

Abstract
High linear energy transfer (LET) radiation including α particles and heavy ions is the major type of radiation find in space and is considered a potential health risk for astronauts. Even though the chance that these high LET particles traversing through the cytoplasm of cells is higher than that through the nuclei, the contribution of targeted cytoplasmic irradiation, to the induction of genomic instability and other chromosomal damages induced by high LET radiation is not known. In the present study, we investigated whether mitochondria are the potential cytoplasmic target of high LET radiation in mediating cellular damage using a mitochondrial DNA (mtDNA) depleted (ρ0) human small airway epithelial (SAE) cell model and a precision charged particle microbeam with a beam width of merely one micron. Targeted cytoplasmic irradiation by high LET α particles induced DNA oxidative damage and double strand breaks in wild type ρ+ SAE cells. Furthermore, there was a significant increase in autophagy, micronuclei, which is an indication of genomic instability, together with the activation of nuclear factor kappa-B (NF-κB) and mitochondrial inducible nitric oxide synthase (iNOS) signaling pathways in ρ+ SAE cells. In contrast, ρ0 SAE cells exhibited a significantly lower response to these same endpoints examined after cytoplasmic irradiation with high LET α particles. The results indicate that mitochondria are essential in mediating cytoplasmic radiation induced genotoxic damage in mammalian cells. Furthermore, the findings may shed some light in the design of countermeasures for space radiation.
Keywords: Mitochondria, genomic instability, autophagy, ROS
1 Introduction
Space radiation contains a complex mixture of high energy and high atomic number particles [1]. Astronauts are constantly exposed to space radiation of various types of particles and energies during long-term stays in space, including low and high LET radiation. Compared with low LET radiation such as x- or gamma-rays, high LET particles are much more effective in triggering diverse biological effects in mammalian cells, including genomic instability and cancer induction [2]. The potential acute and chronic health effects of high LET radiation are due mainly to direct damage to DNA. Although direct nuclear damage and subsequent genetic mutations are likely causal events for cancer, the potential contribution from cytoplasmic damage cannot be ignored. Previous studies have demonstrated that extranuclear target also plays an important role in mediating the genotoxic effects of ionizing radiation [3,4]. However, the role of cytoplasmic damage in promoting genomic instability in the progeny of hit cells is largely unknown. This is mainly due to the technical difficulties in selectively targeting the cytoplasm without affecting the nucleus. The Columbia University charged particle microbeam, which can selectively target the cytoplasm of individual cells with a defined number of α particles, provides a useful tool to investigate the incidence and mechanism of genomic instability and other chromosomal damages induced by extranuclear irradiation.
Mitochondria are important dynamic organelles in the cytoplasm whose major function is to produce ATP through the respiration chain. The unique feature of mitochondria is the presence of its own genetic apparatus, mtDNA. Mammalian mtDNA encodes 13 transcripts of the electron transport chain proteins which are essential to maintain normal mitochondrial function [5]. Mutations in the mtDNA have been associated with several mitochondrial diseases [6], as well as being implicated in human carcinogenesis [7,8]. Mitochondrial dysfunction may lead to impaired cellular energy metabolism and increase mitochondria-derived oxidative stress, thus promoting damage of both nuclear and mtDNA, and activate a variety of signaling cascades involved in regulating cell proliferation and apoptosis [9]. Moreover, our recent studies have shown that targeted cytoplasmic irradiation resulted in mitochondrial fragmentation and reduced respiratory chain function in irradiated human small airway epithelial (SAE) cells, indicating that mitochondria play a potential role in mediating radiation damages in mammalian cells [10]. Since mitochondrial DNA alterations are frequently found in various human cancers [7,8] as well as in directly irradiated and in bystander cells [10,11], our present study elucidating how targeted cytoplasmic stimuli modulates nucleus damage will provide important information on radiation protection pertaining to high LET radiation.
In the present report, telomerase-immortalized SAE cell lines with or without mtDNA were used to investigate the role of mitochondrial function in mediating genomic instability of high LET radiation. Our results show that cytoplasmic irradiation with high LET particles induced an increase in nuclear DNA oxidative damage as well as induction of micronucleus in wild type ρ+ SAE cells. Moreover, there was a significant increase of autophagy that was reversible at 48 hours. In contrast, the biological effects induced by cytoplasmic irradiation with high LET particles were minimal in ρ0 SAE cells, with no obvious nuclear DNA oxidative damage nor micronucleus induction. Similarly, the NF-κB/iNOS signaling pathway was activated in ρ+ SAE cells but not in ρ0 SAE. This data clearly validates the role of mitochondria in modulating the DNA damaging effects of high LET radiation delivered to the cytoplasm of mammalian cells. Our study explores the differential biological effects of high LET radiation on nuclear versus cytoplasmic damage.
2 Material and Methods
2.1 Antibodies
The monoclonal antibodies used for this study included: anti-phospho-ATM (Ser 1981) (Cell Signaling Technology, Inc., Danvers, MA; Cat. #4526); anti-phospho-ATR (Cell Signaling Technology, Inc., Danvers, MA; Cat. #2853); anti-phospho-DNA PKcs (Ser2056) (Abcam, Cambridge, MA; Cat. ab18192); anti-phospho-Histone H2AX (Ser139) (Millipore, Mahopac, NY; Cat. #05-636); anti-8-OHdG (Abcam, Cambridge, MA; Cat. ab10802); LC3B (Invitrogen, Inc., Palo Alto, CA; Cat. L10382); NF-κB p65 (Cell Signaling Technology, Inc., Danvers, MA; Cat.4764S); iNOS (Cell Signaling Technology, Inc., Danvers, MA; Cat.13120).
2.2 Cell culture
The human telomerase reverse transcriptase (h-TERT) immortalized SAE cells generated previously in our laboratory [10] were grown in Small Airway Epithelial Cell Growth Medium (SAGM) medium supplemented with various growth factors supplied by the manufacturer (Lonza., Maryland, USA). Mitochondrial DNA depleted ρ0 SAE cells were generated from the parental ρ+ SAE cells using prolonged exposure to ethidium bromide (50 ng/ml) as described before [12]. The medium was supplemented with 50 μg/ml uridine because cells are auxotrophic to uridine [13]. Cultured cells were maintained at 37 °C in a humidified 5% CO2 incubator.
2.3 High LET cytoplasmic irradiation procedure
High LET α particles cytoplasmic irradiation was conducted using the microbeam at the Radiological Research Accelerator Facility of Columbia University. Approximately, 500–600 cells were plated on microbeam dishes coated with Cell-Tak (BD Biosciences, San Jose, CA) to enhance cell attachment. A defined number of 4He ions at 5.1 MeV were generated using a Singletron and the alpha particles were delivered at two target positions of cytoplasm, which are 8 μm away from each end of the cell nucleus along the major axis of the nucleus. The LET of the alpha particles is ~95 keV/μ and the dose equivalent is ~ 0.3 Gy per particle. Detailed methods for targeted cytoplasmic irradiation with the charged particle microbeam have been described previously [3,14,15]. All control cells were stained and sham-irradiated as well.
2.4 Immunofluorescence
Cells were grown on polypropylene film of the microbeam dishes and rinsed with PBS post high LET cytoplasmic irradiation. Cells were fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature and rinsed three times with PBS. The fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 20 min and stained with 1:200 dilution of the following primary antibodies: γ-H2AX, p-ATM, p-ATR, p-DNA-PKcs and LC3B. Alexa® Fluor 488 or Tetramethyl Rhodamine labeled secondary antibodies were obtained from Jackson Immuno Research (Jackson Immuno Research Labs, INC. West Grove, PA; Cat. 200-542-037 and 200-022-037). DAPI was used for nuclear stain. Images were captured using a laser confocal microscope (Nikon TE200-C1, Japan).
2.5 Immunocytochemistry
Immediately after irradiation, SAE cells were rinsed with PBS and fixed for 15 min in 4% paraformaldehyde at room temperature. The fixed cells were permeabilized (0.2% Triton X-100) for 10 min then rinsed three times with PBS. The cells were immunolabeled with 1:100 dilution of the primary antibody (NF-κB, iNOS) in 2% BSA/PBS for 1 hour; rinsed three times with PBS, before counterstained with secondary antibody (1:200) and the immunocomplex was visualized using the Avidin Biotin Complex (ABC) method. The samples were examined with a Nikon LABOPHOT-2 microscope and images were captured by the SPOT Basic™ software. The staining intensity was measured by image J (NIH) software.
2.6 TUNEL staining
TUNEL staining was performed to determine apoptotic nuclei after cytoplasmic irradiation according to the manufacturer’s instructions (Life Technologies, Inc., Palo Alto, CA). Cells were fixed in 4% paraformaldehyde for 20 min at 24 hours post irradiation and permeabilized (0.2% Triton X-100 and 0.1% sodium citrate) for 15 min on ice. TUNEL reaction mixture was added to cells and incubated for 1 hour at 37 °C in a humidified incubator with occasional mixing. The samples were observed using a Nikon LABO-PHOT-2 fluorescence microscope. DNase I-treated cells were used as a positive control and those without terminal transferase enzyme were used as a negative control.
2.7 Micronuclei scoring
The cytokinesis block technique was used for the micronuclei assay. Briefly, cultures were treated with 1 μg/mL cytochalasin-B for 36 hours and then fixed with 4% paraformaldehide for 20 min. The polypropylene films with cells were covered/stained with DAPI mounting solution. Micronuclei were scored in 200 binucleated cells and classified according to standard criteria [16]. The micronuclei yield was calculated as the ratio of micronuclei number to the scored number of bi-nucleated cells.
Statistics analysis
Data were presented as mean±standard deviation. Statistical analysis was performed by Student’s t test. P<0.05 was considered statistically significant between non-irradiated control and cytoplasmic irradiation groups.
3 Results
3.1 High LET radiation induced nuclear DNA oxidative damage and chromosomal breaks in ρ+ SAE cells
To detect oxidative damage in nuclear DNA, both ρ+ and ρ0 SAE cells were irradiated with α particles through the cytoplasm. The formation of 8-hydroxydeoxyguanosine (8-OHdG), a biomarker for the oxidative damage of nuclear DNA, was quantified by immunocytochemical staining with 8-OHdG polyclonal antibody. As shown in Fig. 1A, 8-OHdG staining was more intense in the nuclei of cytoplasmic irradiated ρ+ SAE cells when compared with untreated control cells. Quantification of the staining intensity demonstrated a two fold increase in 8-OHdG at 0.5 hours post cytoplasmic irradiation and a three fold increase at 24 hours when compared with controls (Fig. 1C). In contrast, the induction of 8-OHdG by cytoplasmic irradiation in ρ0 cells was not obvious (Fig. 1B and D). The baseline levels of 8-OHdG in ρ0 SAE cells were higher than corresponding background levels in ρ+ SAE cells which has been shown in the previous study [12]. These results indicated that high LET cytoplasmic irradiation induced oxidative damage in the nucleus of ρ+ SAE cells.
Figure 1.

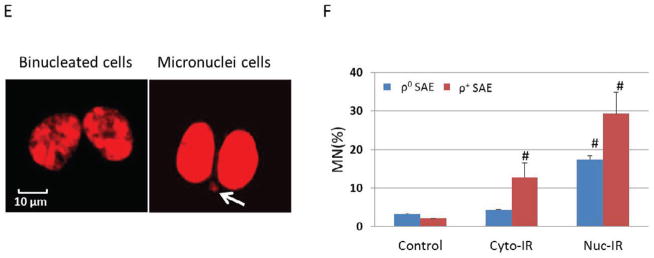
ρ0 SAE cells were less responsive than ρ+ SAE cells to high LET cytoplasmic irradiation induced nuclear DNA oxidative damage and micronucleus formation. A. Detection of 8-OHdG by immunocytochemistry. Representative images of untreated and cytoplasmic irradiated ρ+ SAE cells. B. Representative images of untreated and cytoplasmic irradiated ρ0 SAE cells. C. D. Relative quantification of 8-OHdG staining intensity induced by cytoplasmic irradiation in ρ+ SAE cells (C) and ρ0 SAE cells (D). Images were quantified by Image J software. The average intensity (mean ± SD) per cell was obtained from 200 cells per sample. #, P<0.01 versus the control group. E. Representative images of micronuclei image of ρ+ SAE cells irradiated with ten α particles cytoplasmic irradiation. Arrows indicate micronuclei. F. Nuclear irradiation induced an increased micronuclei level in both ρ+ and ρ0 SAE cells. Cytoplasmic irradiation increased micronuclei in ρ+ SAE cells, however, ρ0 SAE cells did not show significant increase of micronuclei formation. Data represent mean ± SD of three independent experiments. In each experiment, 200 cells were scored. #, P<0.01 versus the non-irradiated control.
Direct DNA damage caused by ionizing radiation has been considered to be the main cause of mutations. In order to ascertain the role of mitochondria in mediating radiation-induced genomic instability, the incidence of micronucleus was examined in both ρ+ and ρ0 SAE cells. Micronuclei were scored in binucleated ρ+ and ρ0 SAE cells at 48 hours post cytoplasmic irradiation. Representative image showed micronuclei formation in cytoplasmic irradiated ρ+ SAE (Fig. 1E). Nuclear irradiation with ten α particles induced 18% and 29% micronuclei frequency in ρ0 and ρ+ SAE cells, respectively. Furthermore, cytoplasmic irradiation with ten α particles also induced a statistically significant increase to 12% in micronuclei yield among ρ+ SAE cells. However, cytoplasmic irradiation had no obvious effect on the frequency of micronuclei in ρ0 SAE cells. These results reveal that the mtDNA-depleted ρ0 SAE cells are less sensitive to nuclear mutations induced by cytoplasmic irradiation.
3.2 High LET cytoplasmic irradiation induced DNA double strand breaks in ρ+ SAE cells
As DNA double strand breaks (DSBs) are the main deleterious lesions induced by radiation, the number of DSBs was scored by examining the foci of phosphorylated H2AX in cytoplasmic irradiated cells. As shown in Fig. 2A, γ-H2AX foci are visualized as bright spots in the nucleus of ρ+ and ρ0 SAE cells. According to distribution of cells with γ-H2AX foci, cells with more than two γ-H2AX foci in the nucleus were quantified as positive cells. We counted the number of foci per cell and the number of cells with foci in each experiment, for example, how many cells had one, two, or even five foci. Two groups were defined in our study: number of cells with less than two γ-H2AX foci, and cells containing more than two γ-H2AX foci using established methodology [17].
Figure 2.


Cytoplasmic irradiation induced DNA DSBs in ρ+ SAE cells. A. Immunofluorescent staining of γ-H2AX foci in ρ+ and ρ0 SAE cells treatment with ten α particles cytoplasmic irradiation at 0.5 hours. B. Percentage of γ-H2AX positive cells after cytoplasmic irradiation in both ρ+ and ρ0 SAE cells. C. Average foci per cell in ρ+ and ρ0 SAE cells post cytoplasmic irradiation. Mean percentage of γ-H2AX foci=cells with more than two γ-H2AX foci/total count cells number
#, P<0.01 versus the control group. Bars indicate ± SD. Results were repeated in three other experiments. In each experiment, 100 cells were scored.
The results indicated that 50% of the ρ+ SAE cells had more than two γ-H2AX foci in the nucleus at 0.5 hours post-irradiation. The foci induction decreased to 30% at 4 hours and the level remained relative stable until 24 hours post irradiation (Fig. 2B). The average number of γ-H2AX foci per cell at the 0.5 hours time point increased to five (Fig. 2C). However, the average foci number per cell was reduced to three in ρ+ SAE cells at 4 hours post irradiation. Unlike the ρ+ SAE cells, there was no significant difference in the percentage of γ-H2AX positive cells and average number of γ-H2AX foci in irradiated ρ0 SAE cells (Fig. 2D and E).
In order to determine whether common DNA repair proteins participated in the repair of DNA double strand breaks induced by targeted cytoplasmic irradiation, phosphorylated ataxia-telangiectasia mutated (ATM), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ataxia telangiectasia and Rad3 related (ATR) were investigated by florescence immunohistochemistry in ρ+ SAE cells. As showed in Fig. 3A and B, the phosphorylated ATM (Ser1981) and DNA-PKcs (Ser2056) were colocalized with γ-H2AX foci indicating ATM and DNA-PKcs may be involved in the DNA damage repair process. However, the phosphorylated ATR (Ser428), which showed a dispersed pattern in the nucleus (Fig. 3C), did not co-localize with γ-H2AX foci induced by cytoplasmic irradiation in ρ+ SAE cells, indicating that ATR might not be a player in repairing the DNA damage induced by cytoplasmic irradiation.
Figure 3.
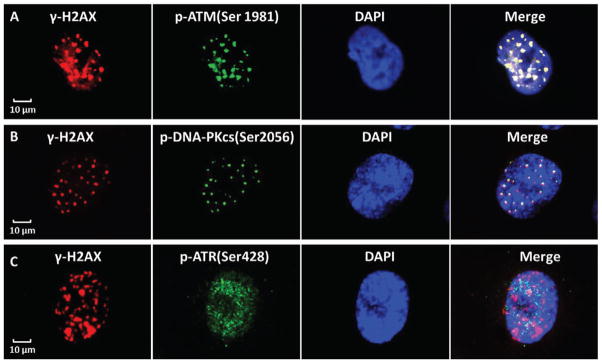
ATM and DNA-PKcs participate in cytoplasmic irradiation induced DNA damage repair in ρ+ SAE cells. A. Immunofluoresent staining of γ-H2AX and p-ATM (Ser1981) in ρ+ SAE cells. The γ-H2AX was labeled in red and p-ATM was labeled in green, the yellow in the merge shows γ-H2AX and p-ATM colocalization in the nucleus. B. The γ-H2AX was labeled in red and p-DNA-PKcs (Ser2056) was labeled in green, the yellow in the merge shows γ-H2AX and p-DNA-PKcs colocalization in the nucleus. C. The γ-H2AX (Red) shows foci in the nucleus, however, ATR (Ser428) staining is diffuse (green) post cytoplasmic irradiation. These representative images show 0.5 hours post cytoplasmic irradiation.
3.3 High LET cytoplasmic irradiation induced apoptosis/autophagy in ρ+ SAE cells
To assess whether cytoplasmic irradiation induces cell death, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) stain was used to detect apoptosis in both ρ+ and ρ0 SAE cell lines. Fig. 4A shows that cytoplasmic irradiation induced 5% TUNEL positive cells in ρ+ SAE cells at 24 hours post-irradiation and the incidence showed a slight but insignificant increase at 48 hours. In contrast, there was no induction of TUNEL positive cells in similarly irradiated ρ0 SAE cells at the 24 hours time point. A delayed apoptosis of 6%, however, was observed at 48 hours post-irradiation (Fig. 4B).
Figure 4.

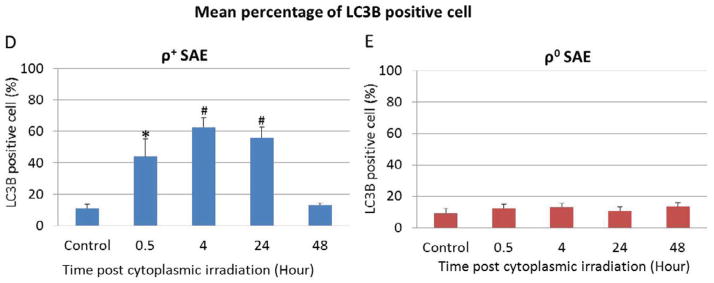
High LET radiation targeted at cytoplasm induces apoptosis and autophagy in ρ+ SAE cells. A. Percentage of apoptosis cells induced by cytoplasmic irradiation in ρ+ and ρ0 SAE cells was detected by TUNEL stain. B. Percentage of the autophagy in ρ+ and ρ0 SAE cells post ten α particles cytoplasmic irradiation. C. Representative image of LC3B staining of autophagy in ρ+ and ρ0 SAE cells post ten α particles cytoplasmic irradiation.
#, P<0.01, *, P<0.05 versus the control group. Bars indicate ± SD. Results are mean of three independent experiments. In each experiment, 100 cells were scored.
Since there was increased oxidative stress and DNA damage, but only 5% cell death at 24 hours post cytoplasmic irradiation in ρ+ SAE cells, in addition to DNA repair, we entertained the idea that autophagy induced by cytoplasmic irradiation may also be a likely effect. The rational being that cytoplasmic irradiation induces mitochondrial fission (14) and autophagy is a major cellular quality control process for the selective degradation of dysfunctional mitochondria. Therefore, we analyzed the level of LC3B, a reliable biomarker of autophagy in irradiated SAE cells. Chloroquine diphosphate treatment was used as a positive control (data not shown). Cytoplasmic irradiation caused LC3B to be recruited to autophagosomes as demonstrated by the appearance of bright punctated structures in ρ+ SAE cells (Fig. 4C). The number of LC3B positive cells increased to 60% at 4 hours post cytoplasmic irradiation and returned to control values at 48 hours (Fig. 4D). However, LC3B stain was diffuse in the cytosol in control and cytoplasmic irradiated ρ0 SAE cells (Fig. 4C). Quantification of LC3B positive ρ0 cells showed no obvious change from 0.5 to 48 hours post cytoplasmic irradiation (Fig. 4E). These results indicated that cytoplasmic irradiation induced autophagy was dependent on the presence or absence of mtDNA.
3.4 Role of NF-κB in response to high LET cytoplasmic irradiation in ρ+ SAE cells
NF-κB is an important transcription factor and triggered by a wide variety of stimuli including oxidative stress. Immunocytochemistry was used to quantify whether cytoplasmic irradiation activates NF-κB protein expression in ρ+ and ρ0 SAE cells. NF-κB p65 staining images of ρ+ and ρ0 SAE cells post cytoplasmic irradiation 24 hours are shown in Fig. 5A. There was more intense of NF-κB p65 staining in cytoplasmic irradiated ρ+ SAE cells when compared with untreated control cells at 24 hours (Fig. 5A, top panel). Quantification of the result showed that NF-κB p65 was increased approximately two-fold compared with control at 0.5 to 24 hours post-irradiation in ρ+ SAE cells (Fig. 5B, top panel); however, there was no obvious NF-κB p65 change in irradiated ρ0 SAE cells (Fig. 5A.B, lower panel).
Figure 5.
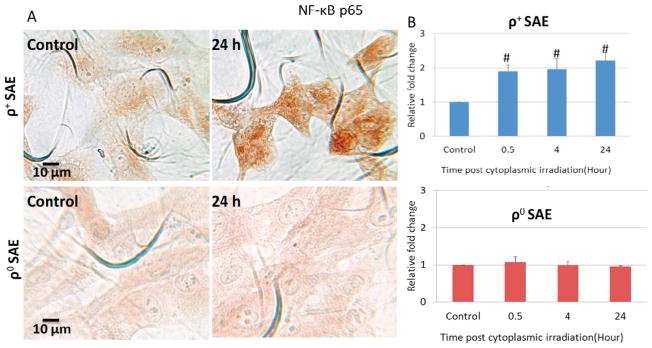
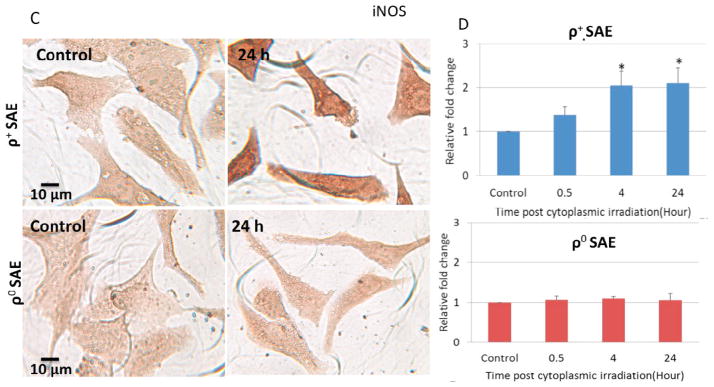
Effects of NF-κB in the regulation of genomic instability induced by cytoplasmic irradiation in ρ+ SAE cells. A. Representative image of NF-κB p65 immunocytochemistry staining of ρ+ and ρ0 SAE cells. B. Quantification of NF-κB p65 by mean gray value of ρ+ and ρ0 SAE cells. Levels of NF-κB p65 were significantly increased post cytoplasmic irradiation with ten α particles in ρ+ SAE cells but not in ρ0 SAE cells. C. Representative image of iNOS immunocytochemistry staining of ρ+ and ρ0 SAE cells. D. iNOS expression were significantly increased induced by cytoplasmic irradiation in ρ+ SAE cells, however, ρ0 SAE cells showed no response.
#, P<0.01, *, P<0.05 versus the control group. Bars indicate ± SD. Results were repeated in three other experiments. In each experiment, 100 cells were scored.
In order to determine whether cytoplasmic irradiation leads to the development of inflammation, the inducible nitric oxide synthase (iNOS) was quantified by immunocytochemistry. iNOS staining images of ρ+ and ρ0 SAE cells post cytoplasmic irradiation at 24 hours were shown in Fig. 5C. There was more intense of iNOS staining in cytoplasmic irradiated ρ+ SAE cells when compared with untreated control cells at 24 hours (Fig. 5C, top panel). The relative iNOS expression in cytoplasmic irradiated cells was increased to two fold at 4 hours and lasted until 24 hours when compared with non-irradiated control (Fig. 5D, top panel). In contrast, there was no significant change of iNOS expression in the time period of observation in ρ0 cells (Fig. 5C. 5D, lower panel).
4 Discussion
In this study, we found a significant increase in 8-OHdG and micronuclei formation in ρ+ SAE, but not in ρ0 SAE, upon exposure to targeted cytoplastmic irradiation, indicating that mitochondria, and specifically mtDNA, play an important role in mediating the response. Micronucleus is often used as a reliable biomarker for genetic damages in mammalian cells exposed to a variety of genotoxic agents [18]. There is evidence that ultraviolet light, x-ray, and γ-ray radiation are efficient inducer of 8-OHdG, a well established biomarker for oxidative DNA damages in various cultured cell lines and animal models [19,20]. Mitochondria are not only the main targets of oxidative damage but the primary site of ROS production [21]. Excessive production of ROS by mitochondria will lead to irreversible alterations of lipids, proteins and DNA resulting in further damage to the mitochondria, cytosol and the nucleus [22]. Furthermore, our previous studies have demonstrated that cytoplasmic irradiated SAE cells show an increase in mitochondrial fission and superoxide production that can be quenched by the radical scavenger, dimethyl sulfoxide [14]. It is likely that oxyradicals induced by membrane lipid peroxidation result in both changes in mitochondrial morphology as well as mtDNA mutations. Taken together with the present studies, our results clearly illustrate the essential role of mtDNA in mediating radiation damages in general, and genomic instability specifically by high LET radiation.
In response to DSBs, members of the kinases in the phosphoinositide three-kinase-related kinase (PIKK) expand family (ATM, ATR, and DNA-PKcs) are recruited first to the DNA damage site and phosphorylate histone H2AX which can be visualized as γ-H2AX foci [23,24]. In the current study, the average γ-H2AX foci increased within 0.5 hours post-irradiation indicating that DNA DSBs in ρ+ SAE cells was a rapid response event, perhaps due to oxidative stress. In contrast, ρ0 SAE cells did not show any DSBs indicating that SAE cells lacking respiratory chain function were less sensitive to DNA damage induced by cytoplasmic irradiation. There is evidence that mtDNA–depleted human skin fibroblasts (ρ0) have a reduced genotoxic response, as determined by mutation at the hprt locus on human x-chromosome [11]. DNA damage triggers phosphorylation of DNA-PKcs at several clusters of serine and threonine residues (including T2609, S2056, and T3950) that regulate the function of DNA-PKcs [25]. Moreover, both DNA-PKcs and the related protein kinase ATM contribute to DNA damage-induced phosphorylation of H2AX on serine 139 to form γ-H2AX [26]. Previous studies showed that ATM was autophosphorylated on Ser1981 in vivo after ionizing radiation in mammalian cells [27]. Our study revealed that γ-H2AX colocalized with ATM (Ser1981) and DNA-PKcs (Ser2056) induced by cytoplasmic irradiation, suggesting that ATM and DNA-PKcs participated in the cytoplasmic irradiation –induced DNA damage response. Although ATR activation was observed after cytoplasmic irradiation, the activated ATR (Ser428) did not colocalize with the well known DSB surrogate marker, γ-H2AX as indicated by the dispersed staining pattern of ATR. Even though ATR is activated primarily in response to single-stranded DNA regions and stalled DNA replication forks [28], the homogenous pattern of ATR staining may probably due to both single strand breaks and DSB at stalled replication forks with extended ss DNA regions, generated by cytoplasmic radiation.
Mitochondria are important dynamic organelles within the cytoplasm that produce ATP through the process of oxidative phosphorylation and are implicated in response to cellular stress, such as apoptosis and immune responses [29,30]. When cells suffer irreparable DNA damage, apoptosis is induced for eliminating damaged cells [31,32]. There are two apoptotic pathways in the cell, the “extrinsic” death-receptor mediated and the “intrinsic” mitochondrial pathways. The intrinsic pathway highlights the role of mitochondria in the regulation of apoptosis [33]. Previous studies have shown that radon exposure increased the apoptotic rate in both human bronchial epithelial cells and to a lesser degree in mtDNA knockdown cells [34]. In agreement with these studies, our results showed that cytoplasmic irradiation induced significant increase in the apoptotic rate in ρ+ SAE cells at 24 and 48 hours. These results demonstrated that the apoptosis observed in ρ+ cells was due to the intrinsic mitochondrial apoptotic pathway and confirmed the key role that mitochondria play in the process. However, ρ0 cells without mtDNA and therefore, without functional mitochondria, also showed increased apoptotic rate, although delayed, after cytoplasmic irradiation at 48 hours. This delayed response may perhaps be due to the induction of the extrinsic death ligand-mediated pathway. Our data clearly indicate that the intrinsic apoptosis pathway is triggered in wild type ρ+ SAE cells exposed to high LET irradiation.
Autophagy is a lysosome-dependent degradation process among eukaryotes which is usually induced in response to stressful conditions including oxidative stress, hypoxia, DNA damage, and anti-cancer therapies [35,36]. Autophagy is also a major cellular quality control process for the selective degradation of organelles, including mitochondria (mitophagy) [37]. Moreover, autophagy itself has a dual role, having pro-death or pro-survival role depending on the specific stimuli [38]. Recently, several studies have provided new insight into p38 and JNK Mitogen Activated Protein Kinase (MAPK) pathway in regulating the balance between autophagy and apoptosis in response to genotoxic stress [39]. Our current studies reveal that targeted cytoplasmic irradiation results in an increased rate of autophagy in ρ+ SAE cells within 24 hours, which returned to control level at 48 hours. However, the ρ0 SAE cells showed no autophagic response to cytoplasmic irradiation. Moreover, our previous report showed that targeted cytoplasmic irradiation with α particles induced mitochondrial fission and respiratory chain dysfunction in ρ+ SAE cells in the immediate post irradiation period [14]. There is also evidence that cells purposefully segregate its dysfunctional mitochondria by fission and eliminate them by autophagy [40]. These results, taken together, suggest that cells can eliminate dysfunctional mitochondria produced as a result of α particle radiation by autophagy in order to maintain cellular homeostasis. Although non-functional mitochondria below the mutational threshold could be eliminated by mitophagy, the accumulation of pathogenic mtDNA mutations in patients demonstrates a failure to remove mitochondria containing pathogenic mtDNA by autophagy above the mutational threshold [37]. This may perhaps be due to the failure of active recruitment of proteins to mitochondria, such as Parkin, which is critically important for the cell to undergo autophagy; and failure of activating general macroautophagy, in order to accomplish robust autophagic sequestration of dysfunctional mitochondria [41].
We also examined the signaling pathways that are triggered by high LET radiation. The NF-κB pathway is highly sensitive to a variety of stimuli and responsible for regulating gene expression of factors that control cell proliferation, adhesion, inflammation and redox status. Furthermore, combined exposure to environmental toxins, such as trichloroethylene and UVB has been shown to induce a toxic response by up regulating NF-κB dependent iNOS expression in early tumor promotion events [42]. Similarly, in the present study, we observed an increased NF-κB p65 and iNOS expression induced by cytoplasmic irradiation only in ρ+ SAE cells but not in ρ0 SAE cells, indicating that NF-κB/iNOS pathways are associated with mitochondrial signaling events. Our finding is consistent with previous reports that NF-κB activation was partially lost in mtDNA-depleted ρ0 HSF in response to α particle irradiation [43], and that iNOS activation is also dependent on transcription of NF-κB [44]. Therefore, it is not surprising that iNOS expression is not induced by cytoplasmic irradiation in ρ0 SAE cells where NF-κB p65 expression is unchanged. Recent studies also highlight the role of NF-κB signaling in regulating autophagy [45]. The possible mechanism is that NF-κB can either activate or inhibit signaling pathways that regulate the transcription of a subset of pro-autophagic-regulating genes leading to the induction of autophagy [46]. Thus, cytoplasmic irradiation is likely to trigger activation of NF-κB/iNOS pathways in a mitochondrial function dependent manner.
In summary, our studies reveal that targeted cytoplasmic irradiation with high LET radiation results in oxidative damage and DNA double strand breaks in ρ+ SAE cells. However, the mtDNA-depleted ρ0 SAE cells show no oxidative stress response to cytoplasmic irradiation. The damaged DNA is correlated with increased micronucleus formation and apoptosis in ρ+ SAE cells but not in ρ0 SAE cells indicating that mitochondrial function is essential in regulating genomic instability induced by high LET radiation. Our data clearly demonstrates the biological consequences of high LET radiation in nuclear versus cytoplasmic damage and the significant role of mitochondrial function in mediating genomic instability. Finally, the findings may provide some insight in the design of countermeasures for space radiation protection.
Acknowledgments
Financial Support: This research was supported by funding from the National Institutes of Health grants 5P01-CA49062-20 and 5R01-ES 12888-06. The Radiological Research Accelerator Facilities is a National Institutes of Health (NIH) sponsored Resource Center through grant EB-002033 (National Institute of Biomedical Imaging and Bioengineering, NIBIB).
The authors thank members of the Radiological Research Accelerator Facility for their assistance with the microbeam irradiations (Drs. Guy Garty, Andrew Harkins, Alan Bigelow and Yanping Xu). This research was supported by the National Institutes of Health grants 5P01-CA49062-20 and 5R01-ES 12888-06. The Radiological Research Accelerator Facilities is a NIH sponsored Resource Center through grant EB-002033 (NIBIB).
Footnotes
Conflict of interest
The authors declare that they have no actual or potential competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Durante M, Cucinotta FA. Heavy ion carcinogenesis and human space exploration. Nat Rev Cancer. 2008;8:465–472. doi: 10.1038/nrc2391. [DOI] [PubMed] [Google Scholar]
- 2.Hada M, Wu H, Cucinotta FA. mBAND analysis for high- and low-LET radiation-induced chromosome aberrations: a review. Mutat Res. 2011;711:187–192. doi: 10.1016/j.mrfmmm.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Hong M, Xu A, Zhou H, Wu L, Randers-Pehrson G, Santella RM, et al. Mechanism of genotoxicity induced by targeted cytoplasmic irradiation. Br J Cancer. 2010;103:1263–1268. doi: 10.1038/sj.bjc.6605888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nugent S, Mothersill CE, Seymour C, McClean B, Lyng FM, Murphy JEJ. Altered mitochondrial function and genome frequency post exposure to γ-radiation and bystander factors. Int J Radiat Biol. 2010;86:829–841. doi: 10.3109/09553002.2010.486019. [DOI] [PubMed] [Google Scholar]
- 5.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 6.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 9.Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res. 2001;488:119–133. doi: 10.1016/s1383-5742(01)00053-9. [DOI] [PubMed] [Google Scholar]
- 10.Piao CQ, Liu L, Zhao YL, Balajee AS, Suzuki M, Hei TK. Immortalization of human small airway epithelial cells by ectopic expression of telomerase. Carcinogenesis. 2005;26:725–731. doi: 10.1093/carcin/bgi016. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H, Ivanov VN, Lien YC, Davidson M, Hei TK. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res. 2008;68:2233–2240. doi: 10.1158/0008-5472.CAN-07-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang SXL, Partridge MA, Ghandhi SA, Davidson MM, Amundson SA, Hei TK. Mitochondria-derived reactive intermediate species mediate asbestos-induced genotoxicity and oxidative stress-responsive signaling pathways. Environ Health Perspect. 2012;120:840–847. doi: 10.1289/ehp.1104287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Partridge MA, Huang SXL, Hernandez-Rosa E, Davidson MM, Hei TK. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007;67:5239–5247. doi: 10.1158/0008-5472.CAN-07-0074. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B, Davidson MM, Zhou H, Wang C, Walker WF, Hei TK. Cytoplasmic Irradiation Results in Mitochondrial Dysfunction and DRP1-Dependent Mitochondrial Fission. Cancer Res. 2013;73:6700–6710. doi: 10.1158/0008-5472.CAN-13-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu LJ, Randers-Pehrson G, Xu A, Waldren CA, Geard CR, Yu Z, et al. Targeted cytoplasmic irradiation with alpha particles induces mutations in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:4959–4964. doi: 10.1073/pnas.96.9.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albertini RJ, Anderson D, Douglas GR, Hagmar L, Hemminki K, Merlo F, et al. IPCS guidelines for the monitoring of genotoxic effects of carcinogens in humans. International Programme on Chemical Safety. Mutat Res. 2000;463:111–172. doi: 10.1016/s1383-5742(00)00049-1. [DOI] [PubMed] [Google Scholar]
- 17.Yoshikawa T, Kashino G, Ono K, Watanabe M. Phosphorylated H2AX foci in tumor cells have no correlation with their radiation sensitivities. J Radiat Res (Tokyo) 2009;50:151–160. doi: 10.1269/jrr.08109. [DOI] [PubMed] [Google Scholar]
- 18.Luzhna L, Kathiria P, Kovalchuk O. Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Front Genet. 2013;4:131. doi: 10.3389/fgene.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemon JA, Rollo CD, Boreham DR. Elevated DNA damage in a mouse model of oxidative stress: impacts of ionizing radiation and a protective dietary supplement. Mutagenesis. 2008;23:473–482. doi: 10.1093/mutage/gen036. [DOI] [PubMed] [Google Scholar]
- 20.Giammarioli S, Filesi C, Sanzini E. Oxidative stress markers: specificity and measurement techniques. Ann DellIstituto Super Sanità. 1999;35:563–576. [PubMed] [Google Scholar]
- 21.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol. 2010;174:212–220. doi: 10.1016/j.resp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol CB. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 24.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 25.Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 26.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 27.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 28.Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair. 2004;3:901–908. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 29.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 30.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 32.Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH. Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol. 2012;83:747–757. doi: 10.1016/j.bcp.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 33.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–2966. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 34.Li BY, Sun J, Wei H, Cheng YZ, Xue L, Cheng ZH, et al. Radon-induced reduced apoptosis in human bronchial epithelial cells with knockdown of mitochondria DNA. J Toxicol Environ Health A. 2012;75:1111–1119. doi: 10.1080/15287394.2012.699841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838–854. doi: 10.4161/auto.6.7.12113. [DOI] [PubMed] [Google Scholar]
- 36.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer Biol Ther. 2011;11:127–137. doi: 10.4161/cbt.11.2.14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174–179. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilkerson RW, De Vries RLA, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali F, Sultana S. Repeated short-term stress synergizes the ROS signalling through up regulation of NFkB and iNOS expression induced due to combined exposure of trichloroethylene and UVB rays. Mol Cell Biochem. 2012;360:133–145. doi: 10.1007/s11010-011-1051-7. [DOI] [PubMed] [Google Scholar]
- 43.Ivanov VN, Ghandhi SA, Zhou H, Huang SX, Chai Y, Amundson SA, et al. Radiation response and regulation of apoptosis induced by a combination of TRAIL and CHX in cells lacking mitochondrial DNA: a role for NF-κB-STAT3-directed gene expression. Exp Cell Res. 2011;317:1548–1566. doi: 10.1016/j.yexcr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song S-H, Min H-Y, Han A-R, Nam J-W, Seo E-K, Park Seoung Woo, et al. Suppression of inducible nitric oxide synthase by (−)-isoeleutherin from the bulbs of Eleutherine americana through the regulation of NF-kappaB activity. Int Immunopharmacol. 2009;9:298–302. doi: 10.1016/j.intimp.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am J Cancer Res. 2011;1:629–649. [PMC free article] [PubMed] [Google Scholar]
- 46.Deruy E, Gosselin K, Vercamer C, Martien S, Bouali F, Slomianny C, et al. MnSOD upregulation induces autophagic programmed cell death in senescent keratinocytes. PloS One. 2010;5:e12712. doi: 10.1371/journal.pone.0012712. [DOI] [PMC free article] [PubMed] [Google Scholar]