

Abstract
The buildup of Abeta and tau is believed to directly cause or contribute to the progressive cognitive deficits characteristic of Alzheimer disease. However, the molecular pathways linking Abeta and tau accumulation to learning and memory deficits remain elusive. There is growing evidence that soluble forms of Abeta and tau can obstruct learning and memory by interfering with several signaling cascades. In this review, I will present data showing that the mammalian target of rapamycin (mTOR) may play a role in Abeta and tau induced neurodegeneration.
Keywords: Alzheimer's disease, Abeta, tau, mTOR, autophagy, AD, PS1, Rapamycin, Learning And Memory, Plaques, Tangles, Review
2. Introduction
Current estimates show that today about 25 million people worldwide have dementia with one new case diagnosed every seven seconds; thus, by 2040 more than 80 million people will be afflicted by a form of dementia (1, 2). Alzheimer disease (AD) is by far the most common form of dementia and accounts for an estimated 60-80% of all cases (2, 3). In the United States (US), 5 million people have AD and with the increase in the aging population, about 16 million people will be afflicted by midcentury in the US alone (1, 3).
Neurophatologically, the AD brain is characterized by the accumulation of plaques, mainly formed of the amyloid-beta peptide and neurofibrillary tangles (NFTs) formed of hyperphosphorylated tau (4). Growing evidence points toward the buildup of soluble Abeta and tau being more proximal to cognitive deficits than plaques and tangles (5). Toward this end, major research efforts have been developed toward understanding alterations in the signaling pathways that may cause changes in neuronal function leading to cognitive decline as a result of Abeta and/or tau accumulation (6-10).
The mammalian target of rapamycin (mTOR) is a conserved protein kinase that plays a key role in controlling a balance between protein synthesis and degradation (11, 12). It interacts with several proteins to form two distinct complexes: the mTOR complex 1 (mTORC1), which controls protein homeostasis, is formed by mTOR, raptor, PRAS40 and mLT8, and its activity is inhibited by rapamycin; the mTORC2, which controls cellular shape by modulating actin function, is formed by mTOR, rictor, mLST8 and hSIN, and is insensitive to rapamycin (11, 12). mTOR signaling is mainly regulated by growth factors, nutrients, energy levels and stress. In turn, mTOR integrates these signals and controls ribosome biogenesis, transcription, translation and macroautophagy (11, 12).
Overwhelming evidence shows a primary role for TOR signaling in aging (13). For example, decreasing TOR signaling in yeast leads to an extension of replicative and chronological lifespan (14, 15). Similar results were obtained in C. elegans and Drosophila (16-18). More recently, results from the National Institute on Aging Interventions Testing Program have shown that pharmacologically reducing mTOR signaling with rapamycin increases median and maximal lifespan in genetically heterogeneous mice (19). The study was conducted at three different sites and involved the use of ∼1900 mice. Notably, these mice were put on rapamycin starting at 600 days of age and despite the late start, rapamycin increased the mean lifespan of males and females mice by 9% and 13%, respectively (19). Consistent with these results, deletion of ribosomal S6 protein kinase 1, a downstream target of mTOR, increased lifespan in female mice (20). Notably, reducing mTOR signaling not only increased lifespan but also reduced age-related pathologies including motor dysfunction and loss of insulin sensitivity (20). These findings are particularly relevant to AD, as they suggest that reducing mTOR signaling may have beneficial effects on age-dependent disorders including neurodegenerative disorders.
3. mTOR Signaling in AD Brains
Evidence from postmortem human AD brains indicates that the levels of phospho-mTOR and two of its downstream targets, p70S6K and the eukaryotic translation factor 4E (eIF4E) are increased compared to age-matched control cases, suggesting higher mTOR activity in AD brains (21-27). Specifically, Jin-Jing Pei and colleagues performed immunohistochemical and biochemical analysis of postmortem AD brains and showed that phosphorylated p70S6K levels were significantly increased in AD brains and correlated with Braak's stage and the levels of total and PHF-tau (21). Indeed, confocal microscopy shows that the levels of activated p70S6K are higher in neurons that are known to later develop NFTs (21). Along these lines, phosphorylated eIF4E levels were found to be 100-fold higher in AD brains compared to age-matched controls (28). The authors concluded that the increase in p70S6K in AD brains could mediate an up-regulation of tau (27). Furthermore, because the activity of both proteins is controlled by mTOR, these data could be interpreted to suggest that mTOR activity is elevated in AD brains. Indeed, the same authors in another study reported a 3-fold increase in the levels of mTOR phosphorylated at Ser2481 in the medial temporal cortex of AD cases compared to control cases (29), which is consistent with increased mTOR activity. Notably, these results were subsequently replicated by another group that reported that the ratio of phosphoSer2448-mTOR/total mTOR is ∼2.6-fold higher in AD brains compared to control cases (23). Overall, these studies show that mTOR signaling is increased in AD brains, and these conclusions are further supported by other studies of human brains (22, 24, 25) and in animal models (see below).
4. mTOR and Abeta
mTOR integrates signaling pathways that respond to growth factors, energy metabolism, nutrients and stress (reviewed in (12)). Among these signaling pathways is the PI3K/AKT pathway, which activates mTOR in response to growth factors such as insulin or insulin-like growth factors (12). Notably, several in vitro studies have shown that Abeta can activate the PI3K/AKT pathway. Along these lines, application of Abeta25-35 to PC12 cells is shown to increase AKT activity in a PI3K-dependent manner (30). Although the authors did not directly measure mTOR signaling, these data, supported by more recent publications (31-34), suggest an increase of mTOR signaling following Abeta administration. This conclusion is consistent with studies showing that the application of Abeta25-35 to N2A cells leads to a transient and significant increase in p70S6K (34). Data further supporting the link between Abeta accumulation and the up-regulation of mTOR signaling come from a study showing that exposed primary neurons to different concentrations of synthetic Abeta monomers and Abeta oligomers for 24 hours (35). It was reported that the levels of AKT phosphorylated at Ser473, mTOR phosphorylated at Ser2448, and its downstream target p4E-BP1 were significantly increased following exposure to Abeta oligomers but not to Abeta monomers (35). Notably, the increase in the levels of phosphorylated mTOR was found when Abeta oligomers were used at a concentration of 3 μg/ml, while lower or higher Abeta concentrations had no significant effects on mTOR phosphorylation (35). In apparent contradiction with these reports, Hugon and colleagues showed that 20 micromolar of Abeta42 decreased the steady-state levels of phosphorylated mTOR and p70S6K (36). However, the over-physiological concentration of Abeta used in this study, which is cytotoxic, makes it hard to compare the results with those reported above showing that a low concentration of Abeta increases mTOR signaling. This is especially true if one considers that when cells are homogenized in preparation for Western blots analysis, one would be dealing with cells at different stages of the neurodegeneration process and with possible secondary changing occurring. Whether the effects of high concentrations of Abeta42 on mTOR signaling are a direct consequence of Abeta administration or secondary events in the neurodegenerative process remains to be determined.
To further elucidate the effect of Abeta on mTOR signaling, we have used Chinese hamster ovary cells stably transfected with a cDNA encoding APP751 containing the Val717-Phe familial AD mutation known as 7PA2 (37). These cells produce high levels of Abeta oligomers, which have been shown to cause LTP and learning and memory deficits (38-40). We reported that the mTOR activity and signaling were significantly increased in 7PA2 cells compared to untransfacted CHO cells as determined by directly measuring mTOR enzymatic activity and by measuring the steady-state levels of phospho p70S6K levels by Western blot (41). We further showed that blocking Abeta production by treating 7PA2 cells with a gamma-secretase inhibitor was sufficient to prevent mTOR hyperactivity (41). Taken together these in vitro data suggest a dose-dependent effect of Abeta on mTOR signaling, with low, more physiological levels of Abeta causing an increase in mTOR signaling, while significantly higher Abeta concentrations decreasing mTOR signaling.
The relationship between Abeta and mTOR has also been analyzed in animal models of AD. Consistent with the cell culture studies, it has been shown that mTOR signaling is sensitive to the levels of Abeta in the brains of transgenic mice. Hugon and colleagues showed that the activation of mTOR and p70S6K was significantly decreased in the cortex of APP/PS1 double transgenic mice expressing human mutant APP751 and PS1M146V showing extracellular Abeta plaques (36). In contrast, these authors reported that in single APP transgenic mice, which have significantly lower Abeta levels compared to the APP/PS1 mice, the activity of mTOR and p70S6K levels were unchanged compared to age-matched control mice (36). To study the relation between Abeta and mTOR in vivo, we have used the 6-month-old 3xTg-AD mice, an animal model that develops age-dependent Abeta and tau accumulation associated with cognitive decline (6, 42-45). At this age, the 3xTg-AD mice show early intraneuronal Abeta accumulation and tau mislocalization, which correlate with the onset of cognitive decline (42-44, 46, 47). Abeta oligomerization is also evident at this age but mice do not yet have extracellular Abeta plaques, which develop around 15 months of age (48). We reported that mTOR enzymatic activity and the levels of phosphorylated p70S6K were significantly increased in the cortex and hippocampus of 6-month-old 3xTg-AD mice compared to age-matched control mice (41, 49). No changes for mTOR and p70S6K were detected in the cerebellum, which at this age has undetectable Abeta levels (41, 49). Furthermore, in the 3xTg-AD mice high Abeta levels are necessary for mTOR hyperactivity, as we showed that genetically or pharmacologically removing Abeta from the brains of the 3xTg-AD mice is sufficient to reduce mTOR activity to levels comparable to those detected in age-matched NonTg mice (49). To determine the effects of Abeta oligomers on mTOR activity, we injected naturally secreted Abeta oligomers into the hippocampi of NonTg mice and showed that this was sufficient to cause mTOR hyperactivity (49). The molecular mechanisms underlying the Abeta-induced cognitive decline appear to be mediated by PRAS-40 phosphorylation (49). PRAS-40 negatively regulates mTOR activity by directly binding to it (50, 51); upon phosphorylation, PRAS-40 detaches from mTOR thereby releasing its inhibitory effect (50, 51). Physiologically, PRAS-40 is phosphorylated by AKT and PIM-1 (31, 34, 52). We recently showed that preventing PRAS-40 phosphorylation by pharmacologically inhibiting AKT and PIM-1 activity blocks the Abeta-induced mTOR hyperactivity in vivo (49).
Overall, there appears to be some inconsistency between our data and the data showing that in APP/PS1 double transgenic mice the levels of mTOR are significantly decreased (36). However, our experiments were conducted in 6-month-old 3xTg-AD mice, which have moderate levels of Abeta oligomers but do not show Abeta plaques. In contrast, the experiments in the APP/PS1 mice were conducted in 12-month-old mice, which have much higher Abeta levels. Indeed, it remains to be determined how mTOR activity and signaling change in the presence of significantly higher Abeta levels and extracellular Abeta plaques in the 3xTg-AD mice. Together these in vivo data are consistent with the data showing that the exposure of primary neurons from wild type mice to increasing concentrations of Abeta oligomers causes an increase in mTOR activity at low micromolar levels but no changes at higher levels of Abeta oligomers.
5. mTOR in Learning and Memory
mTOR plays a key role in controlling protein homeostasis by regulating both protein synthesis and degradation, and it has been directly linked to learning and memory (e.g., (41, 53-56)). Early reports showed that in Aplysia californica stabilization of long-term facilitation requires rapamycin-sensitive protein synthesis (53). These results were subsequently confirmed in mammals where it was reported that bilateral infusions of rapamycin into the auditory cortex of gerbils did not impair the maintenance of the newly acquired auditory cortex-dependent memory, but caused deficits in long-term consolidation of auditory cortex-dependent memory (56). These and other reports (53, 57-61) clearly suggest that complete blockage of mTOR activity is detrimental for basal synaptic plasticity. Nevertheless, there is also a large body of evidence indicating that hyperactive mTOR signaling also has detrimental effects on different forms of learning and memory. For example, it has been shown that mTOR is hyperactive in the hippocampus of an animal model of tuberous sclerosis, a disorder associated with mental retardation, autism, and epilepsy (54). In these mice, mTOR hyperactivity was linked to deficits in late phase long-term potentiation and three independent hippocampal-dependent learning and memory tasks (54). Notably, reducing mTOR hyperactivity with rapamycin, an mTOR inhibitor, rescued the deficits in synaptic plasticity and learning and memory (54). In this study, the authors used a dose of rapamycin that was sufficient to reduce the mTOR signaling in the mutant tuberous sclerosis mice but not in the NonTg mice. Thus, the rapamycin dose is critical because, as discussed above, complete blockage of mTOR has detrimental effects on learning and memory. Consistent with these results, delta9-tetrahydrocannabinol (THC) administration – the primary active component of cannabis – is reported to cause mTOR hyperactivity and hippocampal-dependent learning and memory deficits (55). Notably, low doses of rapamycin rescued the memory deficits associated with cannabinoids consumption (55). Overall, it appears that there may be a window for mTOR signaling that is optimal for learning and memory and that alterations leading to an excessive increase or decrease in mTOR signaling outside such optimal window may have detrimental effects on learning and memory (62-64).
As mentioned above, published data show that Abeta accumulation causes mTOR hyperactivity in vitro and in vivo. Thus, we sought to determine whether mTOR hyperactivity played a role in the Abeta-induced cognitive decline by feeding microencapsulated rapamycin, an mTOR inhibitor, to 3xTg-AD mice for 10 weeks (41). We estimated that each day on average, mice were ingesting 2.24 mg rapamycin per kg of body weight. Remarkably, there is a large body of evidence showing that rapamycin crosses the blood brain barrier (65-69); indeed, at this concentration, mTOR signaling in the brains of the 3xTg-AD mice was reduced to NonTg levels and, more important, early spatial learning and memory deficits were rescued (41). Notably, these findings were independently replicated in a different animal model of AD (70). Although more needs to be done to understand how mTOR hyperactivity causes learning and memory deficits, possible molecular and cellular mechanisms are discussed by Silva and colleagues in (71). It should also be noted that although rapamycin is considered a selective mTOR inhibitor, there is growing evidence that it may also have effects independent of mTOR (72-75). For example, rapamycin suppresses mTOR-dependent translation of some classes of mRNAs but not others (76). Additionally, rapamycin binds to L-type voltage-dependent Ca2+ channels, and is thought that this binding may mediate some of the neuroprotective properties of rapamycin (77). Thus, because of the various effects/targets that rapamycin may have, more needs to be done to fully elucidate the role of mTOR in the cognitive decline associated with AD neuropathology. For example, genetic reduction of mTOR activity may be a more accurate approach to fully elucidate the role of mTOR signaling in AD. Overall, there is growing evidence indicating that the mTOR pathway is one of the pathways by which Abeta exerts its toxicity, further supporting the idea that reducing mTOR signaling in AD may be a valid therapeutic approach.
6. mTOR and Tau
As discussed above, postmortem studies from human AD brains indicate a link between mTOR signaling and tau neuropathology (e.g., (21, 23, 26, 27). This evidence has led to the hypothesis that the chronic increase in mTOR function occurring during aging may facilitate the development of tau pathology (27). Insights into the possible molecular mechanisms underlying the mTOR/tau link have come from numerous laboratories and offer three possible mechanisms of mTOR/tau interaction. First, it is well established that mTOR, via its downstream targets, increases the translation of 5′ top mRNAs (the group to which tau mRNA belongs) suggesting that hyperactive mTOR may facilitate tau accumulation simply by increasing its translation. Supporting this view, it has been shown that rapamycin suppresses the translation of tau, whereas constitutively active p70S6K, a downstream target of mTOR, increases tau translation (78). Second, hyperactive mTOR signaling may facilitate tau accumulation by increasing its phosphorylation, as mTOR directly phosphorylates and inhibits protein phosphatase 2A (PP2A; (79, 80), which plays a primary role in tau de-phosphorylation (81). Indeed, experiments in primary neurons and mice have shown that blocking mTOR activity with metformin, an mTOR inhibitor (82, 83), induces PP2A activity and reduces tau phosphorylation at three PP2A-dependent epitopes, Ser202, Ser356, and Ser262 (84). Notably, metformin had no effect on tau phosphorylation at Ser369, a PP2A-insensitive epitope (84). Third, it has been proposed that mTOR may play a role in the tau-induced neurodegeneration (81) and, a growing body of evidence indicates that tau may induce neurodegeneration by causing cell-cycle re-entry (85). In an elegant study, Feany and colleagues used Drosophila models of tauopathy to determine the role of TOR in the tau-induced neurodegeneration (86). They showed that TOR signaling was increased in flies expressing both mutant and wild type human tau, which develop progressive tau hyperphosphorylation and neurodegeneration (86, 87). Notably, increased TOR signaling was necessary for cell-cycle re-entry in the brain and, genetic and pharmacologic reduction of TOR activity rescued tau-induced neurodegeneration (86). Although the cause of cell death in AD is unclear, different reports have suggested that the accumulation of pathological tau may be the proximal event in the cascade causing cell death in AD (88). Together, these data suggest that mTOR hyperactivity, which can be caused by Abeta or tau accumulation, may mediate neurodegeneration as well as Abeta-induced cognitive decline.
7. mTOR and Autophagy
The autophagic system is a conserved intracellular system designed for the degradation of long-lived proteins and organelles in lysosomes (89-91). Three types of autophagy have been described: macroautophagy, microautophagy, and chaperon-mediated autophagy (CMA). While macro- and microautophagy involve the “in bulk” degradation of regions of the cytosol, CMA is a more selective pathway, and only proteins with a lysosomal targeting sequence are degraded (89, 90, 92). Cumulative evidence suggests that an age-dependent decrease in the autophagy/lysosome system may account for the accumulation of abnormal proteins during aging (93).
Macroautophagy (herein referred to as autophagy) is induced when an isolation membrane is generated surrounding cytosolic components, forming an autophagic vacuole that will eventually fuse with lysosomes for protein/organelle degradation 68-70). Although the molecular mechanisms underlying autophagy induction are not completely understood, an important step in the autophagosome formation is the activation of LC3-I. After its activation, LC3-I is lipidated to form membrane-associated LC3-II, which is incorporated in the growing autophagosome membrane and is often used as a marker of autophagy induction (94, 95). Overall, mTOR negatively regulates autophagy by interfering with its induction (96).
Several neurodegenerative disorders are characterized by the abnormal accumulation of aggregated proteins and are collectively known as proteinopathies. Based on this premise, it has been suggested that alterations in the cellular quality control system, such autophagy, may be involved in disease pathogenesis (97-102). Furthermore, autophagy function decreases with age (the major risk factor for AD and other neurodegenerative disorders) suggesting therefore that the age-dependent decrease in the autophagy function may contribute to the chronic buildup of aggregates in neurons (93, 103). Indeed, genetically reducing autophagy induction leads to profound neurodegeneration and cell loss associated with the accumulation of ubiquitinated inclusions (104-106). Consequently, it has been proposed that inducing autophagy may have beneficial effects in a variety of neurodegenerative disorders (e.g. (99, 107-113)).
The role of autophagy in AD is not well understood and contradicting reports have been published. For example, it has been reported that autophagic vacuoles accumulate in AD brains and in APP/PS1 transgenic mice, and this vacuoles maybe a source of Abeta production, suggesting that an increase in autophagy induction may lead to a further accumulation of Abeta (36, 114, 115). In contrast, other reports show that autophagy protects neurons from Abeta toxicity (41, 70, 116-119). Along these lines, Wyss-Coray and colleagues showed that beclin-1 levels, a key protein involved in autophagy induction, were decreased in AD patients (119). Furthermore, using complementary genetic approaches, the authors showed that decreasing beclin-1 expression in a transgenic mouse model of AD, decreased autophagy induction and increased Abeta accumulation (119). In contrast, increasing beclin-1 expression in the same mice, increased autophagy induction and reduced intracellular and extracellular Abeta pathology (119), clearly indicating that increasing autophagy induction may be beneficial in AD. More recently, it has been shown that parkin mediates the beclin-dependent autophagic clearance of Abeta (120).
We have shown that pharmacologically reducing mTOR hyperactivity in the brains of the 3xTg-AD mice led to a reduction in soluble Abeta and tau levels (41). The effects of rapamycin appear to be mediated by an increase in autophagy induction as we showed that in the brains of rapamycin-treated 3xTg-AD mice there was a significant increase in LC3II and other autophagy related proteins, including Atg5, Atg7 and Atg12 (41). Supporting this view, autophagy induction correlated with the decrease in Abeta levels in another mouse model of AD (70). Furthermore, in cell culture experiments, we directly showed that autophagy induction was necessary for the rapamycin-mediated decrease in Abeta levels (41). A recent report by Paul Greengard's group shows that inducing autophagy by small-molecule enhancer of rapamycin in immortalized cell lines and primary neurons led to an 80% reduction in Abeta40 and Abeta42 levels (121). These data were further supported by recent work showing that genetically increasing autophagic protein turnover ameliorates Abeta pathology and the associated cognitive decline in a mouse model of AD (122). There is an apparent contradiction between the data reported by Nixon and colleagues, who showed that autophagic vacuoles accumulates in AD due to impaired clearance, suggesting that further increase in autophagy may exacerbate the pathology (115), and what our group, Wyss-Coray's, Galvan's, Moussa's and Greengards's groups recently reported (41, 70, 119, 121) –increasing autophagy induction has beneficial effects on AD-like pathology in different animal model of AD. Although the basis of this apparent inconsistency remains to be established, it is tempting to speculate that the relationship between autophagy and Abeta may change with the progression of the disease (Figure 1). At earlier stages of Abeta accumulation, induction of autophagy may facilitate its clearance. As the disease progresses, deficiencies in the clearance of autophagic vacuoles may occur and thus further increasing autophagy may exacerbate the AD phenotype. Indeed, the majority of the studies showing that increasing autophagy induction reduces Abeta and tau accumulation were done in animals with early stages pathology. Thus, when considering the role of mTOR in Abeta and tau pathology and the role of mTOR in autophagy induction, it will be important to determine the effect of increasing autophagy function in mice with established plaques and tangles.
Figure 1.
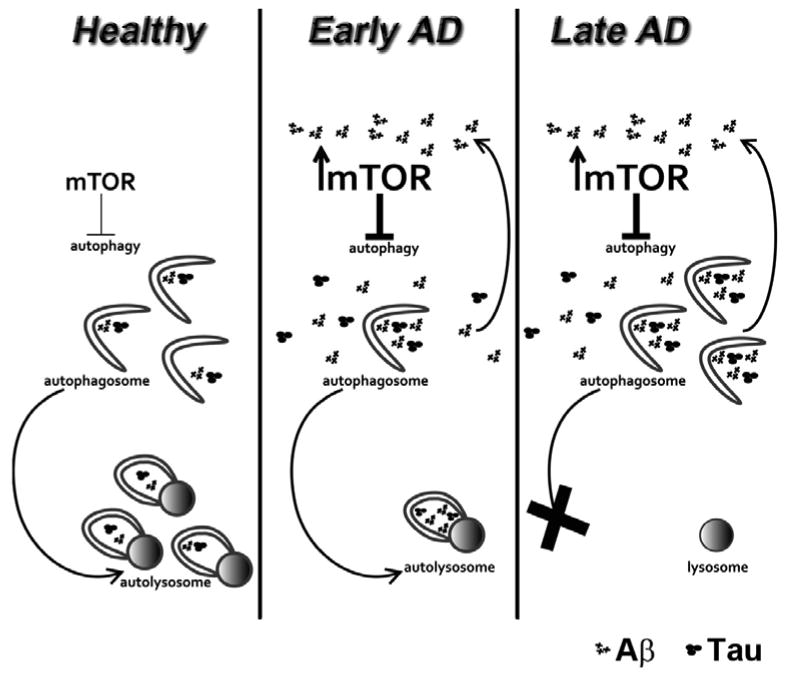
Schematic representation of the involvement of mTOR in AD. In a healthy neuron, mTOR activity is tightly regulated and basal autophagy levels are sufficient to remove Aβ and tau. During early stages AD, an increase in soluble Aβ levels leads to mTOR hyperactivity, which in turn will reduce autophagy induction (represented in the diagram by a reduction in autophagosomes). Lower autophagy function will eventually lead to an increase in the steady-state levels of Aβ and tau. Notably, high Aβ levels will further increase mTOR activity thus creating a vicious cycle that ultimately will promote higher Aβ levels. Increasing autophagy induction in early-AD may represent a valid therapeutic approach as it will facilitate autophagosome formation and thus remove Aβ and tau. During late stages AD, there is evidence that autophagosomes fail to fuse with lysosomes. It is anticipated that increasing autophagy induction in late stages AD may further clog the cells by generating more autophagosomes that will not be cleared, although this has not been directly tested.
8. Conclusions
While there is growing appreciation for a role of mTOR signaling in AD pathogenesis, more needs to be done to understand and fully elucidate the molecular mechanisms linking this key protein kinase to Abeta and tau pathology and the associated cognitive decline. Toward this end, there is a need to better understand how different concentrations of Abeta may have differential effects on mTOR activity and signaling. Additionally, although there is not a general agreement as to the stage of the autophagic process that would be more appropriate to increase (e.g., autophagy induction or autophagy flux), overall there is now clear evidence from several laboratories using different animal models for a role of autophagy in AD.
Acknowledgments
The author thanks Dr. Monica Maldonado for crafting Figure 1. This work was supported by NIA grant awards: K99/R00 AG29729-4 (Oddo, Principal Investigator) and RC2AG036613 (Oddo, Project Co-leader).
References
- 1.2010 Alzheimer's disease facts and figures. Alzheimers Dement. 2010;6:158–94. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the “common” neurologic disorders? Neurology. 2007;68:326–37. doi: 10.1212/01.wnl.0000252807.38124.a3. [DOI] [PubMed] [Google Scholar]
- 4.Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:22687–92. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–8. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, Teter B, Frautschy SA, Cole GM. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer's models. J Neurochem. 2007;103:1594–607. doi: 10.1111/j.1471-4159.2007.04869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu GQ, Mucke L. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 2005;25:9686–93. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–7. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 12.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Sharp ZD, Strong R. The role of mTOR signaling in controlling mammalian life span: what a fungicide teaches us about longevity. J Gerontol A Biol Sci Med Sci. 2010;65:580–9. doi: 10.1093/gerona/glp212. [DOI] [PubMed] [Google Scholar]
- 14.Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 15.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 17.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 19.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer's disease. Neuroreport. 2002;13:2429–32. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- 23.Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–17. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- 24.Onuki R, Bando Y, Suyama E, Katayama T, Kawasaki H, Baba T, Tohyama M, Taira K. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer's disease. Embo J. 2004;23:959–68. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peel AL, Bredesen DE. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol Dis. 2003;14:52–62. doi: 10.1016/s0969-9961(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 26.Pei JJ, Bjorkdahl C, Zhang H, Zhou X, Winblad B. p70 S6 kinase and tau in Alzheimer's disease. J Alzheimers Dis. 2008;14:385–92. doi: 10.3233/jad-2008-14405. [DOI] [PubMed] [Google Scholar]
- 27.Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer's disease. J Cell Mol Med. 2008;12:2525–32. doi: 10.1111/j.1582-4934.2008.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, An WL, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport. 2004;15:2237–40. doi: 10.1097/00001756-200410050-00019. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 30.Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–8. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 31.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 32.Ito S, Kimura K, Haneda M, Ishida Y, Sawada M, Isobe K. Induction of matrix metalloproteinases (MMP3, MMP12 and MMP13) expression in the microglia by amyloid-beta stimulation via the PI3K/Akt pathway. Exp Gerontol. 2007;42:532–7. doi: 10.1016/j.exger.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Ito S, Sawada M, Haneda M, Ishida Y, Isobe K. Amyloid-beta peptides induce several chemokine mRNA expressions in the primary microglia and Ra2 cell line via the PI3K/Akt and/or ERK pathway. Neurosci Res. 2006;56:294–9. doi: 10.1016/j.neures.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Zhang F, Beharry ZM, Harris TE, Lilly MB, Smith CD, Mahajan S, Kraft AS. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol Ther. 2009;8:846–53. doi: 10.4161/cbt.8.9.8210. [DOI] [PubMed] [Google Scholar]
- 35.Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–25. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- 37.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–9. [PubMed] [Google Scholar]
- 38.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 39.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–70. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 40.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 41.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–88. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Oddo S, Caccamo A, Cheng D, Jouleh B, Torp R, LaFerla FM. Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem. 2007;102:1053–63. doi: 10.1111/j.1471-4159.2007.04607.x. [DOI] [PubMed] [Google Scholar]
- 44.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 45.Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, LaFerla FM. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci. 2008;28:12163–75. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Oddo S, Caccamo A, Cheng D, LaFerla FM. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol. 2009;19:421–30. doi: 10.1111/j.1750-3639.2008.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281:1599–604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 49.Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magri A, Oddo S. Naturally secreted amyloid-{beta} increases mTOR activity via a PRAS40-mediated mechanism. J Biol Chem. 2011 doi: 10.1074/jbc.M110.180638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036–44. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 52.Nascimento EB, Snel M, Guigas B, van der Zon GC, Kriek J, Maassen JA, Jazet IM, Diamant M, Ouwens DM. Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates efficient phosphorylation of Ser183 by mTORC1. Cell Signal. 2010;22:961–7. doi: 10.1016/j.cellsig.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–37. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- 54.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–8. doi: 10.1038/nn.2369. [DOI] [PubMed] [Google Scholar]
- 56.Tischmeyer W, Schicknick H, Kraus M, Seidenbecher CI, Staak S, Scheich H, Gundelfinger ED. Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003;18:942–50. doi: 10.1046/j.1460-9568.2003.02820.x. [DOI] [PubMed] [Google Scholar]
- 57.Dash PK, Orsi SA, Moore AN. Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci. 2006;26:8048–56. doi: 10.1523/JNEUROSCI.0671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci. 2006;26:12977–83. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steward O, Schuman EM. Compartmentalized synthesis and degradation of proteins in neurons. Neuron. 2003;40:347–59. doi: 10.1016/s0896-6273(03)00635-4. [DOI] [PubMed] [Google Scholar]
- 60.Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–72. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao R, Li A, Cho HY. mTOR signaling in epileptogenesis: too much of a good thing? J Neurosci. 2009;29:12372–3. doi: 10.1523/JNEUROSCI.3486-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 64.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2:222–32. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 67.Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923–8. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Serkova N, Jacobsen W, Niemann CU, Litt L, Benet LZ, Leibfritz D, Christians U. Sirolimus, but not the structurally related RAD (everolimus), enhances the negative effects of cyclosporine on mitochondrial metabolism in the rat brain. Br J Pharmacol. 2001;133:875–85. doi: 10.1038/sj.bjp.0704142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Supko JG, Malspeis L. Dose-dependent pharmacokinetics of rapamycin-28-N,N-dimethylglycinate in the mouse. Cancer Chemother Pharmacol. 1994;33:325–30. doi: 10.1007/BF00685908. [DOI] [PubMed] [Google Scholar]
- 70.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ehninger D, de Vries PJ, Silva AJ. From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res. 2009;53:838–51. doi: 10.1111/j.1365-2788.2009.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–75. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–72. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 74.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5:725–6. doi: 10.4161/auto.5.5.8504. [DOI] [PubMed] [Google Scholar]
- 76.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruan B, Pong K, Jow F, Bowlby M, Crozier RA, Liu D, Liang S, Chen Y, Mercado ML, Feng X, Bennett F, von Schack D, McDonald L, Zaleska MM, Wood A, Reinhart PH, Magolda RL, Skotnicki J, Pangalos MN, Koehn FE, Carter GT, Abou-Gharbia M, Graziani EI. Binding of rapamycin analogs to calcium channels and FKBP52 contributes to their neuroprotective activities. Proc Natl Acad Sci U S A. 2008;105:33–8. doi: 10.1073/pnas.0710424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 2009;284:27734–45. doi: 10.1074/jbc.M109.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meske V, Albert F, Ohm TG. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3 -dependent phosphorylation of Tau. J Biol Chem. 2008;283:100–9. doi: 10.1074/jbc.M704292200. [DOI] [PubMed] [Google Scholar]
- 80.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–39. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 83.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 84.Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H, Schneider R, Sutherland C, Schweiger S. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.0912793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, Petersen RB, Bajic V, Smith MA. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer's disease. Neurochem Int. 2009;54:84–8. doi: 10.1016/j.neuint.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–41. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 87.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–4. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 88.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–93. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 90.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–44. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 93.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 94.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 96.Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 97.McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals. 2008;16:75–84. doi: 10.1159/000109761. [DOI] [PubMed] [Google Scholar]
- 98.Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691–9. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 100.Oddo S. The ubiquitin-proteasome system in Alzheimer's disease. J Cell Mol Med. 2008;12:363–73. doi: 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li X, Li H, Li XJ. Intracellular degradation of misfolded proteins in polyglutamine neurodegenerative diseases. Brain Res Rev. 2008;59:245–52. doi: 10.1016/j.brainresrev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Caccamo A, Majumder S, Deng JJ, Bai Y, Thornton FB, Oddo S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular weight neurofilament instability. J Biol Chem. 2009 doi: 10.1074/jbc.M109.031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–61. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 104.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 105.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 106.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 108.Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16(2):R115–23. doi: 10.1093/hmg/ddm213. [DOI] [PubMed] [Google Scholar]
- 109.Ouellet M, Emond V, Chen CT, Julien C, Bourasset F, Oddo S, LaFerla F, Bazinet RP, Calon F. Diffusion of docosahexaenoic and eicosapentaenoic acids through the blood-brain barrier: An in situ cerebral perfusion study. Neurochem Int. 2009;55:476–82. doi: 10.1016/j.neuint.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 110.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 111.Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, Modugno N, Siciliano G, Isidoro C, Murri L, Ruggieri S, Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–7. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 113.Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175:736–47. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–37. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hung SY, Huang WP, Liou HC, Fu WM. Autophagy protects neuron from Abeta-induced cytotoxicity. Autophagy. 2009;5:502–10. doi: 10.4161/auto.5.4.8096. [DOI] [PubMed] [Google Scholar]
- 117.Ling D, Salvaterra PM. A central role for autophagy in Alzheimer-type neurodegeneration. Autophagy. 2009;5:738–40. doi: 10.4161/auto.5.5.8626. [DOI] [PubMed] [Google Scholar]
- 118.Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated A{beta} in AD models. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of A{beta} and APP-CTF via Atg5-dependent autophagy pathway. Faseb J. 2011 doi: 10.1096/fj.10-175158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–77. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]