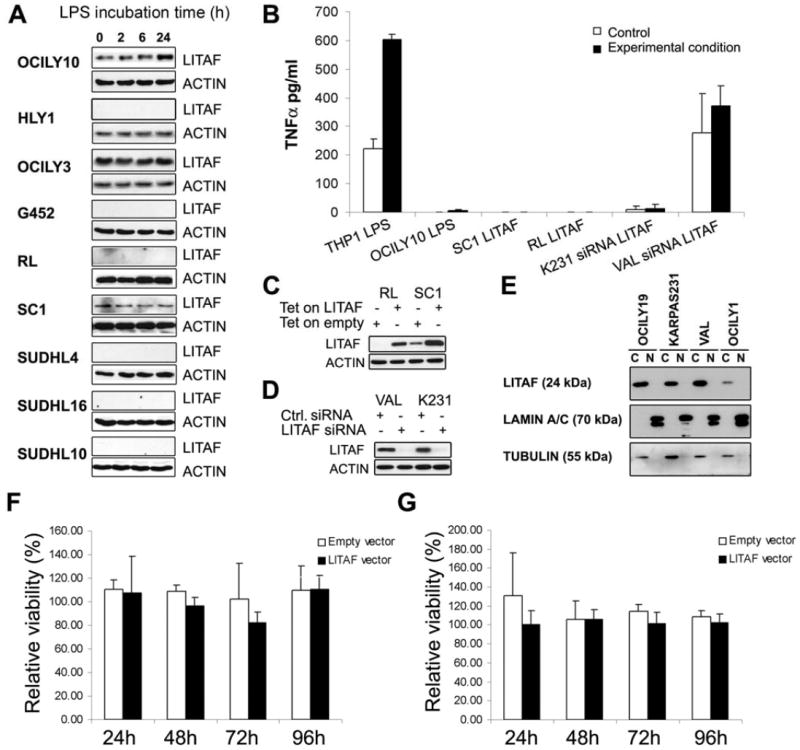
(A) LITAF expression assessed by Western blot in 9 cell lines of Diffuse Large B Cell Lymphoma (DLBCL) incubated with LPS during 24h, using actin detection as loading control. (B) TNFα secretion measured by ELISA in THP1 and OCI-Ly10 cells treated with LPS for 24 hours, in RL and SC-1 cells harboring a tetracycline inducible LITAF expression plasmid (Tet on LITAF) or the corresponding empty vector (Tet on Empty) in presence of 50 ng/mL doxycycline, and in KARPAS-231 (K231) and VAL cells transfected with a LITAF specific siRNA or non-targeting siRNA (Ctrl.). Subcellular fractionation was performed in four DLBCL cell lines as previously described (Beltran, et al 2011) and LITAF was detected by Western blot (C) β-tubulin and LAMIN A/B were also analyzed as markers of the cytoplasmic “C” and nuclear “N” fractions, respectively. The induction of LITAF expression (D) and the silencing of LITAF (E) were confirmed by Western blot. The effect on cell viability of LITAF over-expression induced by doxycycline exposure in SC-1 (F) and RL (G) cells transfected with a tetracycline inducible expression vector was measured by MTS assays. The results are normalized to the corresponding cells not treated with doxycycline. Error bars represent the standard deviation of three independent experiments.
Figure 3. LITAF is involved in autophagy regulation.
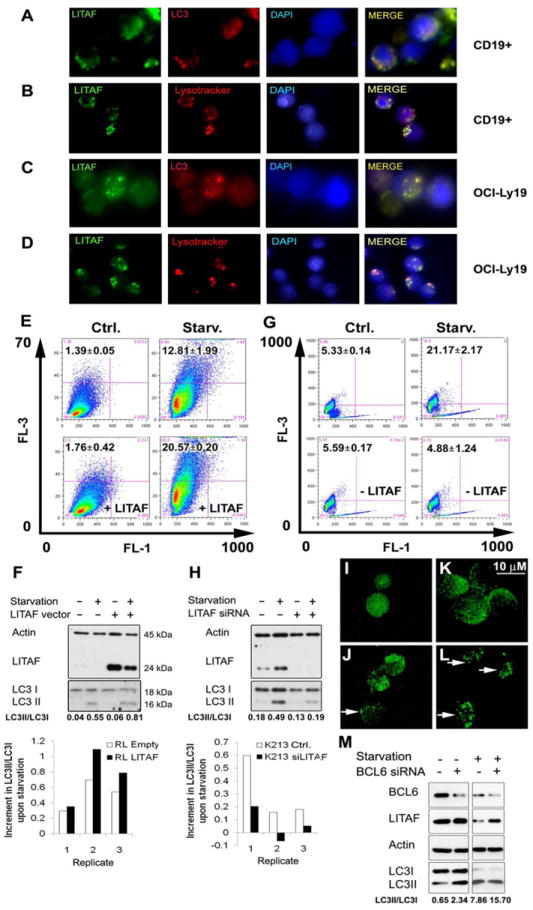
Detection by immunofluorescence of LITAF (Alexa Fluor 488), LC3B (Alexa Fluor 594) and Lysotracker (red) in healthy donor CD19+ B cells (A, B) and OCI-Ly19 cells (C, D) was performed. Nuclei were contrasted with DAPI staining. Images were captured using an epifluorescence inverted microscope Axio Imager 21 (Zeiss). (E) Analysis by flow cytometry with FACSCalibur and the FlowJo software of the FL3/FL1 ratio after acridine orange staining of RL cells stably transfected with a tetracycline inducible LITAF expression vector (lower panels) or the corresponding empty vector (upper panels) in presence of 50 ng/mL doxycycline, in standard culture conditions (left panels) or upon 6 h starvation (right panels). The corresponding mean±SEM of the percentage of cells in the upper left corner of three independent experiments are shown in black numbers in each panel. (F) Analysis by Western blot of LITAF, LC3BI (LC3I) and LC3BII (LC3II) in doxycycline treated RL cells stably transfected with a tetracycline inducible LITAF expression vector or the empty control vector, cultured in standard conditions or upon starvation for 4 h including 2 h incubation with 100 μM leupeptin plus 20 mM NH4Cl to avoid LC3BII degradation by the autophagic flux. Actin was also analyzed as a loading control. Mean values of LC3BII/LC3BI from three independent experiments are displayed below, and the corresponding increments of this ratio in the starved cells compared with non-starved cells in the same three replicate experiments are also shown at the bottom. Data were digitalized with a ScanMaker E900 scanner and quantified with ImageJ 1.46r. The same analyses of FL3/FL1 cytometry, comparing the non-starved cells with the corresponding starved ones (G) and LC3B Western blot (H) were performed in KARPAS-231 cells transfected with a siRNA for LITAF (siLITAF) or with a non-targeting control siRNA (Ctrl.). OCI-LY1 cells were likewise analyzed by Western blot upon BCL6 silencing (M). RL cells with LITAF overexpression and starved exactly as for the Western blot were analyzed by confocal microscopy for LC3B expression (L). Controls including non-starved (I) and starved (J) RL cells harboring the corresponding empty vector, as well as non-starved cells with LITAF overexpression (K) were included. Autophagy vacuoles with intense LC3BII staining (Alexa Fluor 488) coupled to reduced diffuse LC3BI staining in the cytoplasm indicate high autophagy levels (white arrows).
Discussion
In this study, by integrating expression analysis of human B-lymphocyte subsets, ChIP assays and dual luciferase experiments, we demonstrate transcriptional repression of LITAF by BCL6 in B cells, suggesting that LITAF may play a role in mature B-cell development. Despite the fact that LITAF induces TNFα gene expression and secretion upon LPS stimulation in monocytes (Myokai, et al 1999, Tang, et al 2005, Tang, et al 2006), we found that LITAF was rarely induced by LPS in B-cell lymphoma cells and TNFα secretion was not associated with LITAF expression. Furthermore, the location of LITAF in cytoplasmic vesicles observed by IF, as well as its absence in the nucleus of B cells assessed by IHC and subcellular fractionation, also indicated that LITAF was not acting as a transcription factor in B lymphocytes. Rather, our results point to a role of LITAF in promoting autophagy responses in B cells, suggesting a link between autophagy regulation and BCL6. As autophagy is involved in antigen presentation to T cells by B cells within the GCs (Clark, et al 2004, Munz 2009, Strawbridge and Blum 2007, Watanabe, et al 2008), and cognate T-cell/B-cell interactions are known to participate in the selection of the cells with highest affinity for the antigen (MacLennan, et al 1997), the inhibition of LITAF by BCL6 in GC B lymphocytes could hamper antigen presentation during the somatic hypermutation and proliferation processes that are necessary to generate high affinity antibodies. On the other hand, autophagy is essential for T-cell activation due to the high energy requirements of this process (Hubbard, et al 2010), and this could also be the case for B cells at some step of their activation during the humoral immune response. More experiments are warranted to elucidate the significance of the regulation of autophagy by LITAF in B-cell activation and function.
In addition, the regulation of autophagy by LITAF could also have a role in tumorigenesis, as has been reported for other autophagy-related genes. For instance, BECN1, an indispensable gene for autophagy, has been found mono-allelically deleted in 40-75% of sporadic human breast cancers and ovarian cancers (Liang, et al 1999). Furthermore, mono-allelic loss of BECN1 in mice leads to a higher frequency of tumors, including B-cell lymphomas, showing that this gene acts as a haploinsufficient tumor suppressor (Qu, et al 2003, Yue, et al 2003). Thus LITAF, a positive regulator of autophagy like BECN1, might have a similar tumor suppressor role as has been previously suggested by other groups (Liu, et al 2012, Zhou, et al 2011) and constitutive LITAF inhibition by the BCL6 oncoprotein could contribute to B-cell lymphomagenesis. In agreement with this hypothesis, our group previously reported LITAF homozygous deletion and gene expression silencing by promoter hypermethylation in mature GC-derived B-cell lymphomas (Mestre-Escorihuela, et al 2007). Interestingly, we found a tendency for better overall survival in patients of the Type III subgroup of DLBCL with high LITAF expression, supporting the notion of LITAF acting as a tumor suppressor. Nevertheless, when considering the most frequent subgroups of DLBCL, GCB and ABC, high LITAF expression was associated with worse overall survival, most probably because of the higher LITAF expression found in the worse prognosis ABC-DLBCL subgroup. These data encourage further studies to shed light on the possible involvement of LITAF expression inhibition in GC-derived lymphomagenesis.
In summary, our work for the first time provides evidences for the transcriptional repression of LITAF by BCL6 in B cells, whereby LITAF acts as a positive regulator of autophagy. Consequently, BCL6-mediated suppression of LITAF-induced autophagy may play a role in the GC reaction and in GC-derived lymphomagenesis.
Supplementary Material
Acknowledgments
We thank Xabier Morales for excellent confocal imaging acquisition.
This work was supported by Grants from the Spanish Ministries of Science, Innovation and Health (FIS-PI12/0202 and RTICC-RD12/0036/0063 to JAMC; RTICC-RD12/0036/0071to JB and FIS-PI11/00684 to RM); from Fundación Mutua Madrileña (FMM 8553/2011 to RM); from the National Institutes of Health (NIH R01 DE014079, to SA); from AECC (Spanish Association Against Cancer) Scientific Foundation (to RM); from the Government of Navarra (to CB); from the Spanish Ministry of Economy and Competitiveness (the Inncorpora-Torres Quevedo Program PTQ-11-04774 to SR); and by postdoctoral fellowships from the Foundation for Applied Medical Research (to AS and EFR) and the Spanish Ministry of Health ISCIII-FIS (to JIMF).
Footnotes
Authorship contributions
CB, SR, AS, MMV, EFR, JIMF, XS and TT performed experiments; RM, CB and JAMC analyzed and interpreted the data; AM, ISL, AO, YN, JB and SA contributed valuable data, tissues and clinical information. JAMC and RM conceptualized the idea of the study; RM, CB and JAMC wrote the paper. All authors approved the final version of the manuscript.
The authors declare no competing financial interests.
References
- Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol. 2010;105:193–210. doi: 10.1016/S0065-2776(10)05007-8. [DOI] [PubMed] [Google Scholar]
- Beltran E, Fresquet V, Martinez-Useros J, Richter-Larrea JA, Sagardoy A, Sesma I, Almada LL, Montes-Moreno S, Siebert R, Gesk S, Calasanz MJ, Malumbres R, Rieger M, Prosper F, Lossos IS, Piris MA, Fernandez-Zapico ME, Martinez-Climent JA. A cyclin-D1 interaction with BAX underlies its oncogenic role and potential as a therapeutic target in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2011;108:12461–12466. doi: 10.1073/pnas.1018941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Iida S, Louie DC, Dalla-Favera R, Chaganti RS. Heterologous promoters fused to BCL6 by chromosomal translocations affecting band 3q27 cause its deregulated expression during B-cell differentiation. Blood. 1998;91:603–607. [PubMed] [Google Scholar]
- Ci W, Polo JM, Cerchietti L, Shaknovich R, Wang L, Yang SN, Ye K, Farinha P, Horsman DE, Gascoyne RD, Elemento O, Melnick A. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009;113:5536–5548. doi: 10.1182/blood-2008-12-193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ci W, Polo JM, Melnick A. B-cell lymphoma 6 and the molecular pathogenesis of diffuse large B-cell lymphoma. Curr Opin Hematol. 2008;15:381–390. doi: 10.1097/MOH.0b013e328302c7df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MR, Massenburg D, Siemasko K, Hou P, Zhang M. B-cell antigen receptor signaling requirements for targeting antigen to the MHC class II presentation pathway. Curr Opin Immunol. 2004;16:382–387. doi: 10.1016/j.coi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Duy C, Yu JJ, Nahar R, Swaminathan S, Kweon SM, Polo JM, Valls E, Klemm L, Shojaee S, Cerchietti L, Schuh W, Jäck HM, Hurtz C, Ramezani-Rad P, Herzog S, Jumaa H, Koeffler HP, de Alborán IM, Melnick AM, Ye BH, Müschen M. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med. 2010;207:1209–1221. doi: 10.1084/jem.20091299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton HE, Metcalf J, Lacerda AF, Brunetti CR. Accumulation of endogenous LITAF in aggresomes. PLoS One. 2012;7:e30003. doi: 10.1371/journal.pone.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357. doi: 10.4049/jimmunol.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- Lee SM, Chin LS, Li L. Charcot-Marie-Tooth disease-linked protein SIMPLE functions with the ESCRT machinery in endosomal trafficking. J Cell Biol. 2012;199:799–816. doi: 10.1083/jcb.201204137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Liu J, Xing H, Chen Y, Wang L, Wang D, Rao Q, Tang K, Tian Z, He K, Wang M, Wang J. PIG7, transactivated by AML1, promotes apoptosis and differentiation of leukemia cells with AML1-ETO fusion gene. Leukemia. 2012;26:117–126. doi: 10.1038/leu.2011.178. [DOI] [PubMed] [Google Scholar]
- MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM, Luther SA, Orbea HA. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol Rev. 1997;156:53–66. doi: 10.1111/j.1600-065x.1997.tb00958.x. [DOI] [PubMed] [Google Scholar]
- Malumbres R, Chen J, Tibshirani R, Johnson NA, Sehn LH, Natkunam Y, Briones J, Advani R, Connors JM, Byrne GE, Levy R, Gascoyne RD, Lossos IS. Paraffin-based 6-gene model predicts outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Blood. 2008;111:5509–5514. doi: 10.1182/blood-2008-02-136374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, Tibshirani R, Lossos IS. Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood. 2009;113:3754–3764. doi: 10.1182/blood-2008-10-184077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, Rosenwald A, Sugimoto KJ, Wheat LM, Karran EL, Garcia JF, Sanchez L, Prosper F, Staudt LM, Pinkel D, Dyer MJ, Martinez-Climent JA. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki Y, Begum NA, Kobayashi M, Matsumoto M, Toyoshima K, Seya T. Mycobacterium bovis Bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and estrogen-inducible gene, EET-1. J Biol Chem. 2001;276:23065–23076. doi: 10.1074/jbc.M011660200. [DOI] [PubMed] [Google Scholar]
- Munz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- Myokai F, Takashiba S, Lebo R, Amar S. A novel lipopolysaccharide-induced transcription factor regulating tumor necrosis factor alpha gene expression: molecular cloning, sequencing, characterization, and chromosomal assignment. Proc Natl Acad Sci U S A. 1999;96:4518–4523. doi: 10.1073/pnas.96.8.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh S, Polo JM, Shaknovich R, Juszczynski P, Lev P, Ranuncolo SM, Yin Y, Klein U, Cattoretti G, Dalla Favera R, Shipp MA, Melnick A. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood. 2007;110:2067–2074. doi: 10.1182/blood-2007-01-069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- Polo JM, Dell’Oso T, Ranuncolo SM, Cerchietti L, Beck D, Da Silva GF, Prive GG, Licht JD, Melnick A. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med. 2004;10:1329–1335. doi: 10.1038/nm1134. [DOI] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, Melnick A. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol. 2007;8:705–714. doi: 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- Ranuncolo SM, Wang L, Polo JM, Dell’Oso T, Dierov J, Gaymes TJ, Rassool F, Carroll M, Melnick A. BCL6-mediated attenuation of DNA damage sensing triggers growth arrest and senescence through a p53-dependent pathway in a cell context-dependent manner. J Biol Chem. 2008;283:22565–22572. doi: 10.1074/jbc.M803490200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, Calasanz MJ, Panizo C, Richter JA, Hernandez JM, Roman-Gomez J, Prosper F, Martinez-Climent JA. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010;116:2531–2542. doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, Hurt EM, Zhao H, Averett L, Yang L, Wilson WH, Jaffe ES, Simon R, Klausner RD, Powell J, Duffey PL, Longo DL, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Montserrat E, López-Guillermo A, Grogan TM, Miller TP, LeBlanc M, Ott G, Kvaloy S, Delabie J, Holte H, Krajci P, Stokke T, Staudt LM Project, L.L.M.P. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Saito M, Novak U, Piovan E, Basso K, Sumazin P, Schneider C, Crespo M, Shen Q, Bhagat G, Califano A, Chadburn A, Pasqualucci L, Dalla-Favera R. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:11294–11299. doi: 10.1073/pnas.0903854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- Somandin C, Gerber D, Pereira JA, Horn M, Suter U. LITAF (SIMPLE) regulates Wallerian degeneration after injury but is not essential for peripheral nerve development and maintenance: implications for Charcot-Marie-Tooth disease. Glia. 2012;60:1518–1528. doi: 10.1002/glia.22371. [DOI] [PubMed] [Google Scholar]
- Strawbridge AB, Blum JS. Autophagy in MHC class II antigen processing. Curr Opin Immunol. 2007;19:87–92. doi: 10.1016/j.coi.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology. 2003;60:22–26. doi: 10.1212/wnl.60.1.22. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- Tang X, Marciano DL, Leeman SE, Amar S. LPS induces the interaction of a transcription factor, LPS-induced TNF-alpha factor, and STAT6(B) with effects on multiple cytokines. Proc Natl Acad Sci U S A. 2005;102:5132–5137. doi: 10.1073/pnas.0501159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Metzger D, Leeman S, Amar S. LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced LPS-induced cytokine: Evidence for LITAF-dependent LPS signaling pathways. Proc Natl Acad Sci U S A. 2006;103:13777–13782. doi: 10.1073/pnas.0605988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173:1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- Vicente-Dueñas C, Fontán L, Gonzalez-Herrero I, Romero-Camarero I, Segura V, Aznar MA, Alonso-Escudero E, Campos-Sanchez E, Ruiz-Roca L, Barajas-Diego M, Sagardoy A, Martinez-Ferrandis JI, Abollo-Jimenez F, Bertolo C, Peñuelas I, Garcia-Criado FJ, García-Cenador MB, Tousseyn T, Agirre X, Prosper F, Garcia-Bragado F, McPhail ED, Lossos IS, Du MQ, Flores T, Hernandez-Rivas JM, Gonzalez M, Salar A, Bellosillo B, Conde E, Siebert R, Sagaert X, Cobaleda C, Sanchez-Garcia I, Martinez-Climent JA. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci U S A. 2012;109:10534–10539. doi: 10.1073/pnas.1204127109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Ichinose S, Hayashizaki K, Tsubata T. Induction of autophagy by B cell antigen receptor stimulation and its inhibition by costimulation. Biochem Biophys Res Commun. 2008;374:274–281. doi: 10.1016/j.bbrc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Watsuji T, Okamoto Y, Emi N, Katsuoka Y, Hagiwara M. Controlled gene expression with a reverse tetracycline-regulated retroviral vector (RTRV) system. Biochem Biophys Res Commun. 1997;234:769–773. doi: 10.1006/bbrc.1997.6705. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yang Z, Tsuji T, Gong J, Xie J, Chen C, Li W, Amar S, Luo Z. LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene. 2011;30:1892–1900. doi: 10.1038/onc.2010.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.