

Abstract
Background
Morphine stimulates angiogenesis and cancer progression in mice. We investigated whether morphine influences tumour onset, development, and animal model survival, and whether µ-opioid receptor (MOR), lymphangiogenesis, mast cell activation, and substance P (SP) are associated with the tumour-promoting effects of morphine.
Methods
Transgenic mice with a rat C3(1) simian virus 40 large tumour antigen fusion gene which demonstrate the developmental spectrum of human infiltrating ductal breast carcinoma were used. Mice were treated at different ages with clinically relevant doses of morphine or phosphate-buffered saline to determine the effect on tumour development and progression, and on mouse survival. Tumours were analysed for MOR, angiogenesis, lymphangiogenesis, SP, and mast cell activation by immunofluorescent- or laser scanning confocal-microscopy. Cytokine and SP levels were determined by enzyme-linked immunosorbent assay.
Results
Morphine did not influence tumour development when given before the onset of tumour appearance, but significantly promoted progression of established tumours, and reduced survival. MOR-immunoreactivity (ir) was observed in larger but not in smaller tumours. Morphine treatment resulted in increased tumour angiogenesis, peri-tumoural lymphangiogenesis, mast cell activation, and higher levels of cytokines and SP in tumours. SP-ir co-localized with mast cells and elsewhere in the tumours.
Conclusions
Morphine does not affect the onset of tumour development, but it promotes growth of existing tumours, and reduces overall survival in mice. MOR may be associated with morphine-induced cancer progression, resulting in shorter survival. Mast cell activation by morphine may contribute to increased cytokine and SP levels, leading to cancer progression and refractory pain.
Keywords: analgesics, opioid, morphine; angiogenesis, lymphangiogenesis; cancer; mast cells; μ-opioid receptor
Editor's key points.
Morphine activates cellular signalling pathways which can promote angiogenesis in mouse models of cancer.
High levels of MOR expression in metastatic prostate cancer are associated with reduced progression free survival.
This study uses a transgenic mouse model of cancer and investigates the effect of morphine on cancer onset and progresssion.
Morphine does not affect tumour onset, but it promotes development of established tumours with increased MOR expression.
Morphine and other opioids are used in escalating doses to control severe pain in cancer patients.1,2 While morphine exerts analgesia via µ-opioid receptor (MOP/MOR, termed MOR here) in the central nervous system (CNS), it also has direct effects on non-neural cells, including endothelial, tumour, and mast cells.3–6 Previous studies from our laboratory showed that morphine activates mitogen-activated protein kinase/extracellular signal-related kinase and Akt signalling pathways leading to the proliferation and survival of endothelial cells.3 Activation of these signalling pathways and cycloxygenase-2 signalling led to the promotion of angiogenesis, tumour growth, and metastasis and reduced survival in mice.7 Methylnaltrexone (MNTX), an opioid receptor antagonist, inhibits morphine-induced endothelial proliferation, angiogenesis, and disruption of endothelial barrier function.5,8 However, it remains to be determined whether morphine promotes cancer initiation, or whether it stimulates the growth of existing tumours without influencing the development of cancer.
In a retrospective analysis, we found that patients with metastatic prostate cancer requiring higher doses of opioids had shorter progression-free survival (PFS) and overall survival (OS).9 The presence of high MOR-immunoreactivity (ir) in the malignant areas of the prostate biopsies correlated strongly with shorter time to progression, PFS, and OS. Both opioid requirement and MOR-ir retained prognostic significance for PFS and OS in multivariable analyses that adjusted for known prognostic factors.
Opioids are known to stimulate mast cell activation resulting in the release of inflammatory cytokines, neuropeptides such as substance P (SP), and tryptase.6,10–12 High density of tryptase-positive mast cells were found to be associated with advanced stages of colorectal cancer.13 In murine breast cancer models, mast cells modulate the tumour microenvironment and facilitate metastases via their cell surface receptor c-Kit.14
Tryptase and SP released from mast cells activate peripheral nerve terminals leading to the release of more SP.6 In turn, SP further activates the nerve fibres leading to increased pain. Additionally, SP stimulates angiogenesis and promotes cancer progression.15 SP and its high affinity receptor neurokinin 1 (NK1) are highly expressed in HER2+ breast cancer and SP stimulates HER2 signalling and may contribute to drug resistance in breast cancer.16 Thus, mast cell activation by morphine may further exaggerate the pro-inflammatory, pro-nociceptive, and vasoactive tumour microenvironment.
In the present study, we used a transgenic mouse model that mimics the evolutionary spectrum of human breast cancer.17 In this mouse, distinct phases of tumour development, progression, metastasis, and survival can be evaluated, enabling analysis of the effect of opioids on tumour development and progression and also on survival.
Methods
Mouse studies were performed with approval from the University of Minnesota's Institutional Care and Use Committee (IACUC, Protocol #0703A03589 and 1212-30170A). Detailed procedures are described in the Supplementary Appendix.
Mice
Female, transgenic mice with a rat C3(1) simian virus 40 large tumour antigen fusion gene (called C3TAG mice henceforth), which causes highly invasive breast tumours were used.17 These mice develop ductal epithelial atypia around 8 weeks of age that progresses to intraepithelial neoplasia around 12 weeks, and to grossly palpable tumours and invasive carcinoma around 16 weeks. Tumours principally metastasize haematogenously to the lungs, but also to the adrenals, liver, and heart. By 6 months of age, mice die due to universal development of multifocal mammary adenocarcinomas. This model was chosen because the effect of morphine could be distinguished on distinct phases of tumour development (between 8 and 12 weeks), tumour growth (12 weeks onwards), metastases (16 weeks onwards), and survival.18
Drugs and treatments
Mice were injected subcutaneously with either phosphate-buffered saline (PBS) or morphine sulphate (MS; Baxter Esilerderle Healthcare, Cherry Hill, NJ, USA) at 0.5 mg kg−1 day−1 for 2 weeks, followed by dose escalation every 2 weeks as follows: 0.75, 1.0, 1.25 mg kg−1 day−1, and finally to 1.5 mg kg−1 day−1 for the duration of each study as indicated. Mice were divided into three groups to examine the effect of morphine on: (i) tumour development/onset: 6-week-old mice were injected with morphine for 7 weeks; (ii) tumour growth/progression: 3-month-old mice were injected with morphine for 7 weeks; (iii) survival: 3-month-old mice were injected with morphine until they became moribund, which was considered the end of survival as per our animal ethics policy.
Tumour burden
At the end of treatment, mice were euthanized and all visible tumours throughout the body were dissected out. Tumour numbers and tumour weight/mouse were recorded as measures of tumour burden.
Immunofluorescent staining for MOR, CD31, and lymphatic vessel endothelium hyaluranon receptor-1
Tumours were cryosectioned into 6 µm thick sections and immunostained with: goat anti-lymphatic vessel endothelium hyaluranon receptor-1 (LYVE-1) (1:500, R&D Systems, Minneapolis, MN, USA), rat anti-CD31 (1:200, Santa Cruz Biotechnology, Dallas, TX, USA), and rabbit anti-MOR (1:100, Millipore, Billerica, MA, USA) as described by us.6,7,18–20 Anti-MOR antibody was validated for MOR specificity using skin sections from MOR-knockout mice as described earlier (Supplementary Fig. S1).19
Mast cell analysis
Approximately 6 µm thick tumour sections were stained with Toluidine blue and enumerated as described previously.6,21
Laser scanning confocal microscopy of tumour sections
Approximately 6 µm thick tumour sections were fixed in 4% paraformaldehyde, blocked with 3% donkey serum and permeabilized with 0.03% Triton X-100 (all from Sigma-Aldrich, St Louis, MO, USA). Sections were then incubated with primary antibodies, rabbit anti-SP (1:50, AbD Serotec, Raleigh, NC, USA), rat anti-CD31 (1:200, Santa Cruz, CA, USA), goat anti-c-Kit (1:100, BD Bioscience, San Jose, CA, USA), and rabbit anti-FcεR 1 (eBioscience, San Diego, CA, USA) as described by us.6
Enzyme-linked immunosorbent assay for cytokines
Supernatants from tumour lysates were analysed for: tryptase (American Research Products, Inc., Waltham, MA, USA), β-hexosaminidase (Cedarlane Labs, Burlington, NC, USA), granulocyte macrophage colony-stimulating factor (GM-CSF), regulated on activation normal T-cell expressed and secreted (RANTES), SP, and interleukin-6 (IL-6; all from R&D Systems) after the manufacturer's instructions.
Statistical analysis
Data were analysed with Prism software (v 5.0a, GraphPad Prism Inc., San Diego, CA, USA ). A P-value of <0.05 was considered significant. Analysis of variance with Bonferroni's correction was used to correlate the responses between treatments and Student's t-test was used to analyse the significance between PBS and morphine treatment. A Kaplan–Meier analysis was performed to analyse survival data. All data are presented as mean (sem).
Results
Influence of morphine on tumour development and progression, and survival of mice
To examine tumour development/onset, which occurs between 8 and 12 weeks of age in C3TAG mice, we started morphine or PBS injections before this age (at 6 weeks) and continued the treatments for the next 7 weeks (until 13 weeks of age). Morphine treatment did not have a significant effect on tumour burden (weight and number) when compared with PBS treatment (Fig. 1a and b). Both sets of mice developed palpable tumours by 12 weeks of age, as expected.
Fig 1.

Influence of morphine on cancer outcomes in C3TAG mice. Tumour burden (weight and number per mouse) and survival of mice after treatments are shown. (a and b) Six-week-old mice were treated for 7 weeks. n=4 for PBS and 5 for MS. (c and d) Three-month-old mice were treated for 7 weeks with, either PBS, MS, NAL, or MS and NAL. n=6 per treatment. (e) Three-month-old mice with palpable mammary tumours were injected with either MS or PBS until they became moribund (considered end of survival). n=40 for PBS and 17 for morphine. The Kaplan–Meier curve for survival is shown. MS, morphine; NAL, naloxone; PBS, phosphate-buffered saline.
Next, to examine the influence of morphine on the growth of established tumours, we treated 3-month-old mice with morphine or PBS for 7 weeks. Morphine treatment of 3-month-old mice led to significant increases in tumour burden (weight and number) when compared with PBS treatment (Fig. 1c and d). Morphine-induced tumour growth was antagonized by co-treatment with the opioid receptor antagonist naloxone, suggestive of an opioid receptor-mediated mechanism. The Kaplan–Meier analysis showed that continued treatment of 3-month-old mice with morphine resulted in significantly shorter survival when compared with PBS treatment (Fig. 1e).
Together, these data demonstrate that morphine does not influence the onset of tumour development, but promotes the growth of existing tumours via an opioid receptor-mediated mechanism, resulting in shorter survival of tumour-bearing mice.
MOR expression in C3TAG tumours, and effect of morphine on angiogenesis and lymphangiogenesis
MOR-ir was considerably more intense in large (established) tumours when compared with small (just developed) tumours (Fig. 2a). MOR-ir co-localized with endothelial and with non-endothelial cells (Fig. 2a, bottom left panel). Larger tumours also showed appreciably more angiogenesis and lymphangiogenesis when compared with smaller tumours (data not shown).
Fig 2.

Higher MOR expression in growing tumours drives morphine-induced angiogenesis and lymphangiogenesis. (a) Cryosections of large and small tumours showing MOR-ir (green), vasculature (CD31-ir, red), and nuclei (DAPI, blue). Bottom row shows co-localization of MOR-ir with vasculature (yellow). Magnification ×1000; scale bar, 50 µm. (b). Three-month-old mice were treated with morphine or PBS for 7 weeks. Immunostaining of tumour sections co-stained for lymphatic vessel endothelium (LYVE-1, green), blood vessels (CD31, red), and cell nuclei (DAPI, blue) is shown. Magnification ×100; scale bar, 500 µm. Each image in (a) and (b) represents six different tumours.
Morphine stimulated angiogenesis and lymphangiogenesis in the tumours of 3-month-old mice treated for 7 weeks (Fig. 2b). Tumour vasculature in morphine-treated mice was more dense and had more dilated vessels when compared with PBS-treated mice. Lymphatic vessels were organized in a peri-tumoural fashion and also appeared to be more dilated in morphine-treated mice. These data suggest that MOR expression, angiogenesis, and lymphangiogenesis increase as tumours grow, and are further stimulated by morphine treatment in association with morphine-induced tumour growth.
Morphine promotes mast cell activation in tumours
Toluidine blue and FcεR1 and c-Kit staining revealed larger and significantly higher number of mast cells in tumours from mice treated with morphine (Fig. 3a and b). Additionally, tumour sections from morphine-treated mice exhibited intense mast cell degranulation, whereas PBS-treated mice showed few or no degranulating mast cells (Fig. 3a). Mast cells were co-localized with the tumour vasculature or in close proximity to the vasculature in both morphine- and PBS-treated mice. However, the levels of tumour tryptase and β-hexosaminidase, markers of mast cell activation, were significantly higher in morphine-treated mice when compared with PBS-treated mice (Fig. 3d and e). These findings suggest that morphine may influence tumour growth via the activation of mast cells, in addition to promoting angiogenesis and lymphangiogenesis.
Fig 3.

Influence of morphine on mast cell activation in tumours of C3TAG mice. Three-month-old mice were treated with PBS or morphine for 7 weeks. (a) Toluidine blue-stained tumour sections showing intense degranulation in morphine-treated mouse tumour sections. Magnification ×900; scale bar, 200 µm. n=10 per image. (b) Number of mast cells enumerated using Toluidine blue staining is shown as mean (sem) from 10 different tumours. (c) Tumours sections were co-stained with mast cell markers FcεR1 (red) and c-Kit (blue), and CD31 (red, blood vessels). Intense mast cell degranulation is observed in the morphine group. Mast cells are closely associated with the vasculature (magenta) or are in close proximity to the vasculature. Magnification ×1200; scale bar, 20 µm. (d and e) Tryptase and β-hexosaminidase, markers of mast cell activation, were analysed in tumour lysates by ELISA. Each bar shows mean (sem) from 7 to 8 different tumours.
Morphine treatment leads to increased cytokine expression in tumours
Because activated mast cells release cytokines and are also themselves activated by cytokines, we examined whether morphine treatment amplifies the release of GM-CSF, RANTES, and IL-6. Both GM-CSF and RANTES are involved in the recruitment, differentiation, and proliferation of mast cells. An enzyme-linked immunosorbent assay (ELISA) assay showed significantly higher concentrations of GM-CSF, RANTES, and IL6 in tumours of mice treated with morphine when compared with PBS (Fig. 4a–c). These data suggest that morphine stimulates the positive feedback loop of GM-CSF and RANTES release and the recruitment, differentiation, and degranulation of mast cells.
Fig 4.
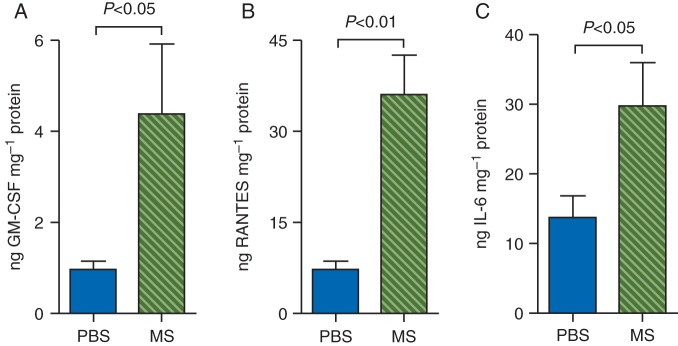
Morphine stimulates cytokine release in the tumours. Three-month-old mice were treated with PBS or morphine for 7 weeks. The concentrations of GM-CSF, RANTES, or IL-6 in tumour lysates were determined using ELISA assays. Each bar represents mean (sem) values from 5 to 8 different tumours.
Morphine triggers the release of SP
We observed greater SP-ir and significantly higher concentration of SP in tumours of morphine-treated mice compared with PBS-treated mice (Fig. 5a and b). SP-ir co-localized with mast cells and with other cells (Fig. 5a). These data suggest that morphine may promote neuro-inflammation leading to cancer progression and pain by stimulating SP release via mast cell degranulation (Fig. 6).
Fig 5.
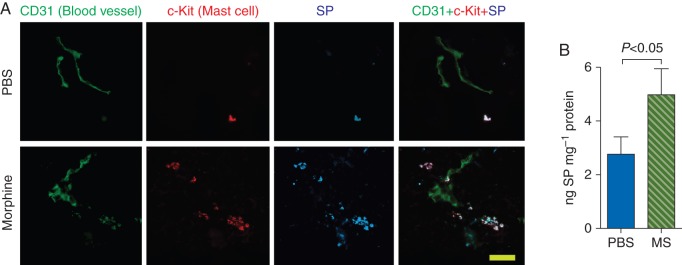
Morphine stimulates SP release in the tumours. Three-month-old mice were treated with PBS or morphine for 7 weeks. (a) Tumour sections immunostained for CD31 (blood vessels, green), c-Kit (mast cells, red), and SP (blue) are shown. Lane 4 shows the co-localization of SP with mast cells on and near the vasculature (white). Each image in each row is from the same field of view. Magnification ×1000; scale bar, 25 µm. n=6 for each image. (b) SP concentration in whole tumour lysates determine by ELISA is shown as the mean (sem) from 7 different tumours. SP, substance P.
Fig 6.

Model showing how morphine may stimulate a vicious cycle of SP release, pain, and inflammation. Morphine treatment stimulates mast cell activation in the tumours. Degranulating mast cells then release SP and tryptase, which may enhance peripheral/intra-tumoural nerve activation leading to pain. Activated nerve fibres may release more SP in turn, thus increasing neuro-inflammation and driving a vicious cycle of mast cell activation, inflammation, disease progression, and pain.
Discussion
We demonstrate that morphine does not influence cancer initiation, but it stimulates the progression of spontaneously developed breast tumours and shortens the survival of tumour-bearing mice. MOR-ir was undetectable in small tumours, but larger tumours demonstrated dense MOR-ir that co-localized with vascular and non-vascular compartments of the tumours. Morphine also promoted lymphangiogenesis, mast cell activation, and degranulation, and in the tumour microenvironment, it increased the levels of inflammatory cytokines, tryptase, and SP; the combination of these effects may underlie morphine-induced tumour progression.
Earlier studies from our laboratory and on the influence of morphine used subcutaneously or orthotopically xenografted tumour cells that develop into localized tumours in mice.3,7,22–24 Owing to distinct time differences (in weeks) between the development and progression phase of tumours in mice used in the present study, we could determine that morphine was not involved in the initiation of breast cancer in this model. This is a critical question with a bearing on the treatment of chronic non-cancer pain. Consistent with our pre-clinical findings, we found that chronic opioid treatment for non-cancer pain was not associated with higher grade or stage of subsequently diagnosed prostate cancer in patients (unpublished observations).
In human studies, it is difficult to separate the possibility that higher opioid exposure causes tumour progression from the possibility that opioid requirement is higher because of tumour progression that increases pain. In the current study, a pre-determined escalating dose schedule of morphine was administered to the entire experimental group starting at the same age. It can therefore confidently be concluded that at least in this mouse model that mimics human breast cancer, morphine itself promotes tumour progression and reduces survival via the cellular and molecular effects identified herein.
It is also apparent from this mouse model that at initiation, tumours do not express MOR, but that MOR is expressed as tumours enlarge. This appears to be due to the cytokines and growth factors, which increase as tumours grow. Earlier, we showed that vascular endothelial growth factor (VEGF) stimulates MOR expression in endothelial cells.25 Other cytokines including IL-6 and TNF-α increase MOR expression in a variety of cells.26,27 Higher MOR-ir in enlarging tumours coincides with the potentiation of tumour growth after morphine treatment. Morphine showed no effect on the initiation of tumour growth, likely because of the lack of MOR in the tumours. Consistent with this phenomenon, we found that patients with advanced (metastatic) prostate cancer expressing higher levels of MOR in prostate cancer tissue had significantly worse disease outcomes than those with lower MOR expression.9
Endogenous opioid levels are increased in the blood of patients with breast cancer and in the tumour tissue in human melanoma.28,29 Therefore, endogenous opioids themselves may interact with MOR in tumours, resulting in disease progression, even without exogenously administered opioids. In MOR knockout mice, lung cancer progression and metastases were reduced when compared with wild-type mice, in the absence of pharmacological opioid administration.22 Similar findings were reported in a mouse model of melanoma.29 In our study, the opioid receptor antagonist naloxone inhibited morphine-induced tumour progression, suggesting that morphine acts via an opioid receptor-mediated mechanism. These findings indicate that MOR expression in tumours is closely associated with disease outcomes, and that endogenous MOR may have a cancer-promoting effect that is further potentiated by treatment with morphine.
Human studies from our group and others have reported that endogenous MOR expression and opioid exposure correlate with disease progression, survival, or both.9,30,31 MOR-ir and MOR binding sites were significantly greater in cancerous tissue when compared with adjacent non-cancerous normal human lung3,22,23,32,33 or prostate9 tissue. Therefore, our observations further underscore the importance of tumour-MOR expression in promoting metastases, leading to reduced survival.
In addition to the tumour vasculature, lymphatic vessels are also involved in tumour metastasis.34 Lymphatic vessel invasion is a strong adverse prognostic factor for survival in patients with breast cancer.35 Since peri-tumoural, rather than intra-tumour, lymphatics are known to be functional in the context of dissemination of tumour cells,34 the higher density of peri-tumoural lymphatics in morphine-treated mice observed in the present study may play a role in dissemination of tumour cells via lymph nodes to distant sites, contributing to the shorter survival of tumour-bearing mice.
Morphine stimulates mast cell degranulation.6 This may have consequences on cancer progression and pain. While mast cells release SP, tryptase released from mast cells stimulates more release of SP via protease-activated receptor 2 (PAR2) on peripheral nerve fibres.6,36 SP stimulates vascular permeability and angiogenesis and also activates mast cells and nerve fibres, leading to vascular dysfunction, inflammation, and pain.37–40 In the present study, we found that morphine treatment led to an increase in GM-CSF and RANTES which are released by mast cells and also act on mast cell differentiation and proliferation.
Increased mast cell tryptase in the periphery, dorsal root ganglia, and spinal cord of mice with paclitaxel-induced neuropathic pain activates PAR2, and mediates the transition from acute to chronic pain in a murine cancer model.41,42 Mast cells promote the growth of pancreatic ductal adenocarcinoma (PDAC) in mice, and correlate with higher recurrence rates and shorter survival in patients.43 Additionally, SP directly influences cancer progression.15,16 SP transactivates HER2 in cancer cells derived from human breast cancer, and an NK1 receptor inhibitor ameliorated tumour growth in vivo in HER2- and EGFR-expressing tumours in mice.16 SP treatment of breast cancer cells inhibited their responsiveness to the EGFR and HER2 tyrosine kinase inhibitors, whereas treatment with an NK1 receptor inhibitor appeared to synergistically augment the therapeutic activity of EGFR/HER2 inhibitors. Therefore, morphine-induced SP in tumours may influence cancer progression, pain, and response to targeted therapies.
Thus, mast cell degranulation releases tryptase, which activates SP release from nerve endings, thereby contributing to sustained neurogenic inflammation and pain in cancer (Fig. 6). Mast cells and MOR may be potential therapeutic targets for simultaneously ameliorating pain and cancer progression.
Supplementary material
Supplementary material is available at British Journal of Anaesthesia online.
Authors’ contributions
J.N. performed mast cell, and cytokine analysis, all data analysis, and wrote the manuscript. K.L., D.V., L.V., S.R., and R.S. co-analysed data and performed ELISA, and neuropeptide analysis and co-wrote the manuscript. W.S. performed angiogenesis and lymphangiogenesis. Y.L. treated the mice and collected data on tumour burden and survival. P.G. advised on clinical relationship, data analysis, editing, and interpretation. K.G. conceived, designed, planned, and supervised the entire study, analysed and interpreted data, and edited the manuscript.
Declaration of interest
None declared.
Funding
This work was supported by NIH RO1 s CA109582, HL68802, 103773, and UO1 HL117664-01 to K.G.
Acknowledgements
We thank Stefan Kren, Katherine N.H. Johnson, and Sugandha Rajput for breeding, genotyping, phenotyping mice, and/or technical assistance; and Carol Taubert for editorial assistance.
References
- 1.Brogan SE, Winter NB. Patient-controlled intrathecal analgesia for the management of breakthrough cancer pain: a retrospective review and commentary. Pain Med. 2011;12:1758–68. doi: 10.1111/j.1526-4637.2011.01262.x. [DOI] [PubMed] [Google Scholar]
- 2.Greco MT, Corli O, Montanari M, et al. Epidemiology and pattern of care of breakthrough cancer pain in a longitudinal sample of cancer patients: results from the Cancer Pain Outcome Research Study Group. Clin J Pain. 2011;27:9–18. doi: 10.1097/AJP.0b013e3181edc250. [DOI] [PubMed] [Google Scholar]
- 3.Gupta K, Kshirsagar S, Chang L, et al. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62:4491–8. [PubMed] [Google Scholar]
- 4.Farooqui M, Geng ZH, Stephenson EJ, et al. Naloxone acts as an antagonist of estrogen receptor activity in MCF-7 cells. Mol Cancer Ther. 2006;5:611–20. doi: 10.1158/1535-7163.MCT-05-0016. [DOI] [PubMed] [Google Scholar]
- 5.Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res. 2006;72:3–11. doi: 10.1016/j.mvr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Vincent L, Vang D, Nguyen J, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122:1853–62. doi: 10.1182/blood-2013-04-498105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooqui M, Li Y, Rogers T, et al. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumour growth, metastasis and mortality, without compromising analgesia. Br J Cancer. 2007;97:1523–31. doi: 10.1038/sj.bjc.6604057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singleton PA, Moreno-Vinasco L, Sammani S, et al. Attenuation of vascular permeability by methylnaltrexone: role of mOP-R and S1P3 transactivation. Am J Respir Cell Mol Biol. 2007;37:222–31. doi: 10.1165/rcmb.2006-0327OC. [DOI] [PubMed] [Google Scholar]
- 9.Zylla D, Gourley BL, Vang D, et al. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer. 2013;119:4103–10. doi: 10.1002/cncr.28345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dybendal T, Guttormsen AB, Elsayed S, et al. Screening for mast cell tryptase and serum IgE antibodies in 18 patients with anaphylactic shock during general anaesthesia. Acta Anaesthesiol Scand. 2003;47:1211–8. doi: 10.1046/j.1399-6576.2003.00237.x. [DOI] [PubMed] [Google Scholar]
- 11.Kolaczkowska E, Seljelid R, Plytycz B. Critical role of mast cells in morphine-mediated impairment of zymosan-induced peritonitis in mice. Inflamm Res. 2001;50:415–21. doi: 10.1007/PL00000264. [DOI] [PubMed] [Google Scholar]
- 12.Veien M, Szlam F, Holden JT, et al. Mechanisms of nonimmunological histamine and tryptase release from human cutaneous mast cells. Anesthesiology. 2000;92:1074–81. doi: 10.1097/00000542-200004000-00026. [DOI] [PubMed] [Google Scholar]
- 13.Malfettone A, Silvestris N, Saponaro C, et al. High density of tryptase-positive mast cells in human colorectal cancer: a poor prognostic factor related to protease-activated receptor 2 expression. J Cell Mol Med. 2013;17:1025–37. doi: 10.1111/jcmm.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy LD, Curry JM, Sahraei M, et al. Arthritis augments breast cancer metastasis: role of mast cells and SCF/c-Kit signaling. Breast Cancer Res. 2013;15:R32. doi: 10.1186/bcr3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz M, Covenas R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides. 2013;48:1–9. doi: 10.1016/j.peptides.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Recio S, Fuster G, Fernandez-Nogueira P, et al. Substance P autocrine signaling contributes to persistent HER2 activation that drives malignant progression and drug resistance in breast cancer. Cancer Res. 2013;73:6424–34. doi: 10.1158/0008-5472.CAN-12-4573. [DOI] [PubMed] [Google Scholar]
- 17.Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci USA. 1994;91:11236–40. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luk K, Boatman S, Johnson KN, et al. Influence of morphine on pericyte–endothelial interaction: implications for antiangiogenic therapy. J Oncol. 2012;2012:458385. doi: 10.1155/2012/458385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber ML, Chen C, Li Y, et al. Morphine stimulates platelet-derived growth factor receptor-β signalling in mesangial cells in vitro and transgenic sickle mouse kidney in vivo. Br J Anaesth. 2013;111:1004–12. doi: 10.1093/bja/aet221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood. 2010;116:456–65. doi: 10.1182/blood-2010-01-260372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arck PC, Handjiski B, Peters EM, et al. Topical minoxidil counteracts stress-induced hair growth inhibition in mice. Exp Dermatol. 2003;12:580–90. doi: 10.1034/j.1600-0625.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 22.Lennon FE, Mirzapoiazova T, Mambetsariev B, et al. Overexpression of the mu-opioid receptor in human non-small cell lung cancer promotes Akt and mTOR activation, tumor growth, and metastasis. Anesthesiology. 2012;116:857–67. doi: 10.1097/ALN.0b013e31824babe2. [DOI] [PubMed] [Google Scholar]
- 23.Mathew B, Lennon FE, Siegler J, et al. The novel role of the mu opioid receptor in lung cancer progression: a laboratory investigation. Anesth Analg. 2011;112:558–67. doi: 10.1213/ANE.0b013e31820568af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tegeder I, Grosch S, Schmidtko A, et al. G protein-independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: involvement of p53 phosphorylation. Cancer Res. 2003;63:1846–52. [PubMed] [Google Scholar]
- 25.Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr Neurovasc Res. 2006;3:171–80. doi: 10.2174/156720206778018767. [DOI] [PubMed] [Google Scholar]
- 26.Kraus J. Regulation of mu-opioid receptors by cytokines. Front Biosci (Schol Ed) 2009;1:164–70. doi: 10.2741/s16. [DOI] [PubMed] [Google Scholar]
- 27.Gupta M, Li Y, Gupta K. Opioids as promoters and regulators of angiogenesis. In: Maragoudakis ME, Papadimitriou E, editors. Angiogenesis: Basic Science and Clinical Applications. Trivandrum, Kerala: Transworld Research Network; 2007. pp. 303–17. [Google Scholar]
- 28.Kajdaniuk D, Marek B, Swietochowska E, Ciesielska-Kopacz N, Buntner B. Is positive correlation between cortisol and met-enkephalin concentration in blood of women with breast cancer a reaction to stress before chemotherapy administration? Pathophysiology. 2000;7:47–51. doi: 10.1016/s0928-4680(00)00028-6. [DOI] [PubMed] [Google Scholar]
- 29.Boehncke S, Hardt K, Schadendorf D, et al. Endogenous mu-opioid peptides modulate immune response towards malignant melanoma. Exp Dermatol. 2011;20:24–8. doi: 10.1111/j.1600-0625.2010.01158.x. [DOI] [PubMed] [Google Scholar]
- 30.Smith TJ, Staats PS, Deer T, et al. Randomized clinical trial of an implantable drug delivery system compared with comprehensive medical management for refractory cancer pain: impact on pain, drug-related toxicity, and survival. J Clin Oncol. 2002;20:4040–9. doi: 10.1200/JCO.2002.02.118. [DOI] [PubMed] [Google Scholar]
- 31.Singleton PA, Mirzapoiazova T, Salgia R, Moss J. Increased μ opioid receptor expression in metastatic lung cancer. Br J Anaesth. 2014;113(S1):i96–i101. doi: 10.1093/bja/aeu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujioka N, Nguyen J, Chen C, et al. Morphine-induced epidermal growth factor pathway activation in non-small cell lung cancer. Anesth Analg. 2011;113:1353–64. doi: 10.1213/ANE.0b013e318232b35a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madar I, Bencherif B, Lever J, et al. Imaging delta- and mu-opioid receptors by PET in lung carcinoma patients. J Nucl Med. 2007;48:207–13. [PubMed] [Google Scholar]
- 34.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–76. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 35.Gujam FJ, Going JJ, Edwards J, Mohammed ZM, McMillan DC. The role of lymphatic and blood vessel invasion in predicting survival and methods of detection in patients with primary operable breast cancer. Crit Rev Oncol Hematol. 2014;89:231–41. doi: 10.1016/j.critrevonc.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Vergnolle N, Wallace JL, Bunnett NW, Hollenberg MD. Protease-activated receptors in inflammation, neuronal signaling and pain. Trends Pharmacol Sci. 2001;22:146–52. doi: 10.1016/s0165-6147(00)01634-5. [DOI] [PubMed] [Google Scholar]
- 37.Rosa AC, Fantozzi R. The role of histamine in neurogenic inflammation. Br J Pharmacol. 2013;170:38–45. doi: 10.1111/bph.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaik-Dasthagirisaheb YB, Varvara G, Murmura G, et al. Vascular endothelial growth factor (VEGF), mast cells and inflammation. Int J Immunopathol Pharmacol. 2013;26:327–35. doi: 10.1177/039463201302600206. [DOI] [PubMed] [Google Scholar]
- 39.Kandere-Grzybowska K, Gheorghe D, Priller J, et al. Stress-induced dura vascular permeability does not develop in mast cell-deficient and neurokinin-1 receptor knockout mice. Brain Res. 2003;980:213–20. doi: 10.1016/s0006-8993(03)02975-5. [DOI] [PubMed] [Google Scholar]
- 40.Amadesi S, Reni C, Katare R, et al. Role for substance p-based nociceptive signaling in progenitor cell activation and angiogenesis during ischemia in mice and in human subjects. Circulation. 2012;125:1774–86. doi: 10.1161/CIRCULATIONAHA.111.089763. S1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440–51. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- 42.Lam DK, Schmidt BL. Serine proteases and protease-activated receptor 2-dependent allodynia: a novel cancer pain pathway. Pain. 2010;149:263–72. doi: 10.1016/j.pain.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang DZ, Ma Y, Ji B, et al. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:7015–23. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]