

Abstract
The Co(II) complex of the D2h-symmetric amidoporphyrin 3,5-DitBu-IbuPhyrin, [Co(P1)], has proven to be an effective metalloradical catalyst for intermolecular amination of C(sp2)–H bonds of aldehydes with fluoroaryl azides. The [Co(P1)]-catalyzed process can employ aldehydes as the limiting reagents and operate under neutral and non-oxidative conditions, generating nitrogen gas as the only byproduct. The metalloradical aldehydic C–H amination is suitable for different combinations of aldehydes and fluoroaryl azides, producing the corresponding N-fluoroaryl amides in good to excellent yields. A series of mechanistic studies support a stepwise radical mechanism for the Co(II)-catalyzed intermolecular C–H amination.
Introduction
Amides represent a class of common functional groups in organic chemistry and exist ubiquitously as key functional and structural motifs in a broad range of important compounds such as natural products, synthetic drugs and polymeric materials.1 Many methods have been established for constructing amide functionalities, including the traditional condensation of carboxylic acids and derivatives with amines,1d transition metal-catalyzed cross-coupling of aryl and alkyl halides with amides,2 direct amidation of alcohols and esters with amines,3 and other approaches.4 As a new development in the field, amide synthesis based on amination of aldehydic C–H bonds via metal-catalyzed nitrene insertion has attracted recent interest due to the wide availability of aldehydes and the advent of new nitrene sources.5 Several transition metal-based catalysts, such as Rh,5e Ru,5f Fe5g,5h and Cu5i complexes, have been shown to be effective in catalyzing C(sp2)–H amination of aldehydes with iminoiodinanes and their in-situ variants as nitrene sources, generating N-sulfonylcarboxamides.6 In view of their many attributes such as structural diversity, wide availability and ease of synthesis, organic azides have been actively pursued as alternative nitrene sources for metal-catalyzed C–H amination.7 To date, only phosphoryl azides8 have been recently demonstrated as effective nitrene sources for Ru-catalyzed amination of aldehydic C–H bonds, forming N-acylphosphoramidates.9–10 Other more common organic azides such as aryl azides7c, 11 have not been demonstrated in a catalytic process. It would be synthetically attractive if N-aryl amides could be prepared directly from aldehydes by catalytic aldehydic C–H amination with aryl azides.
N-Fluoroaryl amide functionalities have been incorporated into many synthetic molecules with demonstrated biological activity and pharmaceutical importance.12 Representative examples include insulin-like growth factor-I (IGF-1) receptor GSK1904529A (I),13 sirtuin-activating compound (STAC) (II),14 androgen receptor (AR) agonist (III)15 and others (Figure 1). In addition, N-fluoroaryl amides have been recently demonstrated as effective directing groups for palladium-catalyzed C–H functionalization.16 The existing synthetic methods for N-fluoroaryl amides typically rely on traditional condensation reactions of carboxylic acids or their derivatives with fluoroanilines. Due to the poor nucleophilicity of polyfluoroanilines, these reactions often suffered from low yields and poor substrate scope, limiting their wide application for preparation of N-fluoroaryl amide derivatives.17 Undoubtedly, a catalytic process based on the C–H amination of aldehydes with fluoroaryl azides would be particularly attractive for the synthesis of N-fluoroaryl amides. However, fluoroaryl azides, which can be conveniently prepared from readily available fluoroanilines, are a class of common aryl azides that have not been previously applied for catalytic C–H amination, including aldehydic C–H amination.18
Figure 1.
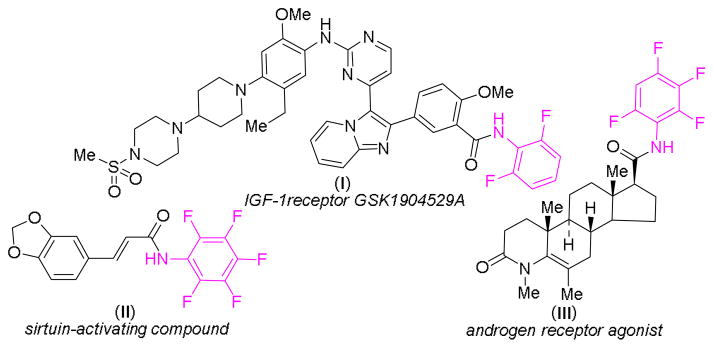
Selected examples of biologically active N-fluoroaryl amides.
As stable metalloradicals with well-defined open-shell doublet d7 electronic structure, cobalt(II) complexes of porphyrins, [Co(Por)], have shown to be effective metalloradical catalysts for amination of different types of C–H bonds with various azides.19–20 Recently, Co(II)-based metalloradical catalysis (MRC) was successfully applied for asymmetric olefin aziridination with fluoroaryl azides as effective nitrene sources.21 The Co(II)-catalyzed aziridination was believed to proceed through Co(III)-nitrene radical A as the key intermediate (Figure 2), which undergoes radical addition to olefin, followed by 3-exo-tet radical cyclization.21 As a further application of the concept of MRC, we anticipated the possibility of a Co(II)-based catalytic process for aldehydic C–H amination with fluoroaryl azides if the Co(III)-supported fluoroaryl nitrene radical A is capable of H-atom abstraction from aldehydes to generate Co(III)-amido complex B, followed by acyl radical substitution (Figure 2). As a result of our studies in this direction, herein we wish to report an effective catalytic system based on Co(II)-MRC that enables selective amination of aldehydic C–H bonds with fluoroaryl azides for amide formation. The Co(II)-based metalloradical amination is suitable for a wide range of aldehydes and can also utilize different fluoroaryl azides, producing the corresponding N-fluoroaryl amides in good to excellent yields. In addition to using aldehydes as the limiting reagent and generating N2 as the only byproduct, the new MRC system is highlighted with a practical protocol that operates under neutral and non-oxidative conditions, permitting high tolerance of other functionalities.
Figure 2.

Proposed pathway for C–H amination of aldehydes with fluoroaryl azides via [Co(Por)]-based metalloradical catalysis.
Results and Discussion
Initially, we examined the reaction of benzaldehyde (1a) with pentafluorophenyl azide (2a) using Co(II) complexes of different D2h-symmetric amidoporphyrins as the catalysts. We were delighted to find that the catalyst [Co(P1)] (P1 = 3,5-DitBu-IbuPhyrin)22 gave the desired amide 3aa in 56% yield in the solvent of benzene (Table 1, entry 1). Subsequent experiments with various solvents revealed that chlorobenzene was the optimal medium for the catalytic reaction, producing the desired product in 77% yield (Table 1, entries 2–6). Low conversion was observed at lower reaction temperature, possibly due to the coordination of the aldehyde to the cobalt center (Table 1, entry 7).23 When [Co(TPP)] (H2TPP = 5,10,15,20-tetraphenylporphyrin) was used as the catalyst, it gave no obvious reaction (Table 1, entry 8), indicating the important role of the possible N–H---F hydrogen bonding as depicted for the Co(III)-supported fluoroaryl nitrene radical A (Figure 2).21 As expected, no reaction occurred without a catalyst (Table 1, entry 9).
Table 1.
Amination of Benzaldehyde (1a) with Pentafluorophenyl Azide (2a) Catalyzed by [Co(Por)]a
| ||||
|---|---|---|---|---|
| entry | catalyst | temp (°C) | solvent | yield (%)b |
| 1 | [Co(P1)] | 80 | PhH | 56 |
| 2 | [Co(P1)] | 80 | PhCl | 77 |
| 3 | [Co(P1)] | 80 | 4-CF3C6H4Cl | 71 |
| 4 | [Co(P1)] | 80 | PhF | 56 |
| 5 | [Co(P1)] | 80 | hexane | 69 |
| 6 | [Co(P1)] | 80 | ClCH2CH2Cl | 65 |
| 7 | [Co(P1)] | 40 | PhCl | trace |
| 8 | [Co(TPP)] | 80 | PhCl | trace |
| 9 | - | 80 | PhCl | NRc |
| ||||
All reactions were carried out with 1.0 equiv. of aldehyde (0.2 M) and 1.2 equiv. of azide using 3 mol % [Co(Por)] as catalyst.
19F-NMR yields.
NR = no reaction.
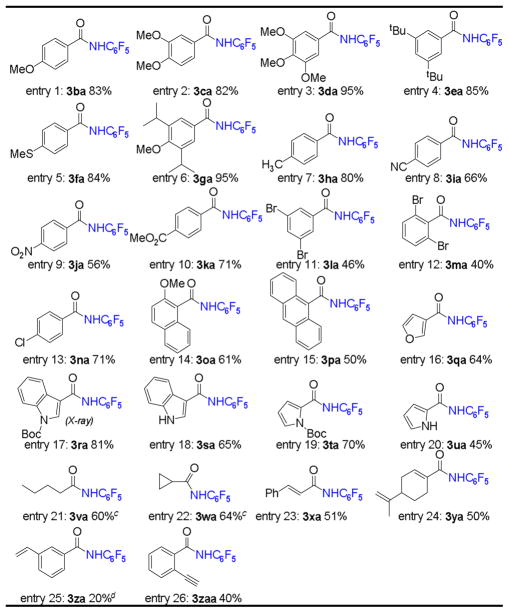
Under the optimized conditions, we then investigated the scope of aldehyde substrate for the catalytic amination by [Co(P1)] with pentafluorophenyl azide (2a). As shown in Table 2, this [Co(P1)]-based MRC system was found to be suitable for a broad range of aldehydes with different steric and electronic properties, including aromatic, heteroaromatic, and aliphatic aldehydes as well as α,β-unsaturated aldehydes. For example, aromatic aldehydes with various electron-donating substituents, including alkyl, alkoxy, and alkylthio groups, could be effectively used as substrates, yielding the desired N-fluoroaryl amides in high yields (Table 2, entries 1–7). It is notable that the easily oxidizable alkylthio functionality was well tolerated because of the neutral and nonoxidative conditions of this metalloradical process (Table 2, entry 5). Interestingly, the Co(II)-based radical amination was shown to be highly chemoselective toward aldehydic C–H bonds in the presence of other C–H bonds, including benzylic, allylic and aliphatic C–H bonds (Table 2, entries 6–7 and 21–22 and 24). As well, electron-poor aromatic aldehydes, such as cyano-, nitro-, and ester-substituted benzaldehydes, could be smoothly aminated in good yields under the MRC system (Table 2, entries 8–10). Moreover, various halogenated benzaldehydes, including the highly sterically demanding 2,6-dibromobenzaldehyde, could be aminated by the Co(II)-MRC system to generate the corresponding N-fluoroaryl halobenzamides (Table 2, entries 11–13), the aryl halide units of which may be further functionalized via palladium-catalyzed amidation.2 In addition to benzaldehyde derivatives, both naphthyl and anthracenyl aldehydes could be employed as the substrates for the amination process (Table 2, entries 14 and 15). Like aromatic aldehydes, heteroaromatic aldehydic C–H bonds could also be aminated. For example, furanyl, pyrrolyl, and indolyl substituted amides could be readily produced in good yields (Table 2, entries 16–20). Importantly, aldehydes with free N–H group could also be used in the transformation, albeit in lower yields compared to that of N-Boc-protected aldehydes (Table 2, entries 17 vs. 18 and entries 19 vs. 20). Aliphatic aldehydes, such as valeraldehyde and cyclopropanecarbaldehyde, also gave the desired products in satisfying yields (Table 2, entries 21 and 22).24 It is worthy to mention that no obvious decarbonylation or ring-opening products were observed, indicating fast rate of the last step of radical substitution. Vinyl aldehydes, such as cinnamaldehyde and perillaldehyde, could produce the corresponding amides without affecting the C=C double bonds (Table 2, entries 23 and 24). Even for substrates containing terminal aromatic C=C and C≡C bonds, the aldehydic C–H amination was still dominated (Table 2, entries 25 and 26). The priority of aldehydic C–H amination over C=C aziridination was further demonstrated by the competition reaction of benzaldehyde (1a) and styrene (1a′) with azide 2a (eq 1). Under the same reaction conditions, the yield of the amide 3aa was found to be double that of aziridine 3a′a (eq 1).21 It is worth mentioning that the metalloradical amination could be readily scaled up, as demonstrated with the gram-scale synthesis of amide 3ba in 79% yield.
Table 2.
|
All reactions were carried out in PhCl using 3 mol % of [Co(P1)] as catalyst with 1.0 equiv. of aldehyde (0.2 M) and 1.2 equiv. of azide for 24 h at 80 °C.
Isolated yields.
At 100 °C.
Small amounts of aziridine and unidentified compounds were observed by NMR.
 |
(1) |
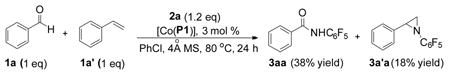
In addition to pentafluorophenyl azide, the [Co(P1)]-based metalloradical catalytic system could effectively enable aldehydic C–H amination with other haloaryl azides (Table 3). As exemplified with p-anisaldehyde (1b), various fluoroaryl azides, including monofluoro-, difluoro-, trifluoro-, and tetrafluoroaryl azides (2b–f), were found to be effective nitrene sources for the metalloradical amination process, giving the corresponding amides in good yields (Table 3, entries 1–5). Fluoroaryl azide with other substituents such as bromotetrafluorophenyl azide (2g) was also a suitable nitrene source for the transformation (Table 3, entry 6). Similar to that of azide 2g, 4-tetrafluoropyridinyl azide (2h) produced the corresponding amide in a high yield as well (Table 3, entry 7). Interestingly, 2,6-dichlorophenyl azide (2i) and 2-pyrimidine azide (2j) could also function as nitrene sources for this type of catalytic transformation, but in much lower efficiency (Table 3, entries 8 and 9). However, 3,4,5-trifluorophenyl azide,21 without ortho-fluorine substituents, gave only trace amount of the desired product under the same condition, again indicating the importance of the possible hydrogen bonding in the catalytic process (Figure 2).25
Table 3.
|
All reactions were carried out in PhCl using 3 mol % of [Co(P1)] as catalyst with 1.0 equiv. of aldehyde (0.2 M) and 1.2 equiv. of azide for 24 h at 80 °C.
Isolated yields.
At 100 °C.
The yield was based on 1H NMR.
Increasing evidence supports that Co(II)-based metalloradical C–H amination and related olefin aziridination reactions with azides undergo a stepwise radical mechanism involving unusual Co(III)-nitrene radical intermediates, which are fundamentally different from metallonitrene intermediates associated with the widely studied Rh2- and Cu-based closed-shell systems.19–20 Accordingly, the current catalytic process of intermolecular aldehydic C–H amination with fluoroaryl azides is supposed to undergo a similar stepwise radical mechanism (Figure 2). To obtain direct evidence for the proposed mechanism, we first sought to measure the deuterium kinetic isotope effect (KIE) for the reaction of benzaldehyde (1a) with pentafluorophenyl azide (2a) by [Co(P1)] (Scheme 1). Since differentiation between protonated (3aa) and deuterated (3aa–d) products by 1H-NMR was problematic for a direct competition experiment between aldehydes 1a and 1a–d due to the acidity of the resulting amide group, indirect competition experiments between them with the use of 4-methylbenzaldehyde (1h) as a reference were conducted to address the issue (Scheme 1). Through separate measurements of kH/kR and kD/kR, the KIE (kH/kD) was derived to be 10.2. This high degree of primary intermolecular KIE is consistent with the proposed mechanism involving the hydrogen-atom abstraction by the key Co(III)-nitrene radical intermediate A (Figure 2).
Scheme 1.
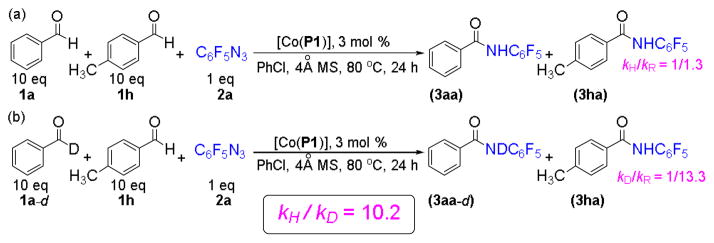
Double Competition Experiments for Measurement of the Kinetic Isotope Effect (KIE).
 |
(2) |
 |
(3) |
 |
(4) |
 |
(5) |
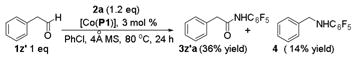
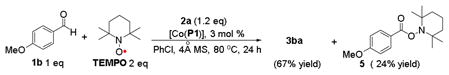
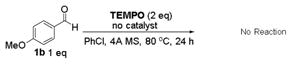
To gain insights on the resulting acyl radical intermediate and its subsequent substitution reaction in the Co(II)-catalyzed process (Figure 2), phenylacetaldehyde (1z′) was employed as the amination substrate since the corresponding phenylacetyl radical is a known acyl radical clock.5a,26 Under the standard conditions, the catalytic reaction of 1z′ afforded N-benzylpentafluoroaniline (4) in 14% yield in addition to the expected aldehydic C–H amination product 3z′a in 36% yield (eq 2).27 The formation of compound 4 is attributed to the decarbonylation of the initially generated acyl radical to give benzyl radical, followed by its substitution reaction with the Co(III)-amido complex. The fact that the amide 3z′a was found to be the major product also suggests that the rate of the last step of radical substitution is greater than that of decarbonylation of the phenylacetyl radical, which is 5.2 × 107 s−1.26 This result is consistent with the extremely low barrier for the radical substitution suggested from the previous computational studies.19 To provide further proof for the existence of the acyl radical intermediate, catalytic amination of aldehyde 1b with azide 2a was carried out in the presence of an excess amount of radical trapping agent TEMPO (eq 3). While the amination product 3ba was still formed as the major product in 67% yield, the corresponding acyl radical was successfully trapped to give compound 5 in 24% yield. In the absence of either catalyst [Co(P1)] (eq 4) or azide 2a (eq 5), no reaction was observed, indicating that TEMPO alone could not trigger the formation of 5.
While the mechanistic studies evidently support the proposed radical mechanism and validate the genuine radical character of the Co(III)-nitrene radical intermediate A (Figure 2), the less than unity value of kH/kR in the competition experiment between benzaldehyde and 4-methylbenzaldehyde (Scheme 1) suggests that the radical intermediate A has some degree of electrophilicity. To verify the electrophilic radical nature of the intermediate A, we carried out systematic competition experiments using a selected set of para-substituted benzaldehydes with wide-ranging electronic properties for their amination reactions with pentafluorophenyl azide. The results revealed a strong linear correlation between the log(kX/kH) and the Hammett constants (σp)28 of the para-substituents with a negative slope of −0.867 (Figure 3).29 This trend signifies the electronic influence of the radical C–H amination with fluoroaryl azides by [Co(P1)], which is also well reflected in the results summarized in Table 2. The strong electron-withdrawing effect of the fluoroaryl group is likely responsible for the electrophilic radical reactivity profile of the catalytic system.
Figure 3.

Correlation of log(kX/kH) versus σp plot for amination of para-substituted benzaldehydes with pentafluorophenyl azide by [Co(P1)].
Conclusions
In summary, we have demonstrated that [Co(P1)] is an effective catalyst for C–H amination of aldehydes with fluoroaryl azides via metalloradical catalysis. The Co(II)-based metalloradical amination system represents the first example of aldehydic C–H amination with aryl azides as the nitrene source. The [Co(P1)]-catalyzed process, which can be operated under neutral and non-oxidative conditions without the need of any additives, proceeds effectively with the use of aldehydes as the limiting reagent and tolerates various functionalities. The resultant N-fluoroaryl amides may find a myriad of potential applications. Efforts are underway to expand the Co(II)/azide-based radical amination system for other types of C–H bonds, including asymmetric intermolecular C–H amination.
Supplementary Material
Acknowledgments
We are grateful for financial support by NSF (CHE-1152767) and NIH (R01-GM098777). We thank Dr. Edwin Rivera for his valuable assistance with NMR measurements.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and analysis data for new compounds; CIF files of compounds 3ra (CCDC 990223) and 3bb (CCDC 990222). (See DOI: 10.1039/b000000x/
References
- 1.Selected recent reviews, see: Bode JW. Top Organomet Chem. 2013;44:13–33.Allen CL, Williams JMJ. Chem Soc Rev. 2011;40:3405–3415. doi: 10.1039/c0cs00196a.Scheidt K. Nature. 2010;465:1020–1022. doi: 10.1038/4651020a.Valeur E, Bradley M. Chem Soc Rev. 2009;38:606–631. doi: 10.1039/b701677h.
- 2.Selected reviews, see: Surry DS, Buchwald SL. Chem Sci. 2011;2:27–50. doi: 10.1039/C0SC00331J.Ma D, Cai Q. Acc Chem Res. 2008;41:1450–1460. doi: 10.1021/ar8000298.Carril M, SanMartin R, Dominguez E, Domnguez E. Chem Soc Rev. 2008;37:639–647. doi: 10.1039/b709565c.Selected examples, see: Vo GD, Hartwig JF. J Am Chem Soc. 2009;131:11049–11061. doi: 10.1021/ja903049z.Martinelli JR, Clark TP, Watson DA, Munday RH, Buchwald SL. Angew Chem Int Ed. 2007;46:8460–8463. doi: 10.1002/anie.200702943.Angew Chem. 2007;119:8612–8615.
- 3.Selected examples, see: Gnanaprakasam B, Milstein D. J Am Chem Soc. 2011;133:1682–1685. doi: 10.1021/ja109944n.Nordstrøm LU, Vogt H, Madsen R. J Am Chem Soc. 2008;130:17672–17673. doi: 10.1021/ja808129p.Gunanathan C, Ben-David Y, Milstein D. Science. 2007;317:790–792. doi: 10.1126/science.1145295.Yoo WJ, Li CJ. J Am Chem Soc. 2006;128:13064–13065. doi: 10.1021/ja064315b.
- 4.a) Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cassidy MP, Raushel J, Fokin VV. Angew Chem Int Ed. 2006;45:3154–3157. doi: 10.1002/anie.200503805. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:3226–3229. [Google Scholar]; c) Cho SH, Yoo EJ, Bae I, Chang S. J Am Chem Soc. 2005;127:16046–16047. doi: 10.1021/ja056399e. [DOI] [PubMed] [Google Scholar]; d) Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]; e) Damkaci F, DeShong P. J Am Chem Soc. 2003;125:4408–4409. doi: 10.1021/ja028694u. [DOI] [PubMed] [Google Scholar]; f) Schäfer G, Matthey C, Bode JW. Angew Chem Int Ed. 2012;51:9173–9175. doi: 10.1002/anie.201204481. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:9307–9310. [Google Scholar]
- 5.For selected reviews, see: Karila D, Dodd RH. Curr Org Chem. 2011;15:1507–1538.Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926–1936. doi: 10.1039/c0cs00095g.Zalatan DN, Du Bois J. Top Curr Chem. 2010;292:347–378. doi: 10.1007/128_2009_19.Chang JWW, Ton TMU, Chan PWH. Chem Rec. 2011;11:331–357. doi: 10.1002/tcr.201100018.For selected examples, see: Chan J, Baucom KD, Murry JA. J Am Chem Soc. 2007;129:14106–14107. doi: 10.1021/ja073872a.Chang JWW, Chan PWH. Angew Chem Int Ed. 2008;47:1138–1140. doi: 10.1002/anie.200704695.Angew Chem. 2008;120:1154–1156.Chen GQ, Xu ZJ, Liu YG, Zhou CY, Che CM. Synlett. 2011:1174–1178.Ton TMU, Tejo C, Tania S, Chang JWW, Chan PWH. J Org Chem. 2011;76:4894–4904. doi: 10.1021/jo200284a.Chang JWW, Ton TMU, Tania S, Taylor PC, Chan PWH. Chem Commun. 2010;46:922–924. doi: 10.1039/b918588g.
- 6.For synthesis of dialkylamides via Rh-catalyzed oxidative amination of aldehydes with N-methylmorpholine N-oxide, see: Tillack A, Rudloff I, Beller M. Eur J Org Chem. 2001;2001:523–528.For a catalyst-free protocol for sulfamidation of aldehydes, see: Borah AJ, Phukan P. Chem Commun. 2012;48:5491–5493. doi: 10.1039/c2cc31258a.
- 7.For reviews, see: Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b.Lu H, Zhang XP. Chem Soc Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a.Driver TG. Org Biomol Chem. 2010;8:3831–3846. doi: 10.1039/c005219c.Fantauzzi S, Caselli A, Gallo E. Dalton Trans. 2009:5434–5443. doi: 10.1039/b902929j.Katsuki T. Chem Lett. 2005;34:1304–1309.
- 8.Xiao W, Zhou CY, Che CM. Chem Commun. 2012;48:5871–5873. doi: 10.1039/c2cc31686b. [DOI] [PubMed] [Google Scholar]
- 9.Alkyl azides were used for amide synthesis through diselenide-promoted ligation with carboxylic acids in the presence of tertiary phosphines, see: Burés J, Martín M, Urpí F, Vilarrasa J. J Org Chem. 2009;74:2203–2206. doi: 10.1021/jo802825e.Alkyl azide for amide synthesis through base-mediated amidation of aldehydes, see: Kulkarni SS, Hu X, Manetsch R. Chem Commun. 2013;49:1193–1195. doi: 10.1039/c2cc37289d.
- 10.Sulfonyl azides have been used for amide synthesis via oxidative hydration of alkynes, see: refs. 4d and 4e.
- 11.For recent reviews, see 7c and Bräse S, Gil C, Knepper K, Zimmermann V. Angew Chem Int Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657.Angew Chem. 2005;117:5320–5374.Cenini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S. Coord Chem Rev. 2006;250:1234–1253.
- 12.a) Zhang MR, Suzuki K. Curr Top Med Chem. 2007;7:1817–1828. doi: 10.2174/156802607782507448. [DOI] [PubMed] [Google Scholar]; b) Li Z, Conti PS. Adv Drug Deliv Rev. 2010;62:1031–1051. doi: 10.1016/j.addr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Sabbatini P, Rowand JL, Groy A, Korenchuk S, Liu Q, Atkins C, Dumble M, Yang J, Anderson K, Wilson BJ, Emmitte KA, Rabindran SK, Kumar R. Clin Cancer Res. 2009;15:3058–3067. doi: 10.1158/1078-0432.CCR-08-2530. [DOI] [PubMed] [Google Scholar]
- 14.Milburn M, Jill M, Auwerx J, Argmann C, Lagouge M, Dipp M. PCT WO2007008548A2. US Patent. 2007
- 15.Wang JM, Carol A. PCT WO 03/092588 A2. U S Patent. 2003
- 16.Selected examples for fluoroaryl amide-directed C–H functionalization, see: Wasa M, Chan KSL, Zhang XG, He J, Miura M, Yu JQ. J Am Chem Soc. 2012;134:18570–18572. doi: 10.1021/ja309325e.He J, Wasa M, Chan KSL, Yu JQ. J Am Chem Soc. 2013;135:3387–3390. doi: 10.1021/ja400648w.
- 17.a) Brotzel F, Chu YC, Mayr H. J Org Chem. 2007;72:3679–3688. doi: 10.1021/jo062586z. [DOI] [PubMed] [Google Scholar]; b) Hoi KH, Çalimsiz S, Froese RDJ, Hopkinson AC, Organ MG. Chem Eur J. 2012;18:145–151. doi: 10.1002/chem.201102428. [DOI] [PubMed] [Google Scholar]
- 18.For selected examples of non-catalytic amination reactions with fluoroaryl azides under thermal and photolytic conditions, see: Keana JFW, Cai SX. J Org Chem. 1990;55:3640–3647.Soundararajan N, Platz MS. J Org Chem. 1990;55:2034–2044.
- 19.For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of Co(III)-nitrene radical intermediate, see: Lyaskovskyy V, Suarez AIO, Lu H, Jiang H, Zhang XP, de Bruin B. J Am Chem Soc. 2011;133:12264–12273. doi: 10.1021/ja204800a.For related DFT studies on the radical mechanism of [Co(Por)]-catalyzed olefin aziridination, see: Olivos Suarez AI, Jiang H, Zhang XP, de Bruin B. Dalton Trans. 2011;40:5697–5705. doi: 10.1039/c1dt10027k.Hopmann KH, Ghosh A. ACS Catal. 2011;1:597–600.
- 20.a) Ragaini F, Penoni A, Gallo E, Tollari S, Gotti CL, Lapadula M, Mangioni E, Cenini S. Chem-Eur J. 2003;9:249–259. doi: 10.1002/chem.200390018. [DOI] [PubMed] [Google Scholar]; b) Ruppel JV, Kamble RM, Zhang XP. Org Lett. 2007;9:4889–4892. doi: 10.1021/ol702265h. [DOI] [PubMed] [Google Scholar]; c) Caselli A, Gallo E, Fantauzzi S, Morlacchi S, Ragaini F, Cenini S. Eur J Inorg Chem. 2008:3009–3019. [Google Scholar]; d) Lu H, Subbarayan V, Tao JR, Zhang XP. Organometallics. 2010;29:389–393. [Google Scholar]; e) Lu H, Tao JR, Jones JE, Wojtas L, Zhang XP. Org Lett. 2010;12:1248–1251. doi: 10.1021/ol100110z. [DOI] [PubMed] [Google Scholar]; f) Lu H, Jiang H, Wojtas L, Zhang XP. Angew Chem Int Ed. 2010;49:10192–10195. doi: 10.1002/anie.201005552. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:10390–10393. [Google Scholar]; g) Lu H, Jiang H, Hu Y, Wojtas L, Zhang XP. Chem Sci. 2011;2:2361–2366. [Google Scholar]; h) Lu H, Hu Y, Jiang H, Wojtas L, Zhang XP. Org Lett. 2012;14:5158–5161. doi: 10.1021/ol302511f. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Zardi P, Intrieri D, Caselli A, Gallo E. J Organomet Chem. 2012;716:269–274. [Google Scholar]
- 21.Jin LM, Xu X, Lu H, Cui X, Wojtas L, Zhang XP. Angew Chem Int Ed. 2013;52:5309–5313. doi: 10.1002/anie.201209599. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:5417–5421. [Google Scholar]
- 22.Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org Lett. 2008;10:1995–1998. doi: 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- 23.The UV-vis spectrum of [Co(P1)] was found no obvious change in the absence or presence of stoichiometric amount PhCHO, indicating that the interaction between [Co(P1)] and PhCHO might be very weak.
- 24.For low-yielding reactions, fluoroanilines and azobenzenes were found to be the major byproducts, which could be easily removed from the desired amides through column chromatography.
- 25.Under the same catalytic reaction conditions, the use of tosyl azide (TsN3), bis(2,2,2-trichloroethyl)phosphoryl azide (TcepN3), and trichloroethoxycarbonyl azide (TrocN3) resulted in no significant formation of the corresponding aldehydic C–H amination products.
- 26.Griller D, Ingold KU. Acc Chem Res. 1980;13:317–323. [Google Scholar]
- 27.19F-NMR also indicated the formation of homo-coupled fluoroazobenzene (~16%) and fluoroaniline (~21%).
- 28.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 29.For plots of the log(kX/kH) data with other Hammett sigma-constants, including electrophilic substituent constant (σρ+), nucleophilic substituent constant (σρ−) and radical substituent constant (σRadical) as well as with bond dissociation energy (BDE) of aldehydic C–H bonds, see Figure S1–5 in the supporting information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.