

Abstract
Since the discovery of nitric oxide (NO) in the 1980s, this cellular messenger has been shown to participate in diverse biological processes such as cardiovascular homeostasis, immune response, wound healing, bone metabolism, and neurotransmission. Its beneficial effects have prompted increased research in the past two decades, with a focus on the development of materials that can locally release NO. However, significant limitations arise when applying these materials to biomedical applications. This Feature Article focuses on the development of NO-releasing and NO-generating polymeric materials (2006–2011) with emphasis on recent in vivo applications. Results are compared and discussed in terms of NO dose, release kinetics, and biological effects, in order to provide a foundation to design and evaluate new NO therapies.
1. Introduction
Nitric oxide (NO) is a signaling molecule that plays a pivotal role in physiological processes such as cardiovascular homeostasis, immune response, bone metabolism, neurotransmission, and cancer.[1-5] Since early research in the 1980s by Furchgott, Zawadzki, Palmer, Ignarro, and others, the molecule once identified as endothelial-derived relaxing factor (EDRF)[6,7] has been the subject of significant research not only in the basic sciences, but also in applied sciences such as the biomaterials field. For instance, recent investigations in this field have demonstrated a key role of NO in phenomena such as the differentiation of mesenchymal stem cells into an osteoblast-like phenotype in silk scaffolds[8] or in acute neural cell death associated with an injured spinal cord.[9]
Over the past two decades, researchers have been working on the development of efficient NO-releasing and -generating materials for clinical therapies.[10-12] Physiological NO concentrations have been estimated to be in the range of 100 pm to 5 nm depending on the situation.[13] Therefore, the effective therapeutic dose of NO may vary greatly and careful assessment of the effects of larger concentrations is required. Although the potential beneficial impact of long-term NO-delivering therapies is significant, challenges such as the short half-lives of most NO donors and the uncertainty regarding safe therapeutic doses of NO for in vivo applications limit commercialization. Nevertheless, once these difficulties are addressed, advances in treatments for atherosclerosis, wound healing, diabetes, and cancer are likely to follow.
In this Feature Article, we will discuss some recent advances in the design and biomedical use of NO-releasing and -generating materials (2006–2011), with emphasis on recent in vivo applications. Molecular therapies without the use of a polymeric matrix are excluded from the present discussion. Since nanometer-scale research in the past two decades has prompted a significant progress in the development of NO-releasing nanoparticles,[14] approaches that incorporate polymers are briefly discussed herein. For strategies not covered in the current Feature Article, readers are referred elsewhere.[4,11,15,16] General concepts behind the molecular signaling of NO and the main types of NO donors available are also introduced.
2. The Molecular Biology of NO Becomes Clear
NO is produced by the enzyme nitric oxide synthase (NOS) from the oxidation of the terminal guanidine nitrogen of l-arginine, generating l-citruline. This enzymatic activity requires the presence of cofactors such as tetrahydrobiopterin (BH4), nicotinamide-adenine-dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), and flavin mononucleotide (FAM). Three main isoforms of this enzyme have been identified: neuronal (nNOS), endothelial (eNOS) and inducible (iNOS). The first isoform is predominantly expressed in neurons and skeletal muscle, while eNOS is expressed mostly in endothelial cells (EC) and iNOS in immune lineage cells and smooth muscle cells.[3,17] Separate chromosomes and genes were identified as being responsible for encoding the different isoforms.[2,18] Once produced and released, NO diffuses to neighbor cells and reaches its target, soluble guanylate cyclase (sGC). The activation of sGC, caused by NO binding to its heme moiety, induces an increase in cyclic guanylate monophosphate (cGMP), a signaling messenger that activates GMP-dependent pathways. Recent findings proved the existence of another lower affinity site in sGC that could account for the phasic activity of the NO-induced activation of sGC.[19] Through cGMP-independent mechanisms, NO also targets sulfhydryl-containing proteins (forming S-nitrosothiols, as in the case of caspases)[20,21] and certain protein kinases.[22] Dose-dependent cellular effects have been largely demonstrated in vitro.[23,24] Low NO levels (10 nm to 1 μm) have been associated with anti-apoptotic effects, whereas high concentrations (>1 μm) induce cell death through necrosis.[25] Given the critical physiological implications of NO, its production is thoroughly regulated by the intracellular calcium concentration, the subcellular localization of NOS, the presence of agonists such as bradykinin and acetylcholine, and several other stimuli such as fluid shear stress, growth factors and pathogens.[26]
New roles and signaling mechanisms for NO are still being discovered and this new biology will help guide the development of more efficient NO delivery therapies. For instance, recent findings have demonstrated a xanthine oxidorreductase-dependent NO production from nitrite in a pulmonary model.[27] More details about the molecular routes involving NO are described elsewhere.[4,28,29]
3. NO Donors: Diazeniumdiolates versus S-nitrosothiols
A large variety of NO donors has been explored for biomedical applications in the past 20 years, such as organic nitrates, oximes and metal-NO complexes such as sodium nitroprusside. However, for the purpose of this Feature Article, we will focus on two classes of donors that have garnered the most attention for applications in the biomedical field and have been utilized widely in combination with biomaterials. These classes are the diazeniumdiolates and the S-nitrosothiols (Table 1).
Table 1.
Diazeniumdiolates versus Nitrosothiols.
| Class | Structure | Release | Properties |
|---|---|---|---|
| Diazeniumdiolates |
|
Proton driven. Spontaneous in physiologic fluids. First-order release kinetics. Reliable half-lives depending on the structure of the nucleophilic adduct | Stable as solids. Potentially carcinogenic nitrosamines may form as by-products. Often light-sensitive |
| Nitrosothiols |
|
Transition metal-mediated catalytic decomposition (e.g., copper). Direct reaction with ascorbate. Homeolytic cleavage by light/temperature. Catalysis by specific enzymes | Stable as solids. Light-sensitive. Endogenously present mainly as nitroso-albumin. Natural NO carrier. Potential for unlimited NO release |
3.1. Diazeniumdiolates
Diazeniumdiolate moieties, of which the distinct chemistry has been recognized for approximately 50 years, are classified as C-, N-, O-, or S-bound.[30] Herein, we will mainly discuss the most biologically relevant N-bound type. Diazeniumdiolates are considered a highly useful class of NO donors, because of their ability to generate NO in a highly predictable manner and their versatility of chemical modifications to tailor NO generation. Moreover, diazeniumdiolated compounds, also known as NONOates (for a discussion on nomenclature, see a previous report[30]), are stable solids. They can spontaneously generate two molecules of biologically active NO per diazeniumdiolate residue when exposed to a physiological fluid, e.g. blood/tissue fluids, by means of hydrolysis when protonated. This release requires no specific metabolites or redox mechanisms, which accommodates facile utilization of these NO donors in medicine. Moreover, the tissue- and metabolite-independent activation of release promises a lack of tolerance development,[31] further increasing their attractiveness.
Typical synthesis of a diazeniumdiolate requires the reaction of a secondary amine, the nucleophile adduct, with highly pressurized NO, usually within a solvent such as acetonitrile or sodium methoxide. This step results in a diazeniumdiolate (NONO) group linked to the amine through its nitrogen atom. Depending on the exact atomic structure of the parent molecule, the half-life of its NO release kinetics can vary from fractions of a second to days or even weeks.[32] Moreover, by adding protective groups to diazeniumdiolate moieties, the normally spontaneous release of NO may be rendered dependent on certain enzymes or metabolites to achieve finer control. One example would be the enzyme-specific cleavage of protective groups to hydrolyze the diazeniumdiolate at specific target sites. For example, the donor V-PYRRO/NO will selectively release its NO load in the liver through cytochrome p450-catalyzed dealkylation of its protective ether group,[33] thereby only inhibiting apoptosis in hepatocytes. Additionally, specific modifications in the chemistry of diazeniumdiolates may alleviate negative side-effects of donors such as the formation of potentially carcinogenic nitrosamines[34] by incorporating lipophilic residues[35] or, prominently addressed in this Feature Article, by covalently linking these NO donors to the backbone of polymers for material coatings or prosthetic grafts.[36-41]
Although no diazeniumdiolated compounds have been approved clinically to date, they have been widely demonstrated as a reliable source of NO for research in a variety of medical fields, with a multitude of animal trials successfully undertaken. Furthermore, two human trials have been conducted, using diazeniumdiolates for the treatment of respiratory stress syndrome[42] and gastroparesis (Amulet Pharmaceuticals, Inc.).
3.2. S-Nitrosothiols
The second widely-investigated class of NO-donating compounds is that of S-nitrosothiols. S-nitrosothiols are currently under consideration as not just a reservoir for NO, but also as natural transporters of NO within biologic systems.[43] Recent investigations indicated that the formation of S-nitrosothiols occurs as an intermediary step during NO intra- and intercellular signaling.[44,45] Several naturally occurring S-nitrosothiols present in both tissue and blood include S-nitrosoglutathione (GSNO), S-nitrosoalbumin, and S-nitrosocysteine. S-nitrosothiols possess several key advantages over other NO donors. For one, the endogenous occurrence of biological S-nitrosothiols suggests a relative lack of toxicity issues compared to those associated with diazeniumdiolates. Similarly to diazeniumdiolates, however, there seems to be no build-up of tolerance over time associated with S-nitrosothiols.[46] Finally, the likely role as a main natural NO carrier presents the opportunity for utilizing those circulating molecules as an unlimited localized NO supply.
S-nitrosothiols are formed by the nitrosylation (or nitrosation) of thiol residues. In vivo, the exact formation mechanisms remain complex and under debate, but in laboratory conditions, they are easily synthesized by the reaction of thiols with nitrous acid in a highly acidic environment. This is only true, however, for low molecular weight thiolic compounds such as glutathione, but not for proteins. For the synthesis of the latter, usually S-transnitrosation reactions are necessary to transfer the nitrosonium ion (NO+) from the low molecular weight thiols to the protein thiols in cysteine.[47]
Contrary to diazeniumdiolates, S-nitrosothiols do not spontaneously release NO, although they may decompose under certain conditions. Although the detailed chemistry behind the release mechanisms from S-nitrosothiols is beyond the objective of this Feature Article (for details, see a previous report[48]), a number of mechanisms for NO release have been identified, including: transition metal-mediated catalytic decomposition (e.g., copper ions), direct reaction with ascorbate, homolytic cleavage of the S-NO bond by light and temperature, or enzyme-mediated release, such as through superoxide dismutase and protein disulphide isomerase. Additionally, reducing agents such as ascorbate can enhance metal ion-mediated release. The role of metal ions in vivo is debated, since such ions are often sequestered, drawing questions on the validity of this pathway. Also, GSNO after oxidation sequesters copper as a disulfide form, preventing further decomposition. Nonetheless, the incorporation of covalently linked metal ions in biomaterials to utilize circulating endogenous S-nitrosothiols for a potentially unlimited source of NO is an active field of research with promising results.[49-51] Other concepts to improve NO delivery through S-nitrosothiols include poly(ethylene glycol) (PEG)-conjugated S-nitrosoalbumin to improve circulation time[52] and the protection in a sol–gel construct for extended release.[53]
Similar to diazeniumdiolates, S-nitrosothiol compounds are not yet approved for clinical practice, but studied intensively for a variety of applications. Significant questions remain about the exact triggers for release and complicate further progress. Nevertheless, despite the complexities of the nature of S-nitrosothiols, these molecules have tremendous potential for clinical therapeutic use. Currently, several clinical trials are underway for the treatment of sexual dysfunction, diabetic ulcers and wound healing (for further details, see Section 5).
4. Novel NO-Releasing and NO-Generating Polymeric Materials
The first successful derivatization of a polymer with diazeniumdiolates as NO donors incorporated in the polymeric backbone was achieved by Smith et al. in the late 1990s.[54,55] Since then, current approaches in the development of NO-releasing and -generating polymers have shown some promise in vivo, but reduced clinical translation so far due to various limitations. For that reason, revolutionary strategies are necessary to advance the field into a more practical direction. The combined use of NO donors with polymeric matrices may not only favour a more controlled administration of the donor, but also regulate NO cellular effects. For example, poly(vinyl alcohol) and poly(vinyl pyrrolidone) polymer solutions reduced the cytotoxicity and enhanced the anti-proliferative effects of GSNO in smooth muscle cell cultures.[56] In this section, recent research on the development of polymer-based NO-releasing and -generating materials without evaluation in vivo are discussed (2006–2011) (Scheme 1). Previous research work on this topic has been reviewed elsewhere.[10,57-64]
Scheme 1.
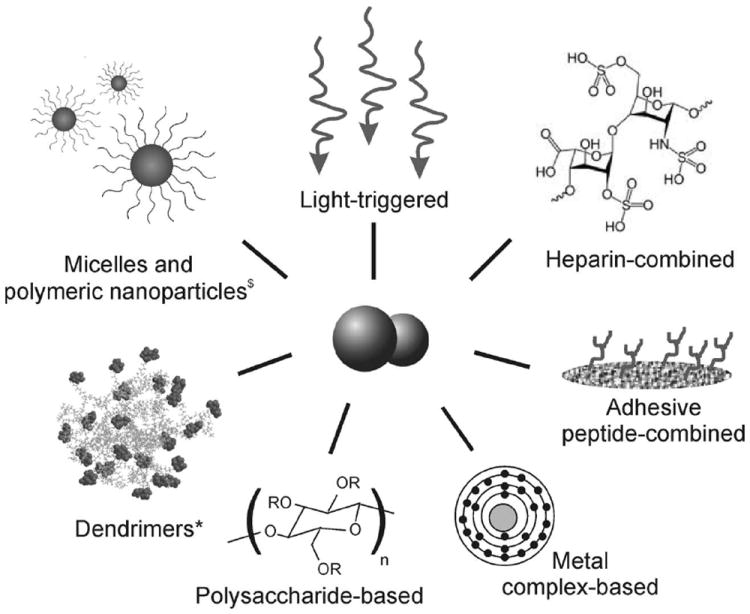
Novel strategies in the design of NO-releasing and generating polymeric materials. $Reproduced with permission.[71] Copyright 2009, American Chemical Society. *Reproduced with permission.[77] Copyright 2010, Elsevier.
4.1. Materials with Enhanced Cell-Adhesive Properties
In an attempt to compensate for the inappropriate functionality and loss of endothelium associated with common cardiovascular diseases such as atherosclerosis, a significant effort has been directed towards the development of materials that mimic endothelial properties and/or promote re-endothelialization. Kushwaha et al. have recently developed native endothelial extracellular matrix-mimicking nanofibrous scaffolds with the ability to release NO.[65] These materials were formed by self-assembling peptide amphiphiles (PA), which contained polylysine sequences as NO-donating residues, endothelial cell-adhesive ligands (YIGSR), and enzyme-degradable sites. In vitro studies demonstrated enhanced EC adhesion and proliferation, as well as reduction of vascular smooth muscle cell (VSMC) proliferation and human platelet adhesion. NO was initially released in a burst (first 48 h), followed by a slow sustained release for over 1 month mediated by diffusion and enzymatic degradation of the matrix. Cumulative NO release values of 0.32 μmol from coated metal stents (surface area 0.396 cm2) were estimated for one-month release. This design was also applied to matrices composed of electrospun polycaprolactone nanofibers (ePCL) coated with self-assembled PA.[66] A total of 3.8 μmol NO was released over 4 weeks from a 16 mm-diameter disc, comparable to the cumulative NO released by endothelial cells at a rate of 10−10 mol cm−2 min−1 reported elsewhere (4.03 μmol).[67] In a similar strategy, Taite et al. incorporated YIGSR ligands into the backbone of a polyurethane-PEG copolymer with lysine-containing peptides as diazeniumdiolate NO donors.[68] NO release studies revealed a biphasic release composed of an initial burst release (70%) followed by a sustained release for over 2 months (~0.13 μmol month−1). Similar to results by Andukuri et al., these materials supported EC adhesion and proliferation; whereas a significant reduction in platelet adhesion and VSMC adhesion and proliferation were found in vitro. The elastomeric mechanical properties of these PU-based materials added an important value for their application in engineering soft tissues.
4.2. Materials Containing Heparin for Enhanced Anti-Thrombogenic Properties
Heparin is commonly used as an anticoagulant due to its non-thrombogenic properties, thus becoming an interesting tool to enhance the inherent anti-thrombogenic properties of NO-releasing materials. Zhou and Meyerhoff prepared polymeric coatings combining both NO release and surface-immobilized active heparin to better mimic the non-thrombogenic properties of the native endothelium.[69] These coatings were designed with a trilayer membrane configuration with a poly(vinyl chloride) or polyurethane coating doped with diazeniumdiolated dibutylhex-anediamine (DBHD/N2O2). Heparin was immobilized into the outer layer via amide bond formation. By modulating the chemical/polymer composition of the NO-releasing layer, the authors fine-tuned NO flux from 0.5 to 60 · 10−10 mol cm−2 min−1 and durations from 24 h to one week. They also demonstrated anti-coagulant properties of the polymer-immobilized heparin by measuring anti-Factor Xa activity. In a different study, this diazeniumdiolate NO donor was combined with surface-bound active thrombomodulin (TM) and heparin to create blood- compatible polymeric coatings for biomedical devices.[70] The active TM and heparin molecules were covalently bound to a CarboSil (a copolymer of silicone and polyurethane) layer by amide bond formation. NO release for up to two weeks was detected.
4.3. Micelle and Microbubble-Based Strategies
Hubbell and co-workers recently hypothesized that the creation of a hydrophobic microenvironment within a micelle core could protect a diazeniumdiolated NO-donor from proton-catalyzed NO liberation (Figure 1).[71] By making use of self-assembling amphiphiles, they designed a diblock copolymer in a nano-particulate structure for long-term NO release. This novel concept relies on the ability of the hydrophilic precursor, poly(N-acryloyl-2,5-dimethylpiperazine) (PAZd), to be converted into a hydrophobic molecule after diazeniumdiolation of the secondary amine groups (PAZd·NONOate). This chemical reaction leads to aggregation and subsequent micellization of the block copolymer that is reverted after NO release. The half-life of these NO-releasing micelles was seven days and NO flux was calculated as 7.9 · 10−13 mol cm−2 min−1. The ability of these NO-loaded micelles to penetrate a rabbit carotid artery ex vivo was also demonstrated, thus opening their use as a therapy for NO delivery in complex tissues.
Figure 1.

Micelle-based strategy for NO delivery. A) PAM-PAZd·NONOate micelles are formed when NO reacts on water-soluble PAZd domains to yield water-insoluble PAZd·NONOate. B) Proton-driven dissociation of diazeniumdiolate groups in order to release NO. Reproduced with permission.[71] Copyright 2009, American Chemical Society.
Kanayama et al. combined the micellization approach, to protect the NO donor, with light-controlled delivery.[72] They used nitrobenzene derivatives as NO donors through a nitro-to-nitrite photo-arrangement followed by the rupture of the O-NO bond when exposed to UV light (330–385 nm). The photomediated NO release was proven to have antitumoral effects on HeLa cell cultures. When exposed to UV-activated NO-releasing micelles, the 50% inhibitory concentration dropped from 4.9 mg mL−1 to 1.9 mg mL−1 and 0.2 mg mL−1 after 3 and 10 min UV exposure, respectively. Unfortunately, the use of UV light may be a potential limitation for the clinical application of these micelles.[73]
4.4. Polysaccharide-Based Strategies
Ethylcellulose, an inert hydrophobic polymer, has been widely used as an interesting tool for controlled drug release, but rarely explored for NO delivery. Wan et al. prepared ethylcellulose membranes doped with either glutaraldehyde-modified glucosamine or diethylenetriamine (DETA/NO) diazeniumdiolates by a monolayer or trilayer membrane configuration.[74] Release rates of 0.1–0.5 · 10−10 (up to 94 days) and 0.2–9 · 10−10 mol cm−2 min−1 (up to 30 days) were obtained, respectively, which are in the range of native endothelial flux. The trilayer structure effectively eliminated the burst release, thus increasing the NO release time. For instance, the release from DETA/NO-doped membranes was significantly extended to 30 days, while the half-life of the pure donor is just ~20 h.[30]
In a more recent study, Meyerhoff and co-workers covalently incorporated methoxymethyl- or sugar-protected diazeniumdiolate groups to chain extender diols in polyurethane backbones.[75] Besides the use of these elastomeric polymers as carriers for NO delivery, this approach allowed for fine control of the surface NO flux by adjusting the number of incorporated diazeniumdiolate groups and offered an alternative to previous NO-releasing polyurethane approaches.[76] Spontaneous hydrolysis of methoxymethyl or carbohydrate moieties under acidic or basic conditions led to NO release from these polymers.
4.5. Dendrimer-Based Strategies
As previously mentioned, NO generation from S-nitrosothiols (RSNO) has also created some interest in the recent years because of its advantage of relying on endogenous components. Johnson et al. recently developed a dendrimer-based system with S-nitroso-N-acetylpenicillamine as NO donor, with release initiated by glutathione (GSH).[77] These novel NO-generating scaffolds successfully minimized ischemia/reperfusion injury in an ex vivo rat heart model with a significant reduction of the optimal therapeutic dose of NO donor under physiological glutathione concentrations.
In a different strategy, Stasko and Schoenfisch described the use of dendrimers made of polypropylenimine for NO release.[78] They compared the effectiveness of primary amine, secondary amine and amide functionalities to store NO as diazeniumdiolates. Secondary amine dendrimers exhibited the higher storage capacity with cumulative NO release values up to 5.6 μmol mg−1 and durations of at least 16 h. The authors further expanded this work by creating S-nitrosothiol-modified dendrimers. These dendrimers, modified with either N-acetyl-d,l-penicillamine or N-acetyl-l-cysteine, were capable of storing ~2.0 μmol NO mg−1. In these studies, S-nitrosothiol-modified dendrimers resulted in a higher reduction in platelet aggregation compared to NO-donor molecules alone.[79]
4.6. Metal Ion-Based Strategies
As metal ions mediate NO release from RSNO, some attempts have focused on the incorporation of these elements on polymeric materials to generate NO from endogenous RSNO present in blood. In this sense, Meyerhoff and co-workers developed polymers with copper-based catalysts immobilized within the polymer network.[50,51] Tecophilic hydrophilic polyurethane and polymethacrylate polymers with covalently linked CuII-cyclen complexes demonstrated to spontaneously generate NO from fresh sheep blood. A steady-state NO flux equivalent to physiological endothelium-derived flux (i.e., 10−10 mol cm−2 min−1) was obtained when the polymer was incubated with saline solutions containing GSNO/GSH or CysSNO/CySH. Cyclen’s square-pyramidal structure and high affinity with CuII, as well as the hydrophilicity of the methacrylate-based polymer, were crucial to facilitate the interactions with RSNO molecules and endogenous reducing agents. These authors also prepared polymeric substrates (e.g., polyurethane catheters and silicon rubber tubings) with the ability to decompose RSNO in the presence of reducing agents.[80] The surfaces were fabricated by the incorporation of organoselenium-modified polyethyleneimine and sodium alginate using a layer-by-layer deposition technique and tested with sheep whole blood. In another approach with the same objective, Meyerhoff and Hwang fabricated organotelluride-tethered polymers.[81] These NO-generating materials contained allylamine hydrochloride to obtain a water soluble polymer capable of crosslinking with a cellulose membrane. In a more recent approach, Puiu et al. derivatized medical polyurethane (Pellethane and Tecophilic) with CuII-cyclen moieties by using a simpler three-step synthetic method.[82] They prepared materials that released NO at levels that exceeded those of endothelial cells by using a methodology feasible for aliphatic- and aromatic-based polyurethanes.[83]
4.7. Light-Triggered Strategies
In the past decades, the research on the design of smart materials has placed a significant emphasis on light as a clean controller stimulus for the design of stimuli-responsive materials. Mascharak and co-workers prepared novel NO delivery systems based on photoactive metal-nitrosyl groups created by reductive nitrosylation of manganese and ruthenium complexes.[84-88] These systems release NO upon illumination with near-infrared light or visible light. In the particular case of Mn[(PaPy3)(NO)] ClO4,[84] illumination with visible light for 30 s resulted in a peak NO concentration of 1.5 μm, while 60 s of illumination resulted in 5 μm NO peak. Continuous illumination for 5 min gave a relatively sustained NO release of approximately 4.5 μm. Studies in vitro revealed effective growth reduction of the bacteria strains Pseudomonas aeruginosa, Escherichia coli, Staphylococcus aureus, and methicillin-resistant Staphylococcus aureus. Moreover, these light-activated manganese-based compounds retained their bactericidal properties when incorporated into methylacrylate-based polymer hydrogels.[89] In a different study, da Silva et al. developed photo-dependent NO-releasing PEG hydrogels containing nitro-ruthenium complexes (e.g., cis-[Ru(NO2)(bpy)2(4-pic)]+ or cis-[Ru(bpy)2(4-pic)(H2O)]2+) as NO donors.[90-93] They used hydroxyethyl cellulose (HEC), polyacrylate, and chitosan to form non-ionic, anionic, and cationic polymeric hydrogels, respectively. The authors attributed NO release from these complexes to reduction of coordinated nitrite by excited nitro-ruthenium complexes after UV–vis irradiation. NO values of 3 nmol cm−2 were estimated to be released in a few minutes. The utility of the HEC hydrogel as a topical skin treatment was investigated in vitro.[91] Nitro-ruthenium complexes penetrated the stratum corneum layer of dermatomed porcine ear skin and released NO after light stimulation. For more details about light-controlled NO dispenser systems including ruthenium and other than polymers, readers are referred elsewhere.[16,94]
4.8. Other Polymeric Approaches
De Oliveira and co-workers synthesized blends of a polynitrosated polyester and poly(methyl methacrylate) for NO release through the incorporation of RSNO covalently attached to the polymer backbone.[95] They obtained NO fluxes of 180 nmol g−1 h−1 when immersed in aqueous solution and demonstrated inhibition of platelet adhesion in vitro. Coneski et al. have synthesized a new group of polyesters based on the condensation of polyols (e.g., glycerol and pentaerythritol) and diacids (e.g., glutaric acid and adipic acid).[96] They functionalized the polymer network for NO delivery by covalent linkage of NO donors (e.g., cysteamine and penicillamine) through NHS/EDC chemistry and subsequent nitrosation through their thiol groups. NO values between 0.01–0.81 μmol cm−2 for up to six days were detected. These biodegradable materials demonstrated tunable thermal and degradation properties and utility as antibacterial materials by reducing up to 80% adhesion of Pseudomonas aeruginosa. When evaluated in L929 fibroblast cultures, cell viability was above 65% for most of the materials tested. The cytotoxic effects detected were mainly attributed to degradation products leached out from the polymer network. Coneski and Schoenfisch also used thiol groups to develop NO-releasing polyurethanes with NO release values up to 0.20 μmol cm−2.[97]
Pasut et al. developed polymer-drug conjugates containing PEG as carriers for a combined therapy of NO and epirubicin, an antitumoral drug.[98] In vitro studies demonstrated selective cytotoxicity against tumoral Caco-2 cells. PEG-based polymers were also used by Kumar et al. to protect nanoparticles containing diazeniumdiolate-based NO pro-drugs and anticancer leads PABA/NO (O2-{2,4-dinitro-5-[4-(N-methylamine)benzoyloxy]phenyl}1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate) and “Double JS-K” (1,5-bis-{1-[(4-ethoxycarbonyl)piperazin-1-yl] diaze-1-ium-1,2-diol-2-ato}-2,4-dinitrobenzene).[99] This approach allowed for the stabilization of the donor against glutathione, one of the main deactivating agents when applied in vivo, and preserved the anti-proliferative activity of the drugs as confirmed with leukemia cell cultures. When compared to non-protected drugs, 50% decomposition was extended from 15 min to 5 h and from 4.5 min to 40 min for protected PABA/NO and “Double JS-K”, respectively. In a different study, Wang and coworkers also used dinitrobenzene in a reaction with different aliphatic diamines for the synthesis of NO-releasing nitrosated polymers.[100] These materials demonstrated an NO concentration-dependent cell survival on neuron-like PC-12 cells.
4.9. Zeolite-Containing Polymeric Materials
In recent years, gas storage in systems such as carbon materials, polymers, and porous materials is attracting much interest for a wide range of applications including energy, environment, and medicine. Particularly, the high-capacity gas storage properties of zeolites have attracted attention for NO delivery.[101,102] Fox et al. developed a bifunctional NO-storing material containing Zn2+-exchanged zeolites (50 wt%) and polytetrafluoroethylene (50 wt%). NO release peaked at 25 μm in the first 10 min and was almost complete after 50 min.[101] The anti-infective properties of these materials were demonstrated in vitro with Gram positive, Gram negative, and certain antibiotic-resistant bacteria strains. In a different approach, Liu and Balkus used electrospun polylactic acid fibers to encapsulate zeolites and effectively release NO following a slower kinetic than plain zeolites.[103]
5. Progress in the in vivo Application of NO-Releasing and NO-Generating Polymeric Materials
5.1. Cardiovascular Applications
Cardiovascular diseases, specifically atherosclerosis, continue to be a major cause of mortality and morbidity in the United States, with coronary disease alone accounting for one out of every six deaths.[104] As a result of atherosclerotic disease, the number of surgical procedures for coronary bypass and percutaneous coronary interventions (PCI) has risen to over one million annually (app. 400 000 and 600 000 procedures, respectively, in 2007).[104] However, despite the advances in surgical techniques and improved pharmaceutical regimens, long-term outcomes are complicated by restenosis, secondary to the development of neointimal hyperplasia (NIH).[105] NIH is characterized by a complex cascade of events which involves platelet adhesion and aggregation, leukocyte chemotaxis, and vascular smooth muscle proliferation and migration. Likewise, problems of platelet adhesion and aggregation leading to thrombosis are a major issue for approximately 340 000 patients with end-stage renal disease, in whom the need for long-term vascular access, often through ePTFE grafts, poses significant risks. Indeed, up to 80% of vascular access dysfunction is caused by graft thrombosis, often accompanied by NIH. Apart from the above-mentioned conditions, thrombosis is also an important risk-factor in all short-term blood exposure to synthetic materials, for example hemodialysis membranes, catheters or intravascular sensors, and devices for extracorporeal circulation (ECC).
NO released by vascular EC has been shown to be a potent anti-thrombotic and anti-NIH agent by inhibiting platelet adhesion/activation and leukocyte chemotaxis, as well as SMC proliferation and migration.[106] Therefore the loss (caused by vascular injury during PCI/bypass) or lack (in artificial grafts/intravascular devices) of endogenous NO is believed to be the main contributor to thrombosis and NIH. This has led to an exponential increase in research on mimicking endothelial NO-release as a therapeutic way to alleviate both conditions after vessel injury and vascular graft implantation.
5.1.1. Inhibition of Thrombus Formation
Antithrombogenic properties have been pursued for blood-contacting medical devices by diverse strategies including inhibition of protein and cell adhesion (e.g., surface coatings with albumin, phosphatidylcholine, pyrolytic carbon, poly(ethylene oxide), and elastin), inhibition of thrombin and fibrin formation (e.g., heparin, thrombin inhibitors, and thrombomodulin) and inhibition of platelet adhesion and activation (e.g., clopidogrel, prostaglandins, and prostacyclin), among others.[61] Limitations such as long-term effectiveness, high-cost of prolonged treatments and side effects derived from systemic administration associated with most of these strategies could be avoided by using efficient NO-based therapies. For instance, Annich et al. suggested an NO-releasing extracorporeal circulation circuit as an alternative to heparin, which elicits abnormal platelet activation, in order to prevent thrombogenesis in a rabbit model.[107] The circuit was coated with a mixture of NO donor, (Z)-1-{N-methyl-N-[6-(N-methylammoniohexyl)amino]}diazen-1-ium-1,2-diolate (MAHMA/N2O2) (16.6 mg) (Figure 2a), and polyvinyl chloride (PVC), with a top PVC coating. NO release was linear and peaked at about 20–25 μmol at day one. NO release was not detected beyond 24 h, thus limiting the applicability of this approach to short-term applications. These coated circuits significantly reduced platelet adhesion/activation and did not exhibit gross thrombus formation compared to controls. Unfortunately, NO donors leached from the polymer coatings into blood circulation raising concerns of possible formation of toxic nitrosamines.[107-109]
Figure 2.

a) (Z)-1-{N-methyl-N-[6-(N-methylammoniohexyl)amino]diazen-1-ium-1,2-diolate (MAHMA/N2O2); b) diazeniumdiolated diamine cross-linked silicone rubber (DACA-6/N2O2-SR). Note the similarity in the NO donor to MAHMA/N2O2. However upon NO release, the diamine remains in the polymer backbone; c) dibutylhexanediamine diazeniumdiolate (DBHD/N2O2). Note the similarity in the NO donor to MAHMA/N2O2. However the methyl group substituents are replaced with butyl groups making DBHD/N2O2 more lipophilic. Reproduced and adapted with permission.[115] Copyright 2003, Elsevier.
To prevent toxicity issues related to nitrosamines, NO donors have been covalently linked to the polymer backbone to avoid leaching.[36-41] For instance, Meyerhoff and co-workers combined the NO donor N-(6-aminohexyl)-3-aminopropyl-trimethoxysilane (DACA-6) with hydroxyl-terminated polydimethylsiloxane to create NO-releasing silicone rubber films (Figure 2b).[40] NO release increased when measured at higher temperatures and could be tailored by modifying film thickness and percentage of DACA-6 loading. Release for up to 20 days was measured from coated silicone rubber tubings (coating thickness 600 μm, DACA-6 15 wt%). When used in a rabbit ECC model, these tubings did not clot while tubings with untreated coatings clotted within 3 h. Platelet activation was apparent only for uncoated tubings and untreated-coated tubings. As hydrophilic diazeniumdiolates are capable of leaching from the polymer matrix and forming toxic nitrosamines, a different strategy to prevent these concerns is the use of lipophilic diazeniumdiolates that can remain in hydrophobic polymers such as PVC. By using an analogue of the previously reported MAHMA/N2O2, Meyerhoff and co-workers developed a lipophilic NO donor, DBHD/N2O2 (Figure 2c), that was incorporated into polymer coatings for the reduction of thrombus formation.[35,110] Commercially available small-diameter polyurethane vascular grafts were coated with DBHD/N2O2 (10 wt%) and mixed into PVC/plasticized dioctyl sebacate (DOS) matrix with potassium tetrakis-4-chlorophenyl borate (KTpClPB) (20 wt%).[35,62,110] The lipophilic salt, KTp-ClPB, buffered the pH and thus increased and prolonged NO release. The authors implanted these grafts in a sheep model bypassing the common carotid artery to the external jugular vein.[110] The thrombus-free surface area significantly increased for NO-releasing polymer grafts (98.2%) compared to polymer-only grafts (79.2%) and uncoated grafts (47.2%). Although the improvement in patency between NO-releasing grafts and control grafts was not statistically significant, control grafts contained significant adherent thrombus and fibrin matrix with inflammatory and red blood cells, an observation not found in NO-releasing grafts (Figure 3a).
Figure 3.

Left: Hematoxylin and eosin stain of sections from uncoated graft (top) and NO-releasing graft (bottom). Note the adherent thrombus and red blood cell infiltration (black arrows) for the uncoated grafts. Reproduced and adapted with permission.[110] Copyright 2004, Elsevier. Center: Scanning electron microscopy images of platelet adherence and activation on DACA-6/N2O2-SR-coated sensors and control sensors. Control sensors exhibited varying extent of platelet adhesion and activation, while the NO-releasing sensors had very little platelet adhesion. Reproduced and adapted with permission.[114] Copyright 2002, American Chemical Society. Right: Images of PVC rods coated with (a) KTpClPB (20 wt%) and without lipophilic NO donor precursor and (b) KTpClPB (20 wt%) with lipophilic precursor (5 wt%). Portion of the rods to the left of dotted lines were exposed to blood flow. Reproduced with permission.[118] Copyright 2008, American Chemical Society.
To estimate the critical flux necessary to preserve platelet count and function and to prevent platelet activation, Annich and co-workers used a polymer coating called Norel-b, which consisted of DBHD/N2O2, PVC/DOS, and KTpClPB.[111,112] Norel-b coating followed by a top coating of PVC/DOS was applied to an ECC circuit of a rabbit arteriovenous shunt model. Compared to basal endothelial NO flux (0.5 · 10−10 mol cm−2 min−1) and bradykinin-stimulated endothelial NO flux (1.6–4.1 · 10−10 mol cm−2 min−1),[67,113] Norel-b coating of 2, 10, 25, and 50 wt% of DBHD/N2O2 had NO fluxes of 2.3, 13.7, 20.6, and 39.1 · 10−10 mol cm−2 min−1, respectively.[112] All Norel-b coatings except 2 wt% were able to retain activated clotting time within baseline and preserve platelet count over the course of 4 h. SEM images showed a significant amount of platelet adhesion and activation for control groups while few platelets adhered to Norel-b coatings. Furthermore, platelet function was preserved for all Norel-b coatings compared to control groups as measured by collagen-induced aggregometry. Based on these results, the critical flux was estimated to be greater than 13.7 · 10−10 mol cm−2 min−1. Annich and co-workers further correlated the inactivation of platelets and monocytes in Norel-b coatings with the attenuation in the expression of P-selectin, a surface glycoprotein indicative of platelet activation, and CD11b, a monocyte surface integrin.[111] Thrombus area after 4 h of blood exposure was significantly reduced in Norel-b coatings (2.8 pixels cm−2) compared to controls (6.7 pixels cm−2).
Because blood gas sensors also encounter complications from platelet adhesion and thrombus formation, Meyerhoff and coworkers used either MAHMA/N2O2 (2 wt%) in a silicone rubber/DOS solution,[109] DACA-6/N2O2-silicone rubber (~100 μm) with a final top-coating of silicone rubber,[114] or DBHD/N2O2 (5% wt/wt) and KTpClPB[115] to coat silicone rubber sensor sleeves of amperometric oxygen sensors. The sensors were implanted in canine carotid and femoral arteries for up to 24 h[109] or in porcine carotid and femoral arteries for up to 16 h.[114,115] The authors reported improved blood compatibility (reduced thrombus formation and platelet adhesion/activation on the surface) and analytical performance of the coated oxygen sensors (Figure 3b). For MAHMA/N2O2-silicone rubber/DOS (7 dip-coatings), NO release was extended beyond 20 h with a cumulative NO release above 0.4 μmol. Sensors coated with DACA-6/N2O2-silicone rubber had an initial flux of ~2.5 · 10−10 mol cm−2 min−1 which decreased to ~1 · 10−10 mol cm−2 min−1 over the course of 20 h. In a separate study, Gifford et al. incorporated DBHD/N2O2 and KTpClPB (2%, 1:1 mol ratio) into polyurethane/polydimethylsiloxane as coatings (~10 μm in thickness) for glucose sensors.[116] NO release lasted approximately 16 h with an average peak NO flux of 4.5 · 10−10 mol cm−2 min−1. When implanted subcutaneously in rats, these NO-releasing sensors had significantly less neutrophil infiltration at day 1, but were not significantly different at day 2 compared to control sensors. Since increasing coating may adversely affect sensor performance, Wu et al. sought to overcome this limitation by using Cu0 catalyst to decompose endogenous RSNO and generate unlimited NO in situ. The authors incorporated Cu0 particles into silicone rubber/polyurethane (2:1) or polyurethane coatings (3 μm, 3 w/v% or 80 nm, 1 w/v%, respectively) to improve the hemocompatibility of oxygen sensors.[117] Cu-coated sensors implanted in the porcine carotid and femoral arteries for up to 20 h measured oxygen levels more accurately and had less thrombus formation than control sensors.
Another concern with the development of NO-releasing materials is the high temperatures commonly used for polymer fabrication which may affect NO donor stability. To address this issue, Xu et al. created a different lipophilic NO donor precursor, O2-(2-hydroxy-5-nitro-4-pentadecyl-benzyl)-1-(N,N-dioctylamino)diazen-1-ium-1,2-diolate (HNBOA/N2O2), that incorporated 2-hydroxy-5-nitrobenzyl as a diazeniumdiolate protecting group for thermal stability.[118] The protected lipophilic precursor was stable up to 90 °C, but only up to 60 °C without protection. Medical-grade polyurethane blended with lipophilic NO donor precursor (5 wt%) and KTpClPB (20 wt%) had an NO flux above 1 · 10−10 mol cm−2 min−1 over 20 h and peaked to ~5 · 10−10 mol cm−2 min−1 at 12 h. These blends were used to dip-coated PVC rods that were implanted in swine femoral and carotid arteries for 8 h. NO-releasing rods had no visible surface clots that were seen in control rods (Figure 3c).
5.1.2. Inhibition of Neointimal Hyperplasia (NIH)
Angioplasty with placement of intravascular stents is a common clinical practice for the treatment of atherosclerosis. Unfortunately, a significant percentage of these procedures fail because of restenosis subsequent to NIH. To improve the success of these interventions, drug-eluting stents loaded with anti-proliferative drugs such as sirolimus and paclitaxel have been introduced into clinical practice.[119] Despite the significant benefits of using this new generation of stents, in-stent restenosis and late stent thrombosis remain as important limitations that prevent successful performance in all patients.[120,121] As NO inhibits the main events leading to NIH, it has been widely studied for preventing NIH progression. Unfortunately, hemo-dynamic effects and the short half-life of NO make systemic administration difficult; therefore prompting the study of NO-releasing materials for localized NO delivery as an alternative strategy. Several studies that utilize perivascular or adventitial NO delivery have shown promise for clinical use by inducing local NIH inhibition. When compared to drug-eluting stents, NO-based therapies also benefit from the absence of synthetic anti-proliferative compounds that might affect local and nonlocal cell targets not involved in NIH.
Kaul et al. used Atrigel, a thermo-sensitive gel that solidifies at 37 °C, to locally deliver NO and inhibit NIH by increasing cGMP levels and suppressing the activity of nuclear factor-κB, a transcription factor involved in inflammatory pathways.[122] They mixed Atrigel with spermine/NO (SPER/NO) (2.5% wt/wt), an NO-releasing diazeniumdiolate, and added magnesium hydroxide (2.5% wt/wt) for control of NO release by neutralization of the acid degrading polymer. Gels were applied periadventitially to rat ileofemoral arteries with balloon injury-induced NIH and allowed to solidify. Subjects treated with polymer + SPER/NO had a greater than 75% reduction in VSMC proliferation by day 3. At day 14, arteries treated with polymer + SPER/NO had significantly smaller intimal area and approximately 75% decrease in the intima/media (I/M) ratio.
To gain insight as to whether a short or long NO donor half-life was superior in preventing NIH, Kibbe and co-workers examined two NO donors, diazeniumdiolated poly(acrylonitrile) (PAN/NO) and 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (PROLI/NO) in a rat carotid artery model.[123] PROLI/NO was previously reported to have a very short half-life of 1.8 s,[124] while PAN/NO has a duration of NO release greater than 80 days.[125] The authors delivered PAN/NO and PROLI/NO perivascularly either alone in powder form (20 mg) or mixed in a gel form (5 wt%) with poloxamer 407 (30 wt%), a thermo-sensitive gel that solidifies at 37 °C.[123] Although all treatments significantly inhibited NIH at day 14 compared to injury alone, PROLI/NO powder resulted in the greatest reduction in intimal area (91%) and I/M ratio (86%). The combined gel therapy of PAN/NO and PROLI/NO performed the second best followed by PROLI/NO gel. All NO-based treatments exhibited antiproliferative effects, but only the powder forms inhibited monocyte/macrophage infiltration. As treatments with poloxamer 407 induced vascular inflammation, Kibbe and co-workers examined nanofiber gels composed of peptide amphiphile and heparin to deliver half the dose of PROLI/NO (10 mg) or 1-[N-(3-aminopropyl)-N-(3-ammoniopropyl]diazen-1-ium-1,2-diolate (DPTA/NO) (10 mg).[126] The nitrite release for PROLI/NO and DPTA/NO nanofiber gels peaked at day 2 (0.6 μmol day−1 and 1.2 μmol day−1, respectively), decreased at day 3, and was not different from control by day 5. Although only half the dose was used, PROLI/NO nanofiber gel treatment had a similar reduction in intimal area and I/M ratio (80% and 77%, respectively) as the PROLI/NO poloxamer gel. PROLI/NO nanofiber gels also inhibited inflammation and adventitial cell proliferation and induced minimal apoptosis in vivo. Despite greater NO release, DPTA/NO nanofiber gel exhibited less inhibition of NIH (45% reduction in I/M ratio) (Figure 4) and did not inhibit inflammation. Both PROLI/NO and DPTA/NO nanofiber gels promoted re-endothelialization following arterial injury. Taken together, these results suggest that longer and greater NO release do not necessarily induce a more significant NIH inhibition.
Figure 4.

A) Hematoxylin and eosin (H&E) and Verhoff-van Gieson (VvG) stains of sections from rat carotid artery. NIH = neointimal hyperplasia. B) I/M area ratios of IA = injury alone, NG = nanofiber gel, PNG = PROLI/NO nanofiber gel, and DNG = DPTA/NO nanofiber gel. Reproduced with permission.[126] Copyright 2008, Elsevier.
Using the same animal injury model but a different NO donor, Lipke and West created a PEG-based hydrogel carrying S-nitrosocysteine (Cys/NO) by covalently binding cysteine to PEG-N-hydroxy-succinimide monoacrylate.[37] These hydrogels reduced platelet adhesion, inhibited VSMC proliferation, and promoted endothelial proliferation in vitro.[37,127] Following in vivo rat arterial injury, PEG-Cys/NO hydrogel precursor (1.25 μmol NO) were mixed with a visible light photoinitiator, applied at the adventitial surface, and polymerized under visible light to form a hydrogel in situ. Compared to control hydrogels, vessels treated with PEG-Cys/NO hydrogels had an 80% decrease in intimal thickness, 77% reduction in I/M ratio, and improved re-endothelialization 14 days post-injury.[37] Furthermore, the authors reported a significant reduction in proliferating medial cells (29%) four days post-injury compared to control hydrogels (51%). In a similar study, West and colleagues mixed PEG-Cys/NO hydrogel precursor with a UV initiator, which led to a faster in situ photocrosslinking.[128] When assessed in vivo, the intimal thickness decreased by 75% at day 14.
In our laboratory, we have recently developed NO-releasing poly(diol-co-citrate) elastomers that incorporate citric acid, aliphatic diols, and N,N’-bis(2-hydroxyethyl)ethylenediamine (diamine diol) as a diazeniumdiolate precursor.[39,41] Varying the length of the aliphatic diol (e.g., 1,8-octanediol or 1,12-dodecanediol) modified the NO release kinetics. As such, poly(1,8-octanediol-co-citrate) containing diamine diol (5 mol%) (POCDA10-NO) had a fast burst release under two days compared to the extended release of up to 21 days for poly(1,12-dodecanediol-co-citrate) containing diamine diol (5 mol%) (PDDCDA10-NO). POCDA10-NO showed a greater total NO release of 10.5 · 10−6 mol cm−2 and a maximum flux of 35.4 · 10−9 mol cm−2 min−1 in comparison to PDDCDA10-NO (4.2 · 10−6 mol cm−2 total release and a maximum flux of 6.6 · 10−9 mol cm−2 min−1). Both polymers supported EC and VSMC adhesion and growth in vitro. However, cellular viability decreased with exposure to increasing NO doses. Since PDDCDA10-NO showed a more extended NO release, films of PDDCDA10-NO (<0.2 mm in thickness) were applied perivascularly in rat carotid arteries after balloon-induced injury (Figure 5). Compared to injury alone, PDDCDA10-NO significantly reduced intimal area and I/M ratio by 45% and 38%, respectively, at 14 days.
Figure 5.

A) Drawing of the common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA), with blood flowing from the CCA to the ICA and ECA (first panel). The drawing depicts how the wraps were placed around the artery and secured with a tie. Also shown are photographs of the actual wraps that were placed and secured around the CCA at the time of surgery (second and third panels). Note the close proximity of the wrap to the adventitial surface of the artery. The last panel shows an intact wrap at harvest, two weeks following arterial injury and placement of the wrap. B) Representative histological haematoxylin and eosin stained sections of the injured carotid arteries with the different treatment groups 14 d after arterial injury (400×). Reproduced and adapted with permission.[39]
In a different approach using NO-releasing stents, Do et al. created NO-containing microspheres made of poly(lactic-co-glycolic acid) (PLGA) (75:25) and PEG (8:1) using a double emulsion technique.[129] The NO donor, N-ethyl-2-(1-ethyl-2-hydroxy-2-nitrosohydrazino)ethanamine (NOC-12), was added to the polymer solution during microsphere formation. Micro-spheres were loaded into machined microchannels on the struts of metal stents, and PEG-based gel covered unfilled regions of the stents. NO-microsphere-loaded stents deployed in the infrarenal abdominal aorta of rabbits induced significantly higher cGMP levels and lower number of proliferating VSMC than control stents loaded with blank microspheres. NO-releasing stents significantly reduced I/M ratio by 46% and 32% at day 7 and 28, respectively.
Unfortunately, not all NO-releasing materials were capable of reducing or preventing NIH. Yoon et al. coated metallic stents with polyurethane and an NO donor, sodium nitroprusside (SNP) (2 mg) (3:2 ratio).[130] A thin (0.7 mg) or thick (2.2 mg) polyurethane barrier layer was applied over the SNP/polyurethane coating. SNP eluted from the thin and thick polyurethane-coated stents for over two and four weeks, respectively, and increased local cGMP levels for up to 14 days. However, when tested in vivo, SNP/polyurethane-coated stents deployed in porcine coronary arteries demonstrated no statistical difference in inhibiting NIH at 28 days compared to controls. Similarly, Buergler et al. examined stents coated with polycaprolactone impregnated with DETA/NO (1 mg).[131] The stents deployed in porcine left anterior descending and right coronary arteries also did not exhibit significant decrease in NIH. Possible reasons include inadequate NO dose as well as poor choice of polymer or stent design and mode of delivery. Furthermore, Yoon et al. noted that stent deployment that penetrated to the adventitia resulted in an intense inflammatory response regardless of coating, while deployment with less arterial injury showed minimal or no inflammation.[130] In a different study, Abbasi et al. applied DETA/NO (1 g) in a silastic elastomer gel around intima-injured aortas of hypercholesterolemic rabbits.[132] Despite a mean difference in vascular injured surface area at 6 weeks (6.7 · 105 μm2 and 3.4 · 105 μm2 for control and DETA/NO groups, respectively), the results were not statistically significant.
5.1.3. Prevention of Cerebral Vasospasms
Subarachnoid hemorrhage is the cause for approximately 10% of stroke cases annually.[133,134] With a high morbidity and mortality rate, delayed cerebral vasospasms occur within days of a subarachnoid hemorrhage. Evidence links the reduction in NO in the early phases after subarachnoid hemorrhage to delayed cerebral vasospasms caused by smooth muscle cell constriction and platelet and leukocyte adhesion to the endothelium. Several studies suggest that the application of exogenous NO has the potential to reverse delayed cerebral vasospasms.
Tamargo and co-workers loaded a ethylene/vinyl acetate copolymer (EVAc) (5 mg) with DETA/NO (20% wt/wt).[135] In vitro pharmacokinetics revealed that 15% of the initial DETA/NO loading amount was released in nine days with a burst release of 7.5% in the first 10 h. To simulate a subarachnoid hemorrhage and induce vasospasms in a rat model, autologous blood clots were applied in the periadventitial space near the femoral arteries. EVAc with or without DETA/NO was applied periadventially one, three, and seven days after the induction of vasospasms. The percentage of lumen patency for arteries treated with DETA/NO-EVAc was significantly higher for all time points than arteries treated only with EVAc (1 day: 94.6% versus 67.6%, three days: 104.6% versus 64.9%, and seven days: 102.4% versus 73.6%, respectively). To further evaluate these materials, the authors conducted a dose-dependent toxicity study with DETA/NO-EVAc implanted into the brain parenchyma of rats.[136] The lethal dose with 20% mortality (LD20) was estimated to be 3.4 mg kg−1. Sudden deaths were caused by intraparenchymal and incisional hemorrhages and occurred by day two post-surgery. In the remaining rats, histological assessments of the implant sites indicated dose-dependent hemorrhage and ischemia at six weeks, with higher doses of DETA/NO causing the more adverse effects. Hemosiderin-laden macrophages were present from 0 through 11.5 mg kg−1 DETA/NO implants but more abundant at the highest dose (13 mg kg−1). Furthermore, Tierney and co-workers examined whether a much lower dose of DETA/NO (0.48 mg kg−1) delivered in EVAc (5 mg; 20% wt/wt) was capable of preventing cerebral vasospasms in a rabbit basilar artery model.[136] To simulate sub-arachnoid hemorrhage and induce vasospasms, they injected autologous non-heparinized blood into the cisterna magna. After 30 min, DETA/NO-EVAc or EVAc only were implanted into the subarachnoid space. At three days post-surgery, rabbits that received DETA/NO-EVAc, EVAc only, or no treatment had basilar artery lumen patency of 93%, 73%, and 71%, respectively, compared to 100% in animals that received the sham operation but did not undergo blood injection.
Because of the hours to days in delay for patients to seek medical attention and surgical intervention after subarachnoid hemorrhage, Tamargo and coworkers examined whether a 24- and 48-hour delay in NO delivery may still prevent cerebral vasospasms in rabbits.[137] Using the method discussed above to induce subarachnoid hemorrhage, DETA/NO at doses of either 0.5 or 1.3 mg kg−1 in EVAc (20% wt/wt) were implanted 24 and 48 h after subarachnoid hemorrhage. A higher dose of DETA/NO (1.3 mg kg−1) significantly increased the basilar artery lumen patency to 97% and 94% for treatments at 24 and 48 h after subarachnoid hemorrhage, respectively. However, treatments with a lower dose of DETA/NO (0.5 mg kg−1) were statistically different from EVAc only at 48 h after subarachnoid hemorrhage (82% versus 68%). The lumen patency for control animals after subarachnoid hemorrhage was 67%.
As most patients who possess the haptoglobin 2-2 genotype are more genetically inclined to develop severe cerebral vasospasms, Tamargo and co-workers implanted DETA/NO-EVAc (0.3 mg; 30% wt/wt) into the subarachnoid space of haptoglobin 2-2 mice upon autologous blood injection.[138] In vitro pharmacokinetics revealed that 6.3% of the initial DETA/NO loading amount was rapidly released in the first 5 min and another 9.3% was slowly released over the course of 36 h. After one day, mice with DETA/NO-EVAc treatment, compared to EVAc-only, had a significant increase in basilar artery lumen patency (96.5% versus 73.3%, respectively), improvement in activity level, and reduction in leukocyte infiltration. In a separate study, the authors extended this work to non-human primates.[139] Subarachnoid hemorrhage was induced by placing autologous blood around the supraclinoid internal carotid, and proximal anterior and middle cerebral arteries. Ten minutes after autologous blood was allowed to clot, five animals received DETA/NO-EVAc (4.3 mg kg−1 dose of DETA/NO; 20% wt/wt) and five animals received EVAc only in the clot-filled space. From the four animals that survived by day seven in the DETA/NO-EVAc group, the angiographic and histological data suggested significant prevention of cerebral vessel narrowing. The only early death in this group was likely produced from hemorrhagic necrosis of the cortical tissue. However, despite the significant decrease in vasospasm, remaining animals treated with DETA/NO-EVAc showed signs of decreased activity for several days, with two out of four showing lack of muscle coordination. No animals in the EVAc-only group had any signs of abnormalities. These physiological abnormalities suggest that the DETA/NO dose used was over the toxic threshold. The maximum tolerable dose of DETA/NO in cynomolgus monkeys was later found to be 1.0 mg kg−1.[140]
5.1.4. Improvement of Blood Perfusion
Because hemoglobin scavenges NO and induces vasoconstriction, Cabrales et al. delivered NO-releasing nanoparticles (NO-np) made of hydrogel/glass composites of tetramethylorthosilicate, PEG, chitosan, glucose, and sodium nitrite to reverse the hemoglobin-induced vasoconstriction and decrease blood pressure.[141] In these particles, nitrite was reduced to NO by electrons generated from glucose. NO-np (1 mg) released approximately 0.32 μmol of NO, had a half-life of 4 h and remained in circulation for up to 6 h.[142] Using hamsters with a dorsal chamber window, infusion of NO-np (10 and 20 mg kg−1) decreased leukocyte immobilization and rolling over 2 h. In a different experiment, hamsters were first infused with polymerized bovine hemoglobin followed by NO-np. Plasma nitrite and nitrate concentrations were higher in animals treated with NO-np. In these animals, mean arterial pressure also decreased compared to control-np and saline only. Furthermore, NO-np treatment restored functional capillary density, arteriolar diameter, and blood flow.
5.2. Female Sexual Dysfunction (FSD)
According to a study conducted by the National Health and Social Life Survey on adult sexual behavior in the United States, the prevalence of sexual dysfunction in women is 43%.[143] A major disorder of FSD is arousal problems, which normally involves vasocongestion or increase in blood flow in the pelvis and swelling of the external genitalia. Current treatment options revolve mainly around altering the hormonal balance through replacement therapy. While hormones such as estrogen and prolactin are under investigation, a testosterone-patch is the only available FDA-approved treatment.[144] At the same time, there has been an increased interest in formulations to increase blood flow locally, such as sildenafil[145] or topical formulations containing l-arginine and alprostadil.[146] As NO is important in regulating the genital blood flow,[147] interest arose in applying the vasodilation properties of NO as a treatment for arousal disorders. One possible approach is to allow NO to diffuse through the vaginal epithelial cells and vasodilate the underlying vaginal blood vessels. In this regard, Yoo et al. delivered GSNO from mucoadhesive polymeric films in a rat vaginal model.[148] To create the NO-releasing films, various amounts of GSNO were added to a polymeric solution of Carbopol, hydroxypropyl methylcellulose (HPMC), and PEG (1.5:1.5:1). Carbopol and HPMC were included for their mucoadhesive properties and dryness relief, while PEG served as a film-forming agent. The NO release from the films under vaginal pH 4.0 was slow for the first hour because of gel-swelling delay and subsequently followed a first-order exponential release kinetic. The first-order rate constant increased with higher loading amounts of GSNO. For instance, for the first 10 h, films with 20 and 30 wt% of GSNO released 50% of NO, while films containing 5 and 10 wt% of GSNO released less than 20% of NO. When inserted into rat vaginal cavities, GSNO-containing films (2 mg of GSNO) did not significantly affect vaginal blood flow until 60 min after application. Vaginal blood flow peaked to 170% of initial at 120 min and was maintained for up to 210 min before returning to baseline (100%). GSNO-containing films with a lower dose (1 mg of GSNO) also significantly increased vaginal blood flow for up to 120 min but were not significantly different from blank films thereafter. Differing significantly from GSNO delivery from films, GSNO-only application had a much faster release and exhibited vaginal blood flow maximum at 4 min, which returned back to baseline within 20 min. As an indication of the potential clinical use of GSNO-containing films, the films remained adhered to isolated porcine vaginal tissue for at least 6 h under a fluid flow rate of 3 ml h−1.
In a more recent study in humans, Souto et al. incorporated GSNO into Pluronic F-127, a triblock copolymer gel of PEG-polypropylene glycol-PEG, and applied it onto the clitoris of sexually active women.[149] Forty subjects were randomly assigned to receive Pluronic F-127 gel (1 mL) with or without GSNO (100 μm). Using Doppler ultrasound, the systolic peak speed, diastolic speed, and resistance rate were measured in the clitoral artery. At 15 min after application (blood flow peak), all parameters measured were statistically higher for all women treated with GSNO gels but not for gels without GSNO.
For a faster increase in vaginal blood flow, Yoo et al. used PLGA microparticles (50:50) to deliver the diazeniumdiolate DETA/NO.[150] Unlike GSNO films, the vaginal blood flow for NO-releasing microparticles (2 mg of DETA/NO) increased immediately after 5 min of application, peaked at 30 min (163%), and was maintained for up to 120 min. On the contrary, the application of the same dose of NO-donor without carrier microparticles increased vaginal blood flow for only 10 min after injection and peaked at 4 min (210%). Compared to hormone replacement treatment, topical application of NO-generating formulations has the advantage of avoiding systemic effects. Moreover, long-term effects of hormone therapy are largely unknown, which raises safety questions. Compared to other vasodilating treatment options, NO may be an interesting and safe alternative.
5.3. Wound Healing
In a study conducted over three years, the cumulative incidence of patients developing foot ulcers amongst diabetic individuals was found to be 5.8%.[151] With a higher mortality than diabetic control group, a significant portion of these patients developed osteomyelitis, a bone infection condition, or underwent lower-extremity amputation. Furthermore, treating diabetic foot ulcers is an economic burden that was estimated to be $28,000 per patient for two years upon diagnosis. Treatment options to improve wound healing vary widely and are based on the exact nature of the wound (i.e., dry, exudative, infected, etc.), but generally involve application of a wound dressing for moisture- and infection-control. Furthermore, many compounds have been investigated over the years for topical formulations to promote vascularization, cell migration, and tissue regeneration in wound lessions, including: growth factors delivery (e.g., PDGF, FGF, and EGF, among others) from polymer carriers, nanoparticles and nanotransporters loaded with opioids, polymer-based vitamin release, nonviral polymeric gene delivery systems, and progenitor cell-based therapies, among others.[14] For further information on topical treatments, see the reviews by White and McIntosh.[152,153] Most complications are related to an impaired wound healing in diabetes, which has been linked to a decrease in NO.[154-156] Although the exact participation of NO in wound healing is still unclear, NO is believed to assist wound repair by re-epithelializiation,[157] increasing collagen synthesis,[155,158,159] and promoting angiogenesis by inducing vascular endothelial growth factor expression.[160,161] As such, many researchers have sought to deliver NO to promote wound healing.[162]
Masters et al. used NO-releasing hydrogels made of poly(vinyl alcohol) (PVA) to treat diabetic mice as an impaired wound healing model.[38] To covalently link the NO donor to the polymer backbone, hydrogels were functionalized with amine groups and treated with NO gas in 1:20 and 1:2 ratios to yield respective theoretical NO concentrations of 0.5 mm and 5 mm. After the addition of an Irgacure initiator, the hydrogels were photocrosslinked using UV light. Close to 80% of the theoretically available NO was released over 48 h when examined at pH 6, as most wound environments are slightly acidic. To evaluate these materials in vivo, diabetic mice wounded dorsally (15 mm in diameter) using surgical scissors were treated with high- or low- dose NO hydrogels (0.5 or 5 mm) or control hydrogel dressings. The time for the wound closure was not different between control and NO treatment. However, at day eight but not further beyond, the control wounds had a significantly smaller wound area compared to NO-treated wounds. Although not statistically significant, granulation tissue thickness was greater for increasing NO concentrations. The mean scar tissue thickness at 29 days for control rats and NO-treated rats (5 mm) were 0.14 and 0.28 mm, respectively. As granulation tissue and scar tissue are important for stable wounds, this study demonstrated that NO-releasing hydrogels improved the quality of the wound tissue.
Using human subjects, de Oliveira and co-workers applied hydrogels (6.8 g) consisting of Synperonic F-127 (23.8 wt%), a triblock copolymer of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide), and GSNO (0.3 mol g−1) or S-nitroso-N-acetylcysteine (SNAC) (0.6 mol g−1) topically on the forearm.[163] They assessed their effects on dermal blood flow and nitrite level for future applications such as wound healing. The first-order decomposition rate constant for SNAC and GSNO were 11 and 3 min−1, respectively. When applied in vivo, SNAC hydrogels induced a 1.2 fold greater maximum blood flow compared to GSNO hydrogels probably caused by their faster NO release. The authors reported an increase in blood flow for both GSNO and SNAC hydrogels that peaked at 30 min and remained elevated throughout the 3 h study. Using dermal microdialysis catheters, the dermal nitrite levels correlated well with dermal blood flow for GSNO hydrogels (maximum dermal nitrite concentration of 28 μm). However, the opposite was observed for SNAC hydrogel treatments, in which nitrite levels were low when blood flow was high and increased when blood flow was decreasing. Such disparity can possibly be explained by NO scavenging by oxyhaemoglobin and serum thiols as a result of the higher blood flow induced by SNAC hydrogels.
Despite the promise of GSNO delivery for wound healing applications, concerns about the induction of nitrosative stress (e.g., nitration of proteins via tyrosine residues) arose. To test this hypothesis, de Oliveira and co-workers further studied the effects of GSNO (0.23 mol g−1 or 0.023 mol g−1) incorporated in Pluronic F-127 hydrogels (26.5 wt%) on the foot sole skins of diabetic rats.[164] High dose of GSNO did not increase the local nitrosotyrosine content on the skin nor blood pressure or heart rate. Furthermore, GSNO gels increased blood flow by two-fold and remained high for over 30 min in both diabetic and healthy rats. This study highlights the therapeutic potential of GSNO hydrogel delivery for local vasodilation. In a separate study, de Oliveira and coworkers also used this GSNO-delivery system to investigate whether application (8 nmol of GSNO) at the early inflammation phase, at the later proliferation phase (granulation tissue formation), or for both phases affected the wound healing process.[165] Hydrogels with or without GSNO were applied to wounds (1 cm2) at the dorsal side of rats daily. At day five and seven, they observed a significant reduction in the lesion area for GSNO-hydrogel treatment for both phases compared to application at either the early or late phases and to control (hydrogel without GSNO) (Figure 6). However, at day 14, the lesion areas were similar for all groups, while significant re-epithelialization was observed only on GSNO-treated groups. Furthermore, GSNO-hydrogel applications for both phases had a significantly lower number of inflammatory cells present at the superficial region of the granulation tissue with organized mature collagen fibers. These results suggested that continual NO availability for both inflammation and proliferation phases is important for improved wound healing.
Figure 6.

Macroscopic images of wounds at 5 (d5) and 14 (d14) days after wounding. GSNO-hydrogel treatment for both phases (GSNOinf+prol) had a significant reduction in lesion area at d5 but was not statistically different at d14. Reproduced and adapted with permission.[165] Copyright 2008, Elsevier.
As strategies described above rely on the use of hydrogels that do not necessarily have the mechanical strength for dermal wound dressings, de Oliveira and coworkers further explored the use of flexible PVA films to release GSNO on the forearm of human subjects. PVA films (~50 mg) absorbed 450 μmol g−1 or 840 μmol g−1 of GSNO with one or five freeze/thawed (F/T) cycles, respectively.[166] Upon one F/T cycle, the crystallinity and physical cross-linking increased and was not different between one and five F/T cycles. However, the GSNO absorption and diffusion coefficient varied with F/T cycle number (one F/T: 2.0 · 10−7 cm2 s−1, five F/T: 5.0 · 10−7 cm2 s−1), possibly because of greater interconnected pores with more cycles. As expected, the peak dermal blood flow was higher with five than one F/T films. Unlike the previous study that absorbed GSNO into a polymer network, Li et al. sought to improve the duration and stability of GSNO by its conjugation to poly(vinyl methyl ether-co-maleic anhydride)/poly(vinyl pyrrolidone) (PVMMA/PVP) (1:1, 6.3 wt%).[36] Under ambient light conditions and at 22 °C, NO release was extended up to 10 days. Compared to wounds treated with PVMMA/PVP, wound sizes were statistically smaller in wounds treated with GSNO-PVMMA/PVP at 4, 7, and 10 days but not further beyond (13 and 16 days). Such results could be attributed to the absence of NO release beyond 10 days, as suggested by in vitro release studies.
Finally, NO-releasing polymeric materials have also been tested on burn wounds. Particularly, Murad and co-workers applied an NO-releasing gel to dorsal burn wounds in rats.[167] Before topical application, cellulose-derived gel containing sodium nitrite (14.6 mm) was mixed with an acidic gel of maleic (14.6 mm) and ascorbic (14.6 mm) acids. NO-releasing gels promoted re-epithelialization, angiogenesis, collagen production, and faster wound recovery. Contrary to results from studies by de Oliviera et al., these NO-releasing gels elicited greater inflammatory cell infiltration. Nevertheless, this cell infiltration is believed to be beneficial for wound healing in order to remove damaged tissue and pathogens. In a different approach by Hardwick et al., sodium nitrite (330 mm) was also incorporated in acidic gels made of ascorbic acid (330 mm) to increase microcirculatory blood flow in the forearm of healthy human subjects.[168] The NO-generating gel was applied on an NO-permeable, polyester membrane (Sympatex(TM)) to prevent pain and tissue damage caused by gel acidity. After application, comparable vasodilatory properties were obtained from these gels with or without membrane use. In a separate study, Schoenfisch and coworkers also examined collagen production and angiogenesis in response to NO-releasing coated silicone implants (8 mm × 8 mm × 2 mm) in subcutaneous pockets of rats.[169] The collagen capsule thickness was significantly reduced at 3 and 6 weeks for coated implants compared to controls. Moreover, tissue surrounding the NO-releasing silicone implants had significantly more blood vessels at one and three weeks. Although the initial inflammatory reduction was not statistically significant for different implants at one week, greater than 30% reduction in the chronic inflammatory response was observed for NO-releasing silicone implants at 3 and 6 weeks.
5.4. Antimicrobial Applications
The overall incidence rate of Staphylococcus aureus infections for inpatients was estimated to be close to 1% or approximately two million cases annually,[170-172] with an associated cost of $14.5 billion annually for inpatient stays.[171] Moreover, with the rise of antibiotic resistant strains, methicillin-resistant S. aureus (MRSA) accounts for approximately half of the S. aureus infections.[173,174] As a consequence, traditional antibiotic-based treatments are necessitating a significant search for alternative bactericidal therapies as well as improved delivery mechanisms to enhance treatment efficacy. Nanotechnology offers a continuously growing palette of antimicrobial systems with promising perspectives such as silver-based nanoparticles and antibiotics-carrying nanoparticles.[14] In a different way, the susceptibility of bacteria to specific molecular moieties has made the research on polymers with inherent antibacterial properties a highly active field.[175] The latter approach, however, may suffer from a lack of sensitivity.
When searching for unconventional antimicrobial treatments, an additional supply of NO, in addition to NO produced from macrophages, is believed to enhance the immune system and kill the bacteria, thus preventing and combating the infection more effectively.[176,177] Previous in vitro studies by Schoenfisch and co-workers suggested that NO-releasing sol–gels reduced bacterial adhesion.[53,177-181] The authors extended this work to examine whether NO release from xerogel-coated silicone rubber was capable of fighting a S. aureus subcutaneous infection in a rat model. They dip-coated silicone squares (8 mm × 8 mm × 2.5 mm) into a sol solution consisting of isobutyltri-methoxysilane (BTMOS) and N-(6 aminohexyl)aminopropyl trimethoxysilane (AHAP3) (40% v/v balance with BTMOS) followed by treatment with NO gas for diazeniumdiolation.[182] The NO flux increased sharply over 2 h to a maximum of approximately 18 · 10−9 mol cm−2 min−1. Roughly 90% of NO was released in the first day, while minimal NO flux was detected up to seven days. The NO flux was tuneable by modifying the AHAP3 content; however the material had poor stability above 40% v/v. Following S. aureus inoculation, these materials were implanted into subcutaneous pockets. Eight days after implantation, the implant infection rate was significantly lower for NO-releasing coated implants (13.3%) compared to uncoated implants (73.3%). A thick tissue capsule, suggesting possible bacterial biofilm, was present around uncoated implants, while capsule thickness was similar for contaminated and non-contaminated coated implants.
Using a newly developed carbon-based NO-releasing coating to prevent nitrosamines, Engelsman et al. dip-coated surgical meshes with poly(ethylene-vinylacetate) (PEVA, 30% acetate) that was converted to carbon-based diazeniumdiolate using dimethylformamide, sodium trimethylsilanolate, and NO gas.[183] Although coated meshes in vitro demonstrated significant antimicrobial properties against S. aureus, E. coli, and P. aeruginosa, no differences in combating S. aureus were observed between coated and uncoated meshes when subcutaneously tested in mice. As NO release from carbon-based coatings was 6 times lower than nitrogen-based coating, insufficient NO release might have led to inappreciable in vivo differences.
In a different approach by using hydrogel/glass NO-np (~10 nm in diameter) containing sodium nitrite, Martinez et al. applied an NO-np powder (5 mg) to excisional wounds (5 mm biopsy punch) on the dorsal side of mice after inoculation with MRSA.[184] Compared to both np-treated and untreated wounds, wounds treated with NO-np had faster wound healing and less bacterial burden, inflammatory cell infiltration, and collagen degradation. As MRSA was commonly found in abscesses from patients with skin and soft tissue infections,[173] Martinez and co-workers further examined whether NO-np were capable of combating MRSA subcutaneous abscesses in mice.[185] One day after inoculating mice with MRSA, they administered NO-np (5 mg/mL) either directly into or topically above the abscess. Both delivery methods significantly lowered the MRSA burden and decreased the abscess size compared to controls. Interestingly, in contrast to studies that link NO to angiogenesis,[160,161,167] a reduction in angiogenesis and higher expression levels of cytokines such as IL-12, TNF-α, MCP-1, and IFN-γ were found in abscesses treated with NO-np. The authors suggested that NO-np prevented bacterial spreading by means of decreasing vascularization and promoting an effective host pro-inflammatory response for bacterial clearance. Taken together, these results demonstrate the efficacy of NO as an antibacterial agent to prevent infections that could derive from materials implantation in vivo.
6. Current Limitations, Future Perspectives and Conclusions
Advances in the biomedical application of NO have prompted its consideration for the treatment of diverse pathologic processes such as neointimal hyperplasia, bacterial infections, and cerebral vasospasms, among others (for summary, see Table 2, 3 and 4). Polymer-based NO therapies offer the potential for local delivery in many applications, thereby reducing the risk of side effects derived from systemic administration. Additionally, the short-term stability of NO and accompanying small action radius further decreases the risk of undesirable effects outside of the intended tissue. Furthermore, the uncertainty regarding the efficacy, complications, and long-term effects of alternative therapies involving gene delivery or stem cell-based therapies continues to make NO an attractive therapeutic molecule. Nevertheless, a considerable number of factors limit the clinical use of NO-releasing and -generating materials for biomedical applications. Firstly, despite a better understanding of the biological range of NO concentrations (100 pm–5 nm), further studies are necessary to address the therapeutic and tolerable ranges of NO doses for better designs of material-based NO therapies. Release profiles should be carefully evaluated such that the burst release is within a safe range, and the sustained release augments the physiological NO levels in the target tissue. Furthermore, administration route of the donors may alter the delivery of the intended doses. Secondly, leaching of undesirable products that are potentially carcinogenic or cytotoxic is an important consideration in the materials design process. Finally, the disparities in the detection methods for measuring NO release prevent direct comparison of results. For example, while the Griess method is still widely used for indirect detection of NO release, it only measures nitrite, an oxidative product of NO.[186] Although it serves as a simple way of determining NO release differences and kinetics, a more direct and accurate detection method to measure absolute NO values, such as chemiluminescence, is recommended before in vivo work. In addition, variations on reporting NO release also complicate the interpretation of the different NO delivery studies. In the reviewed articles, NO release has been reported in terms of flux, maximum flux, total release, concentrations, and average release. Moreover, the normalization over the amount of material is not a common practice. Such inconsistencies not only make comparisons of studies difficult, but also complicate the evaluation of therapeutic levels and outcomes. The often contradictory effects of low and high NO levels warrant the advocacy of a standard.
Table 2.
NO-releasing materials for the inhibition of thrombosis.
| Authors | Ref. | Animal model | Application | NO source | Delivery system | NO detection method | NO release or flux | Time point in vivo |
|---|---|---|---|---|---|---|---|---|
| Annich et al. | [107] | Rabbit | ECC | MAHMA/N2O2 | PVC | Biamperometric method | Cumulative 20–25 μmol (1 d) | 4 h |
| Schoenfisch et al. | [109] | Canine | Oxygen sensors implanted in carotid and femoral arteries | MAHMA/N2O2 | SR | Griess (no specified buffer or temp) | Cumulative > 0.4 μmol | 1 d |
| Zhang et al. | [40] | Rabbit | ECC | DACA-6/N2O2 | SR | Chemiluminescence (23 °C, N2) | >4 · 10−10 mol cm−2 min−1 | 4 h |
| Frost et al. | [114] | Porcine | Oxygen sensors implanted in carotid and femoral arteries | DACA-6/N2O2 | SR | Chemiluminescence (no specified temp) | ~2.5 · 10−10 mol cm−2 min−1 (initial flux) ~1 · 10−10 mol cm−2 min−1 (20 h) | 16 h |
| Batchelor et al. | [35] | Sheep | Arteriovenous grafts from the common carotid artery to the external jugular vein | DBHD/N2O2 | PVC + DOS + KTpClPB | Chemiluminescence | Peak flux ~60 · 10−10 mol cm−2 min−1 and ~110 · 10−10 mol cm−2 min−1 for 4 and 8 wt% donor, respectively (5 h) | 21 d |
| Fleser et al. | [110] | Sheep | Arteriovenous grafts from the common carotid artery to the external jugular vein | DBHD/N2O2 | PVC + DOS + KTpClPB | Chemiluminescence | 1.752 · 10−9 mol cm−2 min−1 (1 week); 3.3 · 10−10 mol cm−2 min−1 (3 weeks) | 21 d |
| Skrzypchak et al. | [112] | Rabbit | ECC | DBHD/N2O2 | PVC + DOS + KTpClPB | Chemiluminescence | 2.33, 13.65, 20.64, and 39.10 · 10−10 mol cm−2 min−1 (2, 10, 25, and 50 wt% donor, respectively) | 4 h |
| Major et al. | [111] | Rabbit | ECC | DBHD/N2O2 | PVC + DOS + KTpClPB | Chemiluminescence | 15 · 10−10 mol cm−2 min−1 | 4 h |
| Wu et al. | [117] | Porcine | Oxygen sensors implanted in carotid and femoral arteries | Cu0 catalyst | SR + PU | Chemiluminescence | >1 · 10−10 mol cm−2 min−1 at physiological GSNO levels | 20 h |
| Xu et al. | [118] | Swine | Rods implanted in carotid and femoral arteries | HNBOA/N2O2 | PU | Chemiluminescence | Peak flux ~5 · 10−10 mol cm−2 min−1 (12 h) | 8 h |
Note: Time points for NO release or flux are the same as the in vivo time points unless otherwise specified. NO detection was conducted at 37 °C in PBS unless otherwise specified.
DACA-6/N2O2: diazeniumdiolated N-(6-aminohexyl)-3-aminopropyl-trimethoxysilane, DBHD/N2O2: diazeniumdiolated dibutylhexanediamine, DOS + KTpClPB: plasticized dioctyl sebacate + potassium tetrakis-4-chlorophenyl borate, ECC: extracorporal circulation, HNBOA/N2O2: O2-(2-hydroxy-5-nitro-4-pentadecyl-benzyl)-1-(N,N-dioctylamino)diazen-1-ium-1,2-diolate, MAHMA/N2O2: (Z)-1-{N-Methyl-N-[6-(N-methylammoniohexyl)amino]}diazen-1-ium-1,2-diolate, PU: polyurethane, PVC: polyvinyl chloride, SR: silicone rubber.
Table 3.
NO-releasing materials for the inhibition of neointimal hyperplasia.
| Authors | Ref. | Animal model | Application | NO source | Delivery system | NO detection method | NO release or flux | Time point in vivo | I/M ratio decrease |
|---|---|---|---|---|---|---|---|---|---|
| Kaul et al. | [122] | Rat | Periadventitially in balloon-injured ileofemoral arteries | SPER/NO | Atrigel | Chemiluminescence | Peak at 4.5 μmol (40 min) 95% released (150 min) | 14 d | ~75% |
| Pearce et al. | [123] | Rat | Perivascularly in balloon-injured carotid arteries | PAN/NO PROLI/NO | Poloxamer | N/A | N/A | 14 d | 86% (PROLI/NO powder) |
| Kapadia et al. | [126] | Rat | Perivascularly in balloon-injured carotid arteries | PROLI/NO DPTA/NO | Peptide amphiphile and heparin (nanofiber gels) | Griess | Peak at 0.6 μmol day−1 and 1.2 μmol day−1, respectively (2 d) | 14 d | 77% and 45%, respectively |
| Lipke et al. | [37] | Rat | Periadventitially in balloon-injured carotid arteries | Cys/NO | PEG | Griess (HEPES buffer) | 35% released (4 h)[127] | 14 d | 77% compared to control hydrogel |
| Serrano et al. | [39] | Rat | Perivascularly balloon-injured carotid arteries | diamine diol/N2O2 | POC, PDDC | Griess | Peak flux 35.4 · 10−9 mol cm−2 min−1 (4 h) and 6.6 · 10−9 mol cm−2 min−1 (24 h), respectively | 14 d | 38% |
| Do et al. | [129] | Rabbit | Microsphere-loaded stent deployed in infrarenal abdominal aorta | NOC-12/N2O2 | PLGA and PEG (microspheres) | Griess | ~18 mM (1 week) | 7 d & 28 d | 46% and 32%, respectively, compared to control microspheres |
| ~8 mm (4 weeks) | |||||||||
| Yoon et al. | [130] | Porcine | Coated stent deployed in coronary arteries | SNP | PU | Famotidine assay to measure SNP (no specified temp) | 85% of total SNP mass eluted from stents (30 d) | 28 d | No improvement |
| Buergler et al. | [131] | Porcine | Coated stent deployed in left anterior descending and right coronary arteries | DETA/NO | Polycaprolactone | Chemiluminescence (flushed with He, no specified temp and buffer) | 9.0 · 10−5 μmol min−1 mg−1 (0 h) and 0.5 · 10−5 μmol min−1 mg−1 (72 h) | 28 d | No improvement |
| Abbasi et al. | [132] | Rabbit | Perivascularly in aortic intimal injured hyper-cholesterolemic rabbits | DETA/NO | Silastic elastomer | N/A | N/A | 6 wks | No improvement |
Note: Time points for NO release or flux are the same as the in vivo time points unless otherwise specified. NO detection was conducted at 37 °C in PBS. I/M ratio decrease is compared to injury alone unless otherwise specified.
Cys/NO: S-nitrosocysteine, DETA/NO: diazeniumdiolated diethylenetriamine, DPTA/NO: 1-[N-(3-aminopropyl)-N-(3-ammoniopropyl] diazen-1-ium-1,2-diolate, I/M: Intima/media, NOC-12/N2O2: diazeniumdiolated N-ethyl-2-(1-ethyl-2-hydroxy-2-nitrosohydrazino)ethanamine, PAN/NO: diazeniumdiolated poly(acrylonitrile), PDDC: poly(1,12-dodecanediol-co-citrate), PEG: poly(ethylene glycol), PLGA: poly(lactic-co-glycolic acid), POC: poly(1,8-octanediol-co-citrate), PROLI/NO: 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate, PU: polyurethane, SNP: sodium nitroprusside, SPER/NO: diazeniumdiolated spermine.
Table 4.
NO-releasing materials for other in vivo applications.
| Authors | Ref. | Animal model | Application | NO source | Delivery system | NO detection method |
NO release or flux |
Time point in vivo |
|---|---|---|---|---|---|---|---|---|
| Tierney et al. | [135] | Rat | CVS (periadventitial space near femoral arteries) | DETA/NO | EVAc | Absorbance (254 nm) | 7.5% (10 h) and 15% (9 d) released of donor | 8 d |
| Gabikian et al. and Pradilla et al. | [136,137] | Rabbit | CVS (subarachnoid space) | DETA/NO | EVAc | N/A | N/A | 3 d |
| Clatterbuck et al. | [139] | Cynomolgus monkey | CVS (space around supraclinoid internal carotid, and proximal anterior and middle cerebral arteries) | DETA/NO | EVAc | N/A | N/A | 7 d |
| Momin et al. | [138] | Mice (haptoglobin 2-2) | CVS (subarachnoid space) | DETA/NO | EVAc | Absorbance (254 nm) | 15.6% (36 h) released of donor | 1 d |
| Cabrales et al. | [141,142] | Hamster | Vasodilation and decrease blood pressure | Sodium nitrite | Hydrogel/glass nanoparticles | Chemiluminescence (N2 in DI water and no specified temp) | Cumulative ~0.32 μmol (5 h) | 2 h & 4 h |
| Yoo et al. | [148] | Rat | Sexual dysfunction (vaginal blood perfusion) | GSNO | Carbopol, HPMC, and PEG | Absorbance (336 nm, citrate buffer pH 4.0) | Cumulative ~225 nmol mg−1 film and ~50 nmol mg−1 film for 20 wt% and 10 wt% of donor, respectively (12 h) | 260 min |
| Yoo et al. | [150] | Rat | Sexual dysfunction (vaginal blood perfusion) | GSNO | PLGA microparticles | N/A | N/A | 4 h |
| Souto et al. | [149] | Human | Sexual dysfunction (clitoral blood flow) | GSNO | Pluronic-F127 | N/A | N/A | 15 min |
| Masters et al. | [38] | Mice | Wound healing | PVA/N2O2 | PVA | Griess (HEPES buffer pH 6, 20 °C) | Cumulative 0.055 μmol and 0.46 μmol for 0.5 mm and 5 mm NO hydrogels, respectively (40 h) | 29 d |
| Seabra et al. | [163] | Human | Wound healing (blood flow) | GSNO and SNAC | Synperonic F-127 | Absorbance (545 nm, no specified buffer) | ~50% decomposition of both donors | 3 h |
| Seabra et al. | [164] | Rat | Wound healing (blood flow) | GSNO | Pluronic F-127 | N/A | N/A | 30 min |
| Amadeu et al. | [165] | Rat | Wound healing | GSNO | Pluronic F-127 | N/A | N/A | 14 d |
| Simões et al. | [166] | Human | Wound healing (blood flow) | GSNO | PVA | Spectrophotometer (336 nm) | Donor diffusion coefficients of 2.0 · 10−7 cm2 s−1 and 5.0 · 10−7 cm2 s−1 for 1 and 5 F/T films, respectively | 98 min |
| Li et al. | [36] | Rat | Wound healing | GSNO | PVMMA/PVP | Griess (22 °C) | Cumulative ~50 nmol mg−1 (12 d) | 16 d |
| Zhu et al. | [167] | Rat | Burn wound healing | Sodium nitrite | Cellulose-derived gel | Amperometric (no specified temp or buffer) | 10 mm (1 h) | 15 d |
| Nablo et al. | [177] | Rat | Antimicrobial effects (S. aureus) | Xerogel/N2O2 | Silicone | Chemiluminescence | 18 · 10−9 mol cm−2 min−1 (2 h), 90% release (1 d) | 8 d |
| Hetrick et al. | [169] | Rat | Foreign body response | Xerogel/N2O2 | Silicone | Chemiluminescence | 6.6 · 10−9 mol cm−2 min−1 (1–2 h), Cumulative 1.35 μmol cm−2 (70 h) | 6 wks |
| Englesman et al. | [183] | Rat | Antimicrobial effects (S. aureus) | Carbon-based diazeniumdiolate | PEVA | Chemiluminescence | Decreased from 61.8 pmol min−1 mesh−1 to 14.9 pmol min−1 mesh−1 in 24 h; not detected beyond 48 h | 6 d |
| Martinez et al. | [184] | Mice | Antimicrobial effects (MRSA) | Sodium nitrite | Hydrogel/glass nanoparticles | Amperometric (no specified temp) | Peaked at ~100 nm (70 min), less ongoing released for ~24 h | 7 d |
| Han et al. | [185] | Mice | Antimicrobial effects (MRSA) | Sodium nitrite | Hydrogel/glass nanoparticles | N/A | Peaked initially at 75 nm; Steady state at 50 nm (no time frame specified) | 4 d |
Note: Time points for NO release or flux are the same as the in vivo time points unless otherwise specified. NO detection was conducted at 37 °C in PBS unless otherwise specified.
CVS: cerebral vasospasms, DETA/NO: diazenium-diolated diethylenetriamine, EVAc: ethylene/vinyl acetate copolymers, GSNO: S-nitrosoglutathione, HPMC: hydroxypropyl methylcellulose, PEG: poly(ethylene glycol), PEVA: poly(ethylene-vinylacetate), PLGA: poly(lactic-co-glycolic acid), PVA: poly(vinyl alcohol), PVMMA/PVP: poly(vinyl methyl ether-co-maleic anhydride)/poly(vinyl pyrrolidone), SNAC: S-nitroso-N-acetylcysteine.
Most of the NO-releasing polymeric systems described contain a finite reservoir of NO. This limitation, including the short half-life of NO and the difficulties for long-term NO storage, complicates the clinical use of those materials. To overcome this drawback, NO-generating materials were developed to utilize a potentially unlimited source of endogenous NO precursors, for example S-nitrosothiols. However, variability among healthy and diseased individuals in the endogenous levels of precursors, substrates, catalysts, or particular blood components makes the outcome of this approach unpredictable and difficult to implement commercially.
In summary, despite the significant progress on the potential therapeutic use of NO, five major aspects introduce complexity to the current clinical application of NO-releasing and -generating materials: 1) the uncertainty of NO concentrations in target tissues, 2) the vagueness about therapeutic NO doses desired for specific applications, 3) the accuracy of NO detection methods, 4) the nonexistence of a standard in NO detection and release reporting, and 5) the absence of efficient long-term NO-delivering and storage materials. In this context, polymer-based materials have proven their efficacy to extend the half-lives of NO donors, facilitate the delivery to the target tissue and reduce the risk of toxic by-products leaching.
Acknowledgments
M.C.J. acknowledges support from the NIH Biotechnology Training Grant. M.C.S. is greatly indebted to the Ministerio de Ciencia e Innovación of Spain for a postdoctoral fellowship “Juan de la Cierva”. R.L. acknowledges support from the AHA Predoctoral Fellowship. G.A.A. acknowledges support by the American Heart Association Established Investigator Award.
Biographies

Guillermo A. Ameer a native of Panama City, Panama, is Professor of Biomedical Engineering and Surgery at Northwestern University. He received his PhD in Chemical and Biomedical Engineering from the Massachusetts Institute of Technology. Dr. Ameer’s research interests include biomaterials, tissue engineering and bio/nanotechnology for improved therapeutics and diagnostics.

Michele C. Jen received her B.S. in Biomedical Engineering from the University of Texas at Austin in 2007. She is a Ph.D. candidate working in the laboratory of Dr. Ameer at Northwestern University and a recipient of the NIH Biotechnology Training Grant. Her research is focused on biomaterials for gene delivery.

María C. Serrano received her PhD in Biochemistry and Molecular Biology from Universidad Complutense de Madrid in 2006 (Spain). After a postdoctoral training period in Dr. Ameer’s research laboratory for over two years, she is now a postdoctoral fellow Juan de la Cierva in the Bioinspired Materials Group leaded by Dr. del Monte in the Materials Science Institute in Madrid (ICMM-CSIC). Her research interests are focused on biomaterials for cardiovascular, bone, and neural tissue regeneration.
Contributor Information
Michele C. Jen, Biomedical Engineering Department, Northwestern University, Evanston IL, 60208, USA
Dr. María C. Serrano, Instituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas Cantoblanco, Madrid 28049, Spain
Robert van Lith, Biomedical Engineering Department, Northwestern University, Evanston IL, 60208, USA
Dr. Guillermo A. Ameer, Email: g-ameer@northwestern.edu, Biomedical Engineering Department, Northwestern University, Evanston IL, 60208, USA; Chemistry of Life Processes Institute, Evanston IL, 60208, USA; Institute for BioNanotechnology in Medicine, Northwestern University, Chicago IL, 60611, USA; Division of Vascular Surgery, Feinberg School of Medicine, Northwestern University, Chicago IL, 60611, USA.
References
- 1.Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric Oxide. 2002;7:1. doi: 10.1016/s1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- 2.Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro LJ. Nitric Oxide. 2006;15:265. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Moncada S. J R Soc Med. 1999;92:164. doi: 10.1177/014107689909200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mocellin S, Bronte V, Nitti D. Med Res Rev. 2007;27:317. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 5.Ignarro LJ, Cirino G, Casini A, Napoli C, Cardiovasc J. Pharmacol. 1999;34:879. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 6.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci USA. 1987;84:9265. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer RM, Ferrige AG, Moncada S. Nature. 1987;327:524. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 8.Damoulis PD, Drakos DE, Gagari E, Kaplan DL. Ann N Y Acad Sci. 2007;1117:367. doi: 10.1196/annals.1402.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu D, Neeley WL, Pritchard CD, Slotkin JR, Woodard EJ, Langer R, Teng YD. Stem Cells. 2009;27:1212. doi: 10.1002/stem.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frost MC, Reynolds MM, Meyerhoff ME. Biomaterials. 2005;26:1685. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Keefer LK. Annu Rev Pharmacol Toxicol. 2003;43:585. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- 12.Ostrovsky AD, Ford PC. Dalton Trans. 2009;28:10660. doi: 10.1039/b912898k. [DOI] [PubMed] [Google Scholar]
- 13.Hall CN, Garthwaite J. Nitric Oxide. 2009;21:92. doi: 10.1016/j.niox.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortivo R, Vindigni V, Iacobellis L, Abatangelo G, Pinton P, Zavan B. Nanomedicine. 2010;5:641. doi: 10.2217/nnm.10.32. [DOI] [PubMed] [Google Scholar]
- 15.Morris RE, Wheatley PS. Angew Chem Int Ed. 2008;47:4966. doi: 10.1002/anie.200703934. [DOI] [PubMed] [Google Scholar]
- 16.Sortino S. Chem Soc Rev. 2010;39:2903. doi: 10.1039/b908663n. [DOI] [PubMed] [Google Scholar]
- 17.Busse R, Mulsch A. FEBS Lett. 1990;275:87. doi: 10.1016/0014-5793(90)81445-t. [DOI] [PubMed] [Google Scholar]
- 18.Napoli C, Ignarro LJ. Nitric Oxide. 2001;5:88. doi: 10.1006/niox.2001.0337. [DOI] [PubMed] [Google Scholar]
- 19.Roy B, Garthwaite J. Proc Natl Acad Sci USA. 2006;103:12185. doi: 10.1073/pnas.0602544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim YM, Talanian RV, Billiar TR. J Biol Chem. 1997;272:31138. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Bombeck CA, Yang S, Kim YM, Billiar TR. J Biol Chem. 1999;274:17325. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 22.Kibbe MR, Li J, Nie S, Watkins SC, Lizonova A, Kovesdi I, Simmons RL, Billiar TR, Tzeng E. J Vasc Surg. 2000;31:1214. doi: 10.1067/mva.2000.105006. [DOI] [PubMed] [Google Scholar]
- 23.Brune B, von Knethen A, Sandau KB. Cell Death Differ. 1999;6:969. doi: 10.1038/sj.cdd.4400582. [DOI] [PubMed] [Google Scholar]
- 24.Kim YM, Bombeck CA, Billiar TR. Circ Res. 1999;84:253. doi: 10.1161/01.res.84.3.253. [DOI] [PubMed] [Google Scholar]
- 25.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG. J Biol Chem. 2000;275:20474. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 26.Peterson TE, Poppa V, Ueba H, Wu A, Yan C, Berk BC. Circ Res. 1999;85:29. doi: 10.1161/01.res.85.1.29. [DOI] [PubMed] [Google Scholar]
- 27.Zuckerbraun BS, Shiva S, Ifedigbo E, Mathier MA, Mollen KP, Rao J, Bauer PM, Choi JJ, Curtis E, Choi AM, Gladwin MT. Circulation. 2010;121:98. doi: 10.1161/CIRCULATIONAHA.109.891077. [DOI] [PubMed] [Google Scholar]
- 28.Francis SH, Busch JL, Corbin JD, Sibley D. Pharmacol Rev. 2010;62:525. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napoli C, Ignarro LJ. Arch Pharm Res. 2009;32:1103. doi: 10.1007/s12272-009-1801-1. [DOI] [PubMed] [Google Scholar]
- 30.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 31.Brilli RJ, Krafte-Jacobs B, Smith DJ, Roselle D, Passerini D, Vromen A, Moore L, Szabó C, Salzman AL. J Appl Physiol. 1997;83:1968. doi: 10.1152/jappl.1997.83.6.1968. [DOI] [PubMed] [Google Scholar]
- 32.Morley D, Keefer LK. J Cardiovasc Pharmacol. 1993;22:S3. [PubMed] [Google Scholar]
- 33.Saavedra JE, Billiar TR, Williams DL, Kim Y-M, Watkins SC, Keefer LK. J Med Chem. 1997;40:1947. doi: 10.1021/jm9701031. [DOI] [PubMed] [Google Scholar]
- 34.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. J Med Chem. 1991;34:3242. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 35.Batchelor MM, Reoma SL, Fleser PS, Nuthakki VK, Callahan RE, Shanley CJ, Politis JK, Elmore J, Merz SI, Meyerhoff ME. J Med Chem. 2003;46:5153. doi: 10.1021/jm030286t. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Lee PI. Mol Pharmaceutics. 2009;7:254. doi: 10.1021/mp900237f. [DOI] [PubMed] [Google Scholar]
- 37.Lipke EA, West JL. Acta Biomater. 2005;1:597. doi: 10.1016/j.actbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Masters KSB, Leibovich SJ, Belem P, West JL, Poole-Warren LA. Wound Repair Regener. 2002;10:286. doi: 10.1046/j.1524-475x.2002.10503.x. [DOI] [PubMed] [Google Scholar]
- 39.Serrano MC, Vavra AK, Jen M, Hogg ME, Murar J, Martinez J, Keefer LK, Ameer GA, Kibbe MR. Macromol Biosci. 2011;11:700. doi: 10.1002/mabi.201000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Annich GM, Miskulin J, Osterholzer K, Merz SI, Bartlett RH, Meyerhoff ME. Biomaterials. 2002;23:1485. doi: 10.1016/s0142-9612(01)00274-5. [DOI] [PubMed] [Google Scholar]
- 41.Zhao H, Serrano MC, Popowich DA, Kibbe MR, Ameer GA. J Biomed Mater Res A. 2010;93:356. doi: 10.1002/jbm.a.32536. [DOI] [PubMed] [Google Scholar]
- 42.Heerden PVv, Sviri S, Ilett KF, Lam C-F. Expert Opinion Investigational Drugs. 2002;11:897. doi: 10.1517/13543784.11.7.897. [DOI] [PubMed] [Google Scholar]
- 43.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Proc Natl Acad Sci. 1992;89:7674. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derakhshan B, Hao G, Gross SS. Cardiovasc Res. 2007;75:210. doi: 10.1016/j.cardiores.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martínez-Ruiz A, Cadenas S, Lamas S. Free Radical Biol Med. 2011;51:17. doi: 10.1016/j.freeradbiomed.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 46.Hanspal IS, Magid KS, Webb DJ, Megson IL. Nitric Oxide. 2002;6:263. doi: 10.1006/niox.2001.0412. [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Rudd MA, Freedman JE, Loscalzo J. J Pharmacol Exp Ther. 1998;284:526. [PubMed] [Google Scholar]
- 48.Hogg N. Ann Rev Pharmacol Toxicol. 2002;42:585. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 49.Cha W, Meyerhoff ME. Biomaterials. 2007;28:19. doi: 10.1016/j.biomaterials.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 50.Hwang S, Cha W, Meyerhoff ME. Angew Chem Int Ed. 2006;45:2745. doi: 10.1002/anie.200503588. [DOI] [PubMed] [Google Scholar]
- 51.Hwang S, Meyerhoff ME. Biomaterials. 2008;29:2443. doi: 10.1016/j.biomaterials.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katsumi H, Nishikawa M, Yamashita F, Hashida M. J Pharmacol Exp Ther. 2005;314:1117. doi: 10.1124/jpet.105.087429. [DOI] [PubMed] [Google Scholar]
- 53.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pulfer SK, Ott D, Smith DJ. J Biomed Mater Res. 1997;37:182. doi: 10.1002/(sici)1097-4636(199711)37:2<182::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 55.Smith DJ, Chakravarthy D, Pulfer S, Simmons ML, Hrabie JA, Citro ML, Saavedra JE, Davies KM, Hutsell TC, Mooradian DL, Hanson SR, Keefer LK. J Med Chem. 1996;39:1148. doi: 10.1021/jm950652b. [DOI] [PubMed] [Google Scholar]
- 56.Sorragi CdL, Shishido SM, Lemos ME, Marcondes S, Antunes E, Krieger MH. Cell Biochem Funct. 2011;29:207. doi: 10.1002/cbf.1738. [DOI] [PubMed] [Google Scholar]
- 57.Eroy-Reveles AA, Mascharak PK. Future Med Chem. 2009;1:1497. doi: 10.4155/fmc.09.111. [DOI] [PubMed] [Google Scholar]
- 58.Friedman A, Friedman J. Expert Opin Drug Deliv. 2009;6:1113. doi: 10.1517/17425240903196743. [DOI] [PubMed] [Google Scholar]
- 59.Halpenny GM, Mascharak PK. Anti-Infective Agents Med Chem. 2010;9:187. [Google Scholar]
- 60.Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 61.Jordan SW, Chaikof EL. J Vasc Surg. 2007;45:104A. doi: 10.1016/j.jvs.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 62.Reynolds MM, Frost MC, Meyerhoff ME. Free Radic Biol Med. 2004;37:926. doi: 10.1016/j.freeradbiomed.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 63.Varu VN, Tsihlis ND, Kibbe MR. Vasc Endovascular Surg. 2009;43:121. doi: 10.1177/1538574408322752. [DOI] [PubMed] [Google Scholar]
- 64.Wu Y, Meyerhoff ME. Talanta. 2008;75:642. doi: 10.1016/j.talanta.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kushwaha M, Anderson JM, Bosworth CA, Andukuri A, Minor WP, Lancaster JR, Jr, Anderson PG, Brott BC, Jun HW. Biomaterials. 2010;31:1502. doi: 10.1016/j.biomaterials.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andukuri A, Kushwaha M, Tambralli A, Anderson JM, Dean DR, Berry JL, Sohn YD, Yoon YS, Brott BC, Jun HW. Acta Biomater. 2010;7:225. doi: 10.1016/j.actbio.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaughn MW, Kuo L, Liao JC. Am J Physiol. 1998;274:H2163. doi: 10.1152/ajpheart.1998.274.6.H2163. [DOI] [PubMed] [Google Scholar]
- 68.Taite LJ, Yang P, Jun HW, West JL. J Biomed Mater Res B: Appl Biomater. 2008;84:108. doi: 10.1002/jbm.b.30850. [DOI] [PubMed] [Google Scholar]
- 69.Zhou Z, Meyerhoff ME. Biomaterials. 2005;26:6506. doi: 10.1016/j.biomaterials.2005.04.046. [DOI] [PubMed] [Google Scholar]
- 70.Wu B, Gerlitz B, Grinnell BW, Meyerhoff ME. Biomaterials. 2007;28:4047. doi: 10.1016/j.biomaterials.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jo YS, van der Vlies AJ, Gantz J, Thacher TN, Antonijevic S, Cavadini S, Demurtas D, Stergiopulos N, Hubbell JA. J Am Chem Soc. 2009;131:14413. doi: 10.1021/ja905123t. [DOI] [PubMed] [Google Scholar]
- 72.Kanayama N, Yamaguchi K, Nagasaki Y. Chem Lett. 2010;39:1008. [Google Scholar]
- 73.Cavalieri F, Finelli I, Tortora M, Mozetic P, Chiessi E, Polizio F, Brismar TB, Paradossi G. Chem Mater. 2008;20:3254. [Google Scholar]
- 74.Wan A, Gao Q, Li H. J Mater Sci Mater Med. 2009;20:321. doi: 10.1007/s10856-008-3511-5. [DOI] [PubMed] [Google Scholar]
- 75.Reynolds MM, Saavedra JE, Showalter BM, Valdez CA, Shanklin AP, Oh BK, Keefer LK, Meyerhoff ME. J Mater Chem. 2010;20:3107. doi: 10.1039/c000152j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reynolds MM, Hrabie JA, Oh BK, Politis JK, Citro ML, Keefer LK, Meyerhoff ME. Biomacromolecules. 2006;7:987. doi: 10.1021/bm060028o. [DOI] [PubMed] [Google Scholar]
- 77.Johnson TA, Stasko NA, Matthews JL, Cascio WE, Holmuhamedov EL, Johnson CB, Schoenfisch MH. Nitric Oxide. 2010;22:30. doi: 10.1016/j.niox.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 78.Stasko NA, Schoenfisch MH. J Am Chem Soc. 2006;128:8265. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 79.Stasko NA, Fischer TH, Schoenfisch MH. Biomacromolecules. 2008;9:834. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang J, Welby JL, Meyerhoff ME. Langmuir. 2008;24:10265. doi: 10.1021/la801466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hwang S, Meyerhoff ME. J Mater Chem. 2007;17:1462. [Google Scholar]
- 82.Puiu SC, Zhou Z, White CC, Neubauer LJ, Zhang Z, Lange LE, Mansfield JA, Meyerhoff ME, Reynolds MM. J Biomed Mater Res B: Appl Biomater. 2009;91B:203. doi: 10.1002/jbm.b.31391. [DOI] [PubMed] [Google Scholar]
- 83.Mitchell-Koch JT, Padden KM, Borovik AS. J Polym Sci Part A: Polym Chem. 2006;44:2282. [Google Scholar]
- 84.Halpenny GM, Gandhi KR, Mascharak PK. ACS Med Chem Lett. 2010;1:180. doi: 10.1021/ml1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eroy-Reveles AA, Leung Y, Mascharak PK. J Am Chem Soc. 2006;128:7166. doi: 10.1021/ja061852n. [DOI] [PubMed] [Google Scholar]
- 86.Eroy-Reveles AA, Leung Y, Beavers CM, Olmstead MM, Mascharak PK. J Am Chem Soc. 2008;130:4447. doi: 10.1021/ja710265j. [DOI] [PubMed] [Google Scholar]
- 87.Rose MJ, Olmstead MM, Mascharak PK. J Am Chem Soc. 2007;129:5342. doi: 10.1021/ja070247x. [DOI] [PubMed] [Google Scholar]
- 88.Halpenny GM, Olmstead MM, Mascharak PK. Inorg Chem. 2007;46:6601. doi: 10.1021/ic700694b. [DOI] [PubMed] [Google Scholar]
- 89.Halpenny G, Steinhardt R, Okialda K, Mascharak P. J Mater Sci : Mater Med. 2009;20:2353. doi: 10.1007/s10856-009-3795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sauaia MG, de Lima RG, Tedesco AC, da Silva RS. J Am Chem Soc. 2003;125:14718. doi: 10.1021/ja0376801. [DOI] [PubMed] [Google Scholar]
- 91.Santana DCAS, Pupo TT, Sauaia MG, da Silva RS, Lopez RFV. Int J Pharm. 2010;391:21. doi: 10.1016/j.ijpharm.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 92.Marquele-Oliveira F, Santana DC, Taveira SF, Vermeulen DM, de Oliveira AR, da Silva RS, Lopez RF. J Pharm Biomed Anal. 2010;53:843. doi: 10.1016/j.jpba.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 93.Sauaia MG, Oliveira FS, Tedesco AC, da Silva RS. Inorg Chim Acta. 2003;355:191. [Google Scholar]
- 94.Rose MJ, Mascharak PK. Coord Chem Rev. 2008;252:2093. doi: 10.1016/j.ccr.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seabra AB, da Silva R, de Souza GFP, de Oliveira MG. Artifi Organs. 2008;32:262. doi: 10.1111/j.1525-1594.2008.00540.x. [DOI] [PubMed] [Google Scholar]
- 96.Coneski PN, Rao KS, Schoenfisch MH. Biomacromolecules. 2010;11:3208. doi: 10.1021/bm1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Coneski PN, Schoenfisch MH. Polym Chem. 2011;2:906. doi: 10.1039/C0PY00269K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pasut G, Greco F, Mero A, Mendichi R, Fante C, Green RJ, Veronese FM. J Med Chem. 2009;52:6499. doi: 10.1021/jm900804m. [DOI] [PubMed] [Google Scholar]
- 99.Kumar V, Hong SY, Maciag AE, Saavedra JE, Adamson DH, Prud’homme RK, Keefer LK, Chakrapani H. Mol Pharm. 2010;7:291. doi: 10.1021/mp900245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang J, Teng Y-H, Hao Y, Oh-Lee J, Mohanty DK. Polym J. 2009;41:715. [Google Scholar]
- 101.Fox S, Wilkinson TS, Wheatley PS, Xiao B, Morris RE, Sutherland A, Simpson AJ, Barlow PG, Butler AR, Megson IL, Rossi AG. Acta Biomater. 2010;6:1515. doi: 10.1016/j.actbio.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 102.Wheatley PS, Butler AR, Crane MS, Fox S, Xiao B, Rossi AG, Megson IL, Morris RE. J Am Chem Soc. 2005;128:502. doi: 10.1021/ja0503579. [DOI] [PubMed] [Google Scholar]
- 103.Liu HA, Balkus KJ. Chem Mater. 2009;21:5032. [Google Scholar]
- 104.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. American Heart Association Statistics Committee and Stroke Statistics Subcommittee, Circulation. 2011;123:e18. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Beathard GA. Adv Ren Replace Ther. 1994;1:131. doi: 10.1016/s1073-4449(12)80044-6. [DOI] [PubMed] [Google Scholar]
- 106.Popowich DA, Varu V, Kibbe MR. Vascular. 2007;15:324. doi: 10.2310/6670.2007.00051. [DOI] [PubMed] [Google Scholar]
- 107.Annich G, Meinhardt J, Mowery K, Ashton B, Merz S, Hirschl R, Meyerhoff M, Bartlett R. Crit Care Med. 2000;28:915. doi: 10.1097/00003246-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 108.Mowery KA, Schoenfisch MH, Saavedra JE, Keefer LK, Meyerhoff ME. Biomaterials. 2000;21:9. doi: 10.1016/s0142-9612(99)00127-1. [DOI] [PubMed] [Google Scholar]
- 109.Schoenfisch MH, Mowery KA, Rader MV, Baliga N, Wahr JA, Meyerhoff ME. Anal Chem. 2000;72:1119. doi: 10.1021/ac991370c. [DOI] [PubMed] [Google Scholar]
- 110.Fleser PS, Nuthakki VK, Malinzak LE, Callahan RE, Seymour ML, Reynolds MM, Merz SI, Meyerhoff ME, Bendick PJ, Zelenock GB, Shanley CJ. J Vasc Surg. 2004;40:803. doi: 10.1016/j.jvs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 111.Major TC, Brant DO, Reynolds MM, Bartlett RH, Meyerhoff ME, Handa H, Annich GM. Biomaterials. 2010;31:2736. doi: 10.1016/j.biomaterials.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Skrzypchak AM, Lafayette NG, Bartlett RH, Zhou Zhengrong, Frost MC, Meyerhoff ME, Reynolds MM, Annich GM. Perfusion. 2007;22:193. doi: 10.1177/0267659107080877. [DOI] [PubMed] [Google Scholar]
- 113.Radomski MW, Palmer RMJ, Moncada S. Biochem Biophys Res Commun. 1987;148:1482. doi: 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- 114.Frost MC, Rudich SM, Zhang H, Maraschio MA, Meyerhoff ME. Anal Chemi. 2002;74:5942. doi: 10.1021/ac025944g. [DOI] [PubMed] [Google Scholar]
- 115.Frost MC, Batchelor MM, Lee Y, Zhang H, Kang Y, Oh B, Wilson GS, Gifford R, Rudich SM, Meyerhoff ME. Microchem J. 2003;74:277. [Google Scholar]
- 116.Gifford R, Batchelor MM, Lee Y, Gokulrangan G, Meyerhoff ME, Wilson GS. J Biomed Mater Res A. 2005;75A:755. doi: 10.1002/jbm.a.30359. [DOI] [PubMed] [Google Scholar]
- 117.Wu Y, Rojas AP, Griffith GW, Skrzypchak AM, Lafayette N, Bartlett RH, Meyerhoff ME. Sens Actuators B. 2007;121:36. doi: 10.1016/j.snb.2006.09.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu H, Reynolds MM, Cook KE, Evans AS, Toscano JP. Org Lett. 2008;10:4593. doi: 10.1021/ol801883f. [DOI] [PubMed] [Google Scholar]
- 119.Wessely R. Nat Rev Cardiol. 2010;7:194. doi: 10.1038/nrcardio.2010.14. [DOI] [PubMed] [Google Scholar]
- 120.Dangas GD, Claessen BE, Caixeta A, Sanidas EA, Mintz GS, Mehran R. J Am Coll Cardiol. 2010;56:1897. doi: 10.1016/j.jacc.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 121.Lemesle G, Maluenda G, Collins SD, Waksman R. Cardiol Clin. 2010;28:97. doi: 10.1016/j.ccl.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 122.Kaul S, Cercek B, Rengstrom J, Xu X-P, Molloy MD, Dimayuga P, Parikh AK, Fishbein MC, Nilsson J, Rajavashisth TB, Shah PK. J Am Coll Cardiol. 2000;35:493. doi: 10.1016/s0735-1097(99)00543-4. [DOI] [PubMed] [Google Scholar]
- 123.Pearce CG, Najjar SF, Kapadia MR, Murar J, Eng J, Lyle B, Aalami OO, Jiang Q, Hrabie JA, Saavedra JE, Keefer LK, Kibbe MR. Free Radical Biol Med. 2008;44:73. doi: 10.1016/j.freeradbiomed.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. J Med Chem. 1996;39:4361. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 125.DeRosa F, Kibbe MR, Najjar SF, Citro ML, Keefer LK, Hrabie JA. J Am Chem Soc. 2007;129:3786. doi: 10.1021/ja0686864. [DOI] [PubMed] [Google Scholar]
- 126.Kapadia MR, Chow LW, Tsihlis ND, Ahanchi SS, Eng JW, Murar J, Martinez J, Popowich DA, Jiang Q, Hrabie JA, Saavedra JE, Keefer LK, Hulvat JF, Stupp SI, Kibbe MR. J Vasc Surg. 2008;47:173. doi: 10.1016/j.jvs.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bohl KS, West JL. Biomaterials. 2000;21:2273. doi: 10.1016/s0142-9612(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 128.Bohl Masters KS, Lipke EA, Rice EEH, Liel MS, Myler HA, Zygourakis C, Tulis DA, West JL. J Biomater Sci Polym Ed. 2005;16:659. doi: 10.1163/1568562053783722. [DOI] [PubMed] [Google Scholar]
- 129.Do YS, Kao EY, Ganaha F, Minamiguchi H, Sugimoto K, Lee J, Elkins CJ, Amabile PG, Kuo MD, Wang DS, Waugh JM, Dake MD. Radiology. 2004;230:377. doi: 10.1148/radiol.2302020417. [DOI] [PubMed] [Google Scholar]
- 130.Yoon JH, Wu CJ, Homme J, Tuch RJ, Wolff RG, Topol EJ, Lincoff AM. Yonsei Med J. 2002;43:242. doi: 10.3349/ymj.2002.43.2.242. [DOI] [PubMed] [Google Scholar]
- 131.Buergler JM, Tio FO, Schulz DG, Khan MM, Mazur W, French BA, Raizner AE, Ali NM. Coron Artery Dis. 2000;11:351. doi: 10.1097/00019501-200006000-00009. [DOI] [PubMed] [Google Scholar]
- 132.Abbasi K, Anvari MS, Mahdanian A, Ahmadi SH, Rabbani S, Karimi A, Marzban M, Shalileh K, Ashrafinia N. Ann Vasc Surg. 2009;23:392. doi: 10.1016/j.avsg.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 133.Pluta R, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller E, Zauner A, Dorsch N, Clark J, Ono S, Kiris T, LeRoux P, Zhang J. Neurolo Res. 2009;31:151. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sehba FA, Bederson JB. In: Early Brain Injury or Cerebral Vasospasm. Feng H, Mao Y, Zhang JH, editors. 110/1. Springer; Vienna: 2011. p. 99. [Google Scholar]
- 135.Tierney TS, Clatterbuck RE, Lawson C, Thai Q-A, Rhines LD, Tamargo RJ. Neurosurgery. 2001;49:945. doi: 10.1097/00006123-200110000-00028. [DOI] [PubMed] [Google Scholar]
- 136.Gabikian P, Clatterbuck RE, Eberhart CG, Tyler BM, Tierney TS, Tamargo RJ. Stroke. 2002;33:2681. doi: 10.1161/01.str.0000033931.62992.b1. [DOI] [PubMed] [Google Scholar]
- 137.Pradilla G, Thai Q-A, Legnani FG, Hsu W, Kretzer RM, Wang PP, Tamargo RJ. Neurosurgery. 2004;55:1393. doi: 10.1227/01.neu.0000143615.26102.1a. [DOI] [PubMed] [Google Scholar]
- 138.Momin EN, Schwab KE, Chaichana KL, Miller-Lotan R, Levy AP, Tamargo RJ. Neurosurgery. 2009;65:937. doi: 10.1227/01.NEU.0000356974.14230.B8. [DOI] [PubMed] [Google Scholar]
- 139.Clatterbuck RE, Gailloud P, Tierney T, Clatterbuck VM, Murphy KJ, Tamargo RJ. J Neurosurg. 2005;103:745. doi: 10.3171/jns.2005.103.4.0745. [DOI] [PubMed] [Google Scholar]
- 140.Tierney TS, Pradilla G, Wang PP, Clatterbuck RE, Tamargo RJ. Neurosurgery. 2006;58:952. doi: 10.1227/01.NEU.0000210182.48546.8F. [DOI] [PubMed] [Google Scholar]
- 141.Cabrales P, Han G, Nacharaju P, Friedman AJ, Friedman JM. Am J Physiol Heart Circ Physiol. 2011;300:H49. doi: 10.1152/ajpheart.00665.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cabrales P, Han G, Roche C, Nacharaju P, Friedman AJ, Friedman JM. Free Radical Biol Med. 2010;49:530. doi: 10.1016/j.freeradbiomed.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Laumann EO, Paik A, Rosen RC. JAMA J Am Med Assoc. 1999;281:537. doi: 10.1001/jama.281.6.537. [DOI] [PubMed] [Google Scholar]
- 144.Jha S, Thakar R. Eur J Obstet Gynecol Reprod Biol. 2010;153:117. doi: 10.1016/j.ejogrb.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 145.Caruso S, Intelisano G, Farina M, Di Mari L, Agnello C. Eur J Obstet Gynecol Reprod Biol. 2003;110:201. doi: 10.1016/s0301-2115(03)00118-0. [DOI] [PubMed] [Google Scholar]
- 146.Dirim A, Goren MR, Peskircioglu L. J Sex Med. 2011;8:800. doi: 10.1111/j.1743-6109.2010.02096.x. [DOI] [PubMed] [Google Scholar]
- 147.Traish AM, Botchevar E, Kim NN. J Sex Med. 2010;7:2925. doi: 10.1111/j.1743-6109.2010.01903.x. [DOI] [PubMed] [Google Scholar]
- 148.Yoo J-W, Acharya G, Lee CH. Biomaterials. 2009;30:3978. doi: 10.1016/j.biomaterials.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 149.Souto S, Palma P, Seabra AB, Fregonesi A, Palma T, Reis LO. J Sex Med. 2011;8:484. doi: 10.1111/j.1743-6109.2010.02045.x. [DOI] [PubMed] [Google Scholar]
- 150.Yoo J-W, Choe E-S, Ahn S-M, Lee CH. Biomaterials. 2010;31:552. doi: 10.1016/j.biomaterials.2009.09.058. [DOI] [PubMed] [Google Scholar]
- 151.Ramsey SD, Newton K, Blough D, McCulloch DK, Sandhu N, Reiber GE, Wagner EH. Diabetes Care. 1999;22:382. doi: 10.2337/diacare.22.3.382. [DOI] [PubMed] [Google Scholar]
- 152.White R, McIntosh C. J Wound Care. 2008;17:426. doi: 10.12968/jowc.2008.17.10.31305. [DOI] [PubMed] [Google Scholar]
- 153.White R, McIntosh C. J Wound Care. 2009;18:335. doi: 10.12968/jowc.2009.18.8.43633. [DOI] [PubMed] [Google Scholar]
- 154.Luo J-D, Wang Y-Y, Fu W-L, Wu J, Chen AF. Circulation. 2004;110:2484. doi: 10.1161/01.CIR.0000137969.87365.05. [DOI] [PubMed] [Google Scholar]
- 155.Schäffer MR, Tantry U, Efron PA, Ahrendt GM, Thornton FJ, Barbul A. Surgery. 1997;121:513. doi: 10.1016/s0039-6060(97)90105-7. [DOI] [PubMed] [Google Scholar]
- 156.Witte MB, Kiyama T, Barbul A. Br J Surg. 2002;89:1594. doi: 10.1046/j.1365-2168.2002.02263.x. [DOI] [PubMed] [Google Scholar]
- 157.Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J, Frank S. J Invest Dermatol. 1999;113:1090. doi: 10.1046/j.1523-1747.1999.00784.x. [DOI] [PubMed] [Google Scholar]
- 158.Thornton FJ, Schäffer MR, Witte MB, Moldawer LL, MacKay SLD, Abouhamze A, Tannahill CL, Barbul A. Biochem Biophys Res Commun. 1998;246:654. doi: 10.1006/bbrc.1998.8681. [DOI] [PubMed] [Google Scholar]
- 159.Witte MB, Thornton FJ, Efron DT, Barbul A. Nitric Oxide. 2000;4:572. doi: 10.1006/niox.2000.0307. [DOI] [PubMed] [Google Scholar]
- 160.Frank S, Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J. FASEB J. 1999;13:2002. [PubMed] [Google Scholar]
- 161.Lee PC, Salyapongse AN, Bragdon GA, Shears LL, Watkins SC, Edington HDJ, Billiar TR. Am J Physiol Heart Circ Physiol. 1999;277:H1600. doi: 10.1152/ajpheart.1999.277.4.H1600. [DOI] [PubMed] [Google Scholar]
- 162.Luo J-d, Chen AF. Acta Pharmacol Sin. 2005;26:259. doi: 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- 163.Seabra AB, Fitzpatrick A, Paul J, De Oliveira MG, Weller R. Br J Dermatol. 2004;151:977. doi: 10.1111/j.1365-2133.2004.06213.x. [DOI] [PubMed] [Google Scholar]
- 164.Seabra AB, Pankotai E, Fehér M, Somlai Á, Kiss L, Bíró L, Szabó C, Kollai M, De Oliveira MG, Lacza Z. Br J Dermatol. 2007;156:814. doi: 10.1111/j.1365-2133.2006.07718.x. [DOI] [PubMed] [Google Scholar]
- 165.Amadeu TP, Seabra AB, de Oliveira MG, Monte-Alto-Costa A. J Surg Res. 2008;149:84. doi: 10.1016/j.jss.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 166.Simões MMdSG, de Oliveira MG. J Biomed Mater Res Part B: Appl Biomater. 2010;93B:416. doi: 10.1002/jbm.b.31598. [DOI] [PubMed] [Google Scholar]
- 167.Zhu H, Ka B, Murad F. World J Surg. 2007;31:624. doi: 10.1007/s00268-007-0727-3. [DOI] [PubMed] [Google Scholar]
- 168.Hardwick JB, Tucker AT, Wilks M, Johnston A, Benjamin N. Clin Sci. 2001;100:395. [PubMed] [Google Scholar]
- 169.Hetrick EM, Prichard HL, Klitzman B, Schoenfisch MH. Biomaterials. 2007;28:4571. doi: 10.1016/j.biomaterials.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jarvis WR. Infection Control Hosp Epidemiol. 1996;17:552. doi: 10.1086/647371. [DOI] [PubMed] [Google Scholar]
- 171.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Jacobson C, Smulders M, Gemmen E, Bharmal M. Clin Infect Dis. 2007;45:1132. doi: 10.1086/522186. [DOI] [PubMed] [Google Scholar]
- 172.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Smulders M, Lapetina E, Gemmen E. Arch Intern Med. 2005;165:1756. doi: 10.1001/archinte.165.15.1756. [DOI] [PubMed] [Google Scholar]
- 173.Anderson DJ, Arduino JM, Reed SD, Sexton DJ, Kaye KS, Grussemeyer CA, Peter SA, Hardy C, Choi YI, Friedman JY, Fowler VG., Jr Infection Control Hosp Epidemiol. 2010;31:701. doi: 10.1086/653205. [DOI] [PubMed] [Google Scholar]
- 174.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. N Engl J Med. 2006;355:666. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 175.Timofeeva L, Kleshcheva N. Appl Microbiol Biotechnol. 2011;89:475. doi: 10.1007/s00253-010-2920-9. [DOI] [PubMed] [Google Scholar]
- 176.Lowenstein CJ, Dinerman JL, Snyder SH. Ann Intern Med. 1994;120:227. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- 177.Nablo BJ, Prichard HL, Butler RD, Klitzman B, Schoenfisch MH. Biomaterials. 2005;26:6984. doi: 10.1016/j.biomaterials.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 178.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 179.Nablo BJ, Rothrock AR, Schoenfisch MH. Biomaterials. 2005;26:917. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 180.Nablo BJ, Schoenfisch MH. J Biomed Mater Res A. 2003;67A:1276. doi: 10.1002/jbm.a.20030. [DOI] [PubMed] [Google Scholar]
- 181.Nablo BJ, Schoenfisch MH. Biomacromolecules. 2004;5:2034. doi: 10.1021/bm049727w. [DOI] [PubMed] [Google Scholar]
- 182.Nablo BJ, Schoenfisch MH. Biomaterials. 2005;26:4405. doi: 10.1016/j.biomaterials.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 183.Engelsman AF, Krom BP, Busscher HJ, van Dam GM, Ploeg RJ, van der Mei HC. Acta Biomater. 2009;5:1905. doi: 10.1016/j.actbio.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 184.Martinez LR, Han G, Chacko M, Mihu MR, Jacobson M, Gialanella P, Friedman AJ, Nosanchuk JD, Friedman JM. J Invest Dermatol. 2009;129:2463. doi: 10.1038/jid.2009.95. [DOI] [PubMed] [Google Scholar]
- 185.Han G, Martinez LR, Mihu MR, Friedman AJ, Friedman JM, Nosanchuk JD. PLoS ONE. 2009;4:e7804. doi: 10.1371/journal.pone.0007804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Giustarini D, Rossi R, Milzani A, Dalle-Donne I. Meth Enzymol. 2008;440:361. doi: 10.1016/S0076-6879(07)00823-3. [DOI] [PubMed] [Google Scholar]