

Abstract
Inhibition of the catalytic subunit of the heterodimeric methionine S-adenosyl transferase-2 (MAT2A) with fluorinated N,N-dialkylaminostilbenes (FIDAS agents) offers a potential avenue for the treatment of liver and colorectal cancers where upregulation of this enzyme occurs. A study of structure–activity relationships led to the identification of the most active compounds as those with (1) either a 2,6-difluorostyryl or 2-chloro-6-fluorostyryl subunit, (2) either an N-methylamino or N,N-dimethylamino group attached in a para orientation relative to the 2,6-dihalostyryl subunit, and (3) either an N-methylaniline or a 2-(N,N-dimethylamino)pyridine ring. These modifications led to FIDAS agents that were active in the low nanomolar range, that formed water-soluble hydrochloride salts, and that possessed the desired property of not inhibiting the human hERG potassium ion channel at concentrations at which the FIDAS agents inhibit MAT2A. The active FIDAS agents may inhibit cancer cells through alterations of methylation reactions essential for cancer cell survival and growth.
Introduction
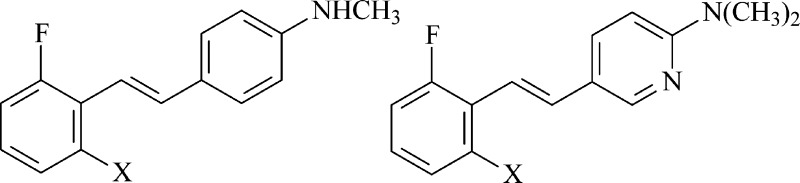
The development of antineoplastic agents with novel molecular targets opens the door to new, potentially valuable treatment strategies. We previously reported the effect of difluorinated N,N′-dialkylaminostilbenes (FIDAS agents) on the proliferation of colon and liver cancer cells. We identified (E)-4-(2′,6′-difluorostyryl)-N,N-dimethylaniline (FIDAS 1a, Figure 1) as a lead structure with in vitro activity in LS174T cells in the low micromolar range1−3 and utilized a biotinylated analogue of this FIDAS agent to identify the catalytic subunit of the heterodimeric enzyme, methionine S-adenosyl transferase-2 (MAT2A), as the sole binding partner.2 We validated MAT2A as the target by suppression using shRNA, which demonstrated in vitro depression in S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH) levels in cells exposed to FIDAS agents. Also, we demonstrated in vivo activity of these FIDAS agents on human colon cancer using mouse xenograft studies1 and in a three-dimensional culture model of primary CRC organoids that mimics the microenvironment of tumors (Supporting Information Figure S2). Other reports describe hydroxy- or methoxy-substituted stilbenes as potential antineoplastic agents,4−10 but experience directed our study of structure–activity relationships (SAR) away from oxygenated stilbenes because of their potential for redox reactions and off-target biological effects.
Figure 1.

FIDAS agents: Top row contains FIDAS agents in the aniline family; second row contains FIDAS agents in the 5-aminopyridine family; third row contains FIDAS agents in the 2-aminopyridine family; and bottom row contains FIDAS agents in the 2-aminopyrimidine family.
Since FIDAS agents offer a potentially new avenue for the treatment of liver and colorectal cancers where MAT2 levels are upregulated,11−14 we sought analogues of FIDAS 1a with improved (nanomolar) potency, water solubility, and the desired property of not interacting with the human ether-à-go-go-related (hERG) potassium channel. In general, drug candidates must avoid hERG activation to progress toward investigational new drug (IND) status, and our commitment to moving FIDAS agents down this pathway led us to incorporate hERG testing in our evaluation of candidates produced in this SAR study. Binding to the hERG channel results in QT interval prolongation in the electrocardiogram and adverse cardiac events.15 Drug-induced ventricular fibrillation, in these cases, may lead to sudden death and hence the need to identify drug candidates that do not bind hERG at concentrations of the FIDAS agents that inhibit MAT2A.
Results
Synthesis of FIDAS Agents
A Wadsworth–Emmons condensation of 2,6-dihalobenzyl diethyl phosphonates with the appropriate 4-(N,N-dimethylamino)benzaldehydes secured the (E)-4-(2′,6′-dihalostyryl)-N,N-dimethylanilines (1a–3a) (Figure 2).17 Demethylation of 1a–3a using cyanogen bromide18 secured the (E)-4-(2′,6′-dihalostyryl)-N-methylanilines (1b–3b). Synthesis of the corresponding pyridine and pyrimidine analogues in the N,N-dimethylamino series also employed the Wadsworth–Emmons condensation of the 2,6-dihalobenzyl diethyl phosphonates with the appropriate pyridine-2-carboxaldehyde, pyridine-3-carboxaldehyde, and pyrimidine-5-carboxaldehyde and secured (E)-2-(2′,6′-dihalostyryl)-5-(N,N-dimethylamino)pyridines (4a–6a), (E)-5-(2′,6′-dihalostyryl)-2-(N,N-dimethylamino)pyridines (7a–9a), and (E)-5-(2′,6′-dihalostyryl)-2-(N,N-dimethylamino)pyrimidines (10a–12a), respectively (Figure 2). Synthesis of the corresponding pyridine and pyrimidine analogues in the N-methylamino series employed the Wadsworth–Emmons condensation of the 2,6-dihalobenzyl diethyl phosphonates with the appropriate pyridine-3-carboxaldehyde and pyrimidine-5-carboxaldehyde and secured (E)-5-(2′,6′-dihalostyryl)-2-(N-methylamino)pyridines (7b–9b) and (E)-5-(2′,6′-dihalostyryl)-2-(N-methylamino)pyrimidines (10b–12b), respectively (Figure 2). Synthesis of 1c involved the N-monoethylation of (E)-4-(2′,6′-difluorostyryl)aniline3 (Figure 3). Synthesis of 7c involved the initial Wadsworth–Emmons condensation of 2,6-difluorobenzyl diethyl phosphonate with 2-(tert-butyloxycarbonyl)aminopyridine-5-carboxaldehyde, N-ethylation, and deprotection of the tert-butoxycarbonyl group with trifluoracetic acid (Figure 3). Synthesis of 10c involved the Wadsworth–Emmons condensation of 2,6-difluorobenzyl diethyl phosphonate with 2-(N-ethylamino)pyridine-5-carboxaldehyde (Figure 3). No E/Z isomerization of these FIDAS agents, as determined by 1H NMR of freshly prepared solutions, occurred when samples were stored as solids in the dark at low temperatures. For biological experiments, freshly prepared DMSO stock solutions were prepared, diluted with buffer, and used immediately as described below.
Figure 2.

Synthetic route to FIDAS agents with N-methylamino or N,N-dimethylamino groups. Reagents: (a) NaH, DMF followed by 4-(CH3)2N(C6H4)CHO to give 1a–3a; (b) CNBr, acetone, 16 h, 56 °C followed by conc. HCl, 3 h, reflux; (c) NaH, DMF followed by 5-(CH3)2N(C6H3N)-2-CHO to give 4a–6a; (d) NaH, DMF followed by 2-(CH3)2N(C6H3N)-5-CHO to give 7a–9a; (e) NaH, DMF followed by 2-CH3NH(C6H3N)-5-CHO to give 7b–9b; (f) NaH, DMF followed by 2-(CH3)2N(C6H2N2)-5-CHO to give 10a–12a; (g) NaH, DMF followed by 2-CH3NH(C6H2N2)-5-CHO to give 10b–12b.
Figure 3.

Synthetic route to FIDAS agents with N-ethylamino groups. Reagents: (a) NaH, DMF followed by 4-O2N(C6H4)CHO; (b) SnCl2, HOAc, conc. HCl; (c) K2CO3, CH3CH2I, acetone; (d) NaH, DMF followed by 4-tBuOCONH(C6H4)CHO; (e) NaH, C2H5I; (f) CF3CO2H; (g) NaH, DMF followed by 2-CH3CH2NH(C6H2N2)-5-CHO.
MAT2A Inhibition Assays
Inherent fluorescence of the 2′,6′-difluorostyryl- and 2′-chloro-6′-fluorostyryl-substituted FIDAS agents at 454 nm facilitated the development of a fluorescence anisotropy assay to evaluate the binding between MAT2A and FIDAS agents.19 Restriction of this assay to only those FIDAS agents that displayed inherent fluorescence led to the application of another, previously reported, MAT2A inhibition assay to evaluate the activity of FIDAS agents.2 Recombinant MAT2A holoenzyme was purified and used to synthesize SAM from methionine and ATP.16 For this assay, each FIDAS analogue was preincubated with MAT2A holoenzyme and then mixed with methionine and ATP. Phosphate Pi concentrations were analyzed to determine the level of MAT2A activity.20 Final 30 μM concentrations of FIDAS agents were used for these experiments, which were repeated in triplicate. The ratio of MAT2A inhibition for selected FIDAS agents relative to FIDAS 1a is summarized in Figure 4.
Figure 4.

Ratio of MAT2A inhibition for selected FIDAS agents relative to FIDAS 1a. Recombinant MAT2A holoenzyme was purified and used to synthesize SAM from methionine and ATP. For inhibition assay, each FIDAS analogue was preincubated with MAT2A holoenzyme and then mixed together with methionine and ATP. The reaction products are SAM and Pi. Pi concentration was analyzed to determine the MAT2A activity. Inhibition activity of each compound was compared with that of FIDAS 1a.
In Vitro Testing of FIDAS Agents with LS174T Colorectal Cancer Cells
We treated LS174T colon cancer cells with FIDAS agents 1–3 in the aniline family, 4–6 in the 5-aminopyridine family, 7–9 in the 2-aminopyridine family, and 10–12 in the 2-aminopyrimidine family, and we determined the IC50 value of each compound on LS174T cell proliferation (Table 1 and Supporting Information Figure S1). The IC50 values were calculated using Prism 5 analysis of the data from the concentration–response curves for each compound. Concentrations of FIDAS agents ranging from 1 to 300 nM were used for these experiments, which were repeated in triplicate at each concentration. Among these four families, only compound 5a in the 5-aminopyridine family showed an IC50 value comparable to that of FIDAS 1a. Because this outcome did not represent a significant advance, further work with the 5-aminopyridine family was not pursued. Similarly, the 2-aminopyrimidines 10–12 displayed IC50 values in the 200–900 nM range and also were not pursued in detail.
Table 1. Comparison of IC50 Values for the Inhibition of LS174T Cancer Cells, IC50 for hERG Inhibition Values, and the Ratio of IC50 for hERG Inhibition to the IC50 Values for the Inhibition of LS174T Cancer Cells.
| FIDAS | inhibition of LS174T cell proliferation IC50 (nM) | hERG inhibition IC50 (μM) | ratio of IC50 for hERG inhibition to IC50 for inhibition of LS174T cell proliferation (× 103) |
|---|---|---|---|
| 1a | 26.9 ± 6.5 | 32.1.1 ± 4.4 | 1.2 |
| 1b | 7.6 ± 1.2 | 49.1 ± 11.1 | 6.5 |
| 1c | 199.7 ± 3.3 | ||
| 2a | 19.2 ± 6.4 | ||
| 2b | 7.6 ± 0.5 | 49.8 ± 5.1 | 6.6 |
| 3a | 213.8 ± 7.3 | ||
| 3b | 50.1 ± 6.7 | ||
| 4a | 57.9 ± 11.6 | 21.1 ± 1.1 | 0.4 |
| 5a | 20.4 ± 4.1 | 20.7 ± 1.8 | 1.0 |
| 6a | 59.5 ± 10.2 | ||
| 7a | 13.3 ± 7.8 | 22.0 ± 3.6 | 1.7 |
| 7b | 30.1 ± 5.8 | ||
| 7c | 31.3 ± 5.5 | ||
| 8a | 4.7 ± 0.6 | 59.5 ± 17.8 | 12.7 |
| 8b | 4.0 ± 0.2 | 19.9 ± 4.1 | 5.0 |
| 9a | 30.1 ± 3.1 | ||
| 9b | 19.6 ± 8.5 | ||
| 10a | >200 | 40.5 ± 9.7 | 0.5 |
| 10b | >200 | ||
| 10c | >200 | ||
| 11a | >200 | ||
| 11b | >200 | ||
| 12a | >200 | ||
| 12b | >200 |
Replacing the 2′,6′-difluoro-substituents in FIDAS 1a with 2′-chloro-6′-fluoro substituents led to compounds with comparable or increased inhibitory activity: IC50 value for 1a ≈ IC50 value for 2a; IC50 value for 5a < IC50 value for 4a; and IC50 value for 8a ≈ IC50 value for 7a. Replacing the 2′,6′-difluoro substituents in FIDAS 1a with 2′,6′-dichloro substituents led, in general, to relatively inactive compounds in comparison with that of their difluorinated counterparts: IC50 value for 3a > IC50 value for 1a; IC50 value for 6a ≈ IC50 value for 4a; IC50 value for 9a > IC50 value for 7a; and IC50 value for 12a > IC50 value for 10a.
Replacing the N,N-(dimethylamino)-substituent in FIDAS 1a with an N-(methylamino) substituent led, generally, to compounds with increased inhibitory activity: IC50 value for 1b < IC50 value for FIDAS 1a; IC50 value for 2b ≈ IC50 value for 2a; and IC50 value for 3b < IC50 value for 3a. Replacing the N-methylamino substituent in 1b with the sterically larger N-ethylamino-substituent in 1c led to inactive compounds in some cases (e.g., IC50 for 1c > IC50 for 1b) or compounds with comparable activity (e.g., IC50 for 7b ≈ IC50 for 7c; IC50 for 10b ≈ IC50 for 10c). In summary, the introduction of either the N-methylamino substituent in 1b and 2b or the introduction of the 2′-chloro-6′-fluorostyryl moiety in 8a and 8b led to the most potent compounds with IC50 values less than 10 nM (Table 1).
[3H]-Dofetilide Binding to Plasma Membranes Overexpressing the hERG Channel
[3H]-Dofetilide competition binding assays using HEK-293 cell membranes stably expressing the hERG channel (hERG-HEK) correlate well with results from voltage-clamp assays and provide useful predictive screening assays for QT prolongation.21 We utilized a [3H]-dofetilide binding assay to evaluate FIDAS agent interaction with hERG. Amitriptyline (final concentration, 1 mM) was used as the positive control and exhibited an IC50 value (10.7 ± 2.25 μM) that was in agreement with published values.22 We selected a subset of the FIDAS agents in Figure 1 that possessed potent in vitro inhibition with LS174T colorectal cancer cells and that represented the structural subtypes in this SAR study (i.e., (E)-4-(2′,6′-difluorostyryl)-N,N-(dimethyl)aniline (FIDAS 1a); (E)-2-(2′,6′-difluorostyryl)-5-N,N-(dimethylamino)pyridine (4a); (E)-5-(2′,6′-difluorostyryl)-2-N,N-(dimethylamino)pyridine (7a); and (E)-5-(2′,6′-difluorostyryl)-2-N,N-(dimethylamino)pyrimidine (10a) as well as variants with different halogenation and methylation patterns). Concentrations of FIDAS agents ranging from 10–9 to 10–4 M were assayed in duplicate for these experiments (n = 3 experiments/analogue). IC50 values for hERG inhibition of [3H]-dofetilide binding ranged from 20 to 60 μM. Ratios of the IC50 values for the hERG inhibition and IC50 values for the inhibition of LS174T cell proliferation (Table 1) ranged from 2 to 4 orders of magnitude for the subset of FIDAS agents that were studied.
Discussion
We reported that (E)-4-(2′,6′-difluorostyryl)-N,N-dimethylaniline (FIDAS 1a) selectively bound to the catalytic subunit of methionine S-adenosyltranferase-2 (MAT2A), an enzyme upregulated in various liver and colorectal cancers.1−3FIDAS 1a possessed in vitro and in vivo activity against various liver and colorectal cancer cell lines, possessed good oral bioavailability and a reasonable half-life in vivo, and displayed minimal gross toxicity in terms of body weight loss/death at doses exceeding those that produced in vivo tumor reduction in a xenograft model.1,2 In seeking analogues with increased potency and improved water solubility relative to that of FIDAS 1a, we undertook a SAR study that included pyridine and pyrimidine rings into the FIDAS platform (Figure 1). We also focused on the modification of substituents, such as the halogen and the N,N-dimethylamino substituents in FIDAS 1a, as a means of improving potency. Finally, potency alone is no longer sufficient to guide translational drug development. In developing these FIDAS agents as potential drug candidates, we sought FIDAS agents that avoided binding to the hERG potassium channel associated with drug-induced, adverse cardiac events.15 This study describes our success in meeting these requirements (i.e., nanomolar potency, water solubility, and absence of hERG activation) into the FIDAS platform.
We synthesized and evaluated four families of FIDAS agents, as shown in Figure 1: (E)-4-(2′,6′-dihalostyryl)-N-alkyl- and (E)-4-(2′,6′-dihalostyryl)-N,N-(dialkyl)anilines (1–3), which we refer to as the aniline family; (E)-2-(2′,6′-dihalostyryl)-5-(N-alkylamino)- and (E)-2-(2′,6′-dihalostyryl)-5-(N,N-dialkylamino)pyridines (4–6), which we refer to as the 5-aminopyridine family; (E)-5-(2′,6′-dihalostyryl)-2-(N-alkylamino)- and (E)-5-(2′,6′-dihalostyryl)-2-(N,N-dialkylamino)pyridines (7–9), which we refer to as the 2-aminopyridine family; and (E)-5-(2′,6′-dihalostyryl)-2-(N-alkylamino)- and (E)-5-(2′,6′-dihalostyryl)-2-(N,N-dialkylamino)pyrimidines (10–12), which we refer to as the 2-aminopyrimidine family. Syntheses relied principally on the Wadsworth–Emmons coupling of arylphosphonates with aryl aldehydes (Figures 2 and 3). FIDAS agents were characterized fully, and their purity was established by a combination of 13C nuclear magnetic resonance (NMR) and combustion analyses.
We explored variations in the location, number, and nature of the two halogen substituents within FIDAS 1a. As reported previously, alterations in the location of the two halogens, other than the 2,6-dihalostyryl arrangement, invariably led to diminished activity in a cell proliferation in vitro assay using LS174T cells.1−3 The inclusion of halogen substituents larger than chlorine or the introduction of more than two halogen substituents diminished or produced no appreciable increase in activity (data not shown). We found, however, that the substitution of one, but not both, of the fluorines in FIDAS 1a with a chlorine substituent resulted in slightly improved levels of in vitro activity. For example, the 2′-chloro-6′-fluoro analogue, 2a, was comparable in activity to that of FIDAS 1a and was 10-fold better than that of 3a (Table 1).
Modification of the alkyl groups in the N,N-(dialkylamino)phenyl subunit of FIDAS 1a produced an even more dramatic improvement in activity than alteration in just the halogen substituents. Prior studies1−3 had established that only the para orientation of the N,N-dimethylamino group and the 2,6-dihalostyryl group possessed good biological activity; consequently, we did not explore ortho or meta orientations of these groups in this study. Within the para series, the removal of one of the methyl groups, as in the monomethyl analogues, 1b, 2b, and 3b, produced an active series with in vitro potencies in the 7–50 nM range (Table 1). Additional modifications involving substitution of a larger N-alkyl group than N-methyl, such as N-ethyl in FIDAS 1c, led to diminished in vitro activity. Inclusion of either N-alkylamino- or N,N-(dialkylamino)pyridine or pyrimidine rings in place of the N,N-(dimethylamino)phenyl ring in FIDAS 1a offered an attractive option for addressing the water-solubility issue and improving potency. There are some inconsistencies between MAT2A inhibition and cell proliferation inhibition, particularly in the case of 8b, suggesting that other characteristics, such as stability and solubility, may also contribute to the efficacy of FIDAS agents in cell or in vivo assays. Although the hydrochloride salt of FIDAS 1a was readily absorbed in a mouse bioavailability study, the in vitro IC50 was only 27 nM in LS174T cells. We found experimentally that the N-methyl analogues, 1b and 2b, had IC50 values for the inhibition of LS174T cells that were 4-fold greater than that of 1a. We also performed a proof-of-concept experiment using primary CRC cells. We found that 2b significantly inhibited the growth of CRC organoids (Supporting Information Figure 2), which further suggests that these agents are potential candidates for CRC treatment.
With the objective of developing antineoplastic drugs with limited affinity for human hERG, we utilized a [3H]-dofetilide binding assay to evaluate the interaction of a subset of these FIDAS agents with hERG. Ratios of the IC50 values for the inhibition of LS174T cell proliferation and the IC50 values for hERG inhibition (Table 1) were, as desired, larger than 2–4 orders of magnitude, with the exceptions of 4a and 10a. Because these latter two FIDAS agents were also in families that were the least active of the four families studied, namely, the 5-aminopyridines (4–6) and the 2-aminopyrimidines (10–12), they were set aside for further SAR development. The most active compounds (i.e., 1b, 2b, 8a, and 8b) did not interact appreciably with hERG and possess ratios of IC50 values for hERG inhibition relative to IC50 values for inhibition of LS174T cell proliferation (Table 1) that were greater than that of FIDAS 1a and that are well within the selectivity range for drugs entering preclinical development.23
In summary, four key findings emerged from these studies: (1) FIDAS agents in the aniline family (1–3) and in the 2-aminopyridine family (7–9) possess lower IC50 values in the inhibition of LS174T cell proliferation than that of the 5-aminopyridines (4–6) and the 2-aminopyrimidines (10–12), (2) the most active FIDAS agents possessed either 2,6-difluorostyryl or 2-chloro-6-fluorostyryl subunits, (3) the most active FIDAS agents possessed small N-alkyl groups, specifically the N-methylamino or the N,N-dimethylamino groups, in a para orientation relative to the 2,6-dihalostyryl subunit, and (4) 2-aminopyridines 8a and 8b not only displayed IC50 values less than 10 nM but also formed water-soluble hydrochloride salts. In summary, we developed potent analogues of FIDAS 1a that exhibited significantly increased ratios of IC50 for hERG inhibition to IC50 for inhibition of LS174T cell proliferation, thereby opening the door to further development of these compounds as potential antineoplastic drugs.
Experimental Procedures
Chemistry
Chemicals were purchased from Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) or were synthesized according to literature procedures. Solvents were used from commercial vendors without further purification unless otherwise noted. Nuclear magnetic resonance spectra were determined on a Varian instrument (1H, 400 MHz; 13C, 100Mz). High-resolution electrospray ionization (ESI) mass spectra were recorded on a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Resolution was set at 100 000 (at 400 m/z). Samples were introduced through direct infusion using a syringe pump with a flow rate of 5 μL/min. Purity of compounds was greater than 95%, as established using combustion analyses determined by Atlantic Microlabs, Inc., Norcross, GA. Compounds were chromatographed on preparative layer Merck silica gel F254 unless otherwise indicated.
General Procedure for the Synthesis of FIDAS Agents
To a solution of 1.65 mmol (1.1 equiv) of diethyl phosphonate in 5 mL of anhydrous DMF at 0 °C was added 2.25 mmol (1.5 equiv) of sodium hydride (washed with anhydrous hexanes to remove oil). The mixture was stirred for 15 min, and 1.5 mmol (1 equiv) of appropriate aldehyde dissolved in 1 mL of anhydrous DMF was added dropwise at 0 °C. The mixture was stirred 12 h at 25 °C and quenched by pouring into 30 mL of water with stirring. A precipitate was collected by filtration and purified by recrystallization and/or chromatography as noted for individual compounds described below. Several compounds in this study were reported previously: FIDAS 1a,1−31b,2,32a,2,32b,2,33a,3 and 3b.3
(E)-4-(2′,6′-Difluorostyryl)-N-ethylaniline (1c)
To a mixture of 150 mg (0.65 mmol) of (E)-4-(2′,6′-difluorostyryl)aniline3 and 100 mg (0.72 mmol, 1.1 equiv) of K2CO3 in 3 mL of acetone was added 101 mg (0.65 mmol, 1 equiv) of iodoethane. The mixture was refluxed for 12 h, poured into water, and extracted with CH2Cl2. The combined organic phases were dried over anhydrous MgSO4 and evaporated to give a product that was purified by chromatography using 1:7 ethyl acetate–hexane (Rf = 0.43) to afford 103 mg (61%) of 1c as a white solid: mp 50–51 °C. 1H NMR (CDCl3): δ 7.38 (d, 2H, J = 8.4 Hz), 7.36 (d, 1H, J = 16.4 Hz), 7.12–7.03 (m, 1H), 6.94–6.84 (m, 3H), 6.59 (d, 2H, J = 8.8 Hz), 3.71 (br s, 1 H), 3.19 (q, 2H, J = 7.2 Hz), 1.27 (t, 3H, J = 7.2 Hz). 13C NMR (CDCl3): δ 160.80 (dd, J1 = 248.9 Hz, J2 = 8.4 Hz, two C), 148.55, 135.33 (t, J = 8.4 Hz), 128.02 (two C), 127.71, 126.61 (t, J = 10.0 Hz), 115.58 (t, J = 15.6 Hz), 112.63 (two C), 111.41 (dd, J1 = 19.8 Hz, J2 = 6.8 Hz, two C), 110.61, 38.30, 14.80. HRMS (ESI) calcd for C16H16F2N [MH+], 260.12453; found, 260.12384. Anal. Calcd for C16H15F2N: C, 74.11; H, 5.83. Found: C, 74.20; H, 5.90.
(E)-2-(2′,6′-Difluorostyryl)-5-(N,N-dimethylamino)pyridine (4a)
Yield 81%, mp 90–92 °C (from hexane). 1H NMR (DMSO-d6): δ 8.17 (d, 1H, J = 3.2 Hz), 7.42–7.28 (m, 4H), 7.18–7.11 (m, 2H), 7.06 (dd, 1H, J1 = 8.4 Hz, J2 = 3.2 Hz), 2.98 (s, 6H). 13C NMR (DMSO-d6): δ 160.24 (dd, J1 = 248.2 Hz, J = 8.4 Hz, two C), 145.47, 142.08, 134.78, 134.42 (t, J = 8.0 Hz), 128.49 (t, J = 11.0 Hz), 123.50, 118.19, 114.20 (t, J = 15.2 Hz), 112.62, 112.00 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 39.56 (two C). HRMS (ESI) calcd for C15H15F2N2 [MH+], 261.11978; found, 261.11884. Anal. Calcd for C15H14F2N2: C, 69.22; H, 5.42. Found: C, 69.33; H, 5.59.
(E)-2-(2′-Chloro-6′-fluorostyryl)-5-(N,N-dimethylamino)pyridine (5a)
Yield 80%, Rf = 0.44 (ethyl acetate–hexane 1:5), mp 78–80 °C. 1H NMR (DMSO-d6): δ 8.20 (d, 1H, J = 2.4 Hz), 7.53 (d, 1H, J = 16.4 Hz), 7.39–7.25 (m, 5H), 7.06 (dd, 1H, J1 = 8.4 Hz, J2 = 3.2 Hz), 2.98 (s, 6H). 13C NMR (DMSO-d6): δ 160.61 (d, J = 249.7 Hz), 145.52, 141.91, 135.26 (d, J = 12.9 Hz), 134.88, 133.39 (d, J = 6.1 Hz), 128.64 (d, J = 9.9 Hz), 125.96 (d, J = 3.8 Hz), 123.72, 123.55 (d, J = 14.4 Hz), 118.12, 116.88 (d, J = 2.3 Hz), 115.18 (d, J = 23.5 Hz), 39.54 (two C). HRMS (ESI) calcd for C15H15ClFN2 [MH+], 277.09023; found, 277.08939. Anal. Calcd for C15H14ClFN2: C, 65.10; H, 5.10. Found: C, 65.04; H, 5.20.
(E)-2-(2′,6′-Dichlorostyryl)-5-(N,N-dimethylamino)pyridine (6a)
Yield 86%, mp 85–86 °C (from hexane). 1H NMR (DMSO-d6): δ 8.19 (d, 1H, J = 3.2 Hz), 7.52 (d, 2H, J = 7.6 Hz), 7.42–7.36 (m, 2H), 7.30–7.26 (m, 1H), 7.15 (d, 1H, J = 16.0 Hz), 7.07 (dd, 1H, J1 = 8.8 Hz, J2 = 3.2 Hz), 2.98 (s, 6H). 13C NMR (DMSO-d6): δ 145.56, 141.62, 136.25, 134.78, 134.03, 133.53 (two C), 129.00 (two C), 128.78, 123.39, 120.27, 118.20, 39.58 (two C). HRMS (ESI) calcd for C15H15Cl2N2 [MH+], 293.06068; found, 293.05986. Anal. Calcd for C15H14Cl2N2: C, 61.45; H, 4.81. Found: C, 61.55; H, 4.75.
(E)-5-(2′,6′-Difluorostyryl)-2-(N,N-dimethylamino)pyridine (7a)
Yield 87%, mp 103–104 °C (from hexane). 1H NMR (DMSO-d6): δ 8.23 (d, 1H, J = 2.4 Hz), 7.89 (dd, 1H, J1 = 9.0 Hz, J2 = 2.4 Hz), 7.33–7.24 (m, 2H), 7.16–7.09 (m, 2H), 6.89 (d, 1H, J = 16.8 Hz), 6.68 (d, 1H, J = 8.8 Hz) 3.06 (s, 6H). 13C NMR (DMSO-d6): δ 159.97 (dd, J1 = 247.0 Hz, J = 7.2 Hz, two C), 158.70, 148.03, 133.79, 132.60 (t, J = 8.0 Hz), 128.13 (t, J = 10.6 Hz), 120.48, 114.54 (t, J = 15.6 Hz), 111.97 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 110.14, 106,04, 37.65 (two C). HRMS (ESI) calcd for C15H15F2N2 [MH+], 261.11978; found, 261.11885. Anal. Calcd for C15H14F2N2: C, 69.22; H, 5.42. Found: C, 69.32; H, 5.44.
(E)-5-(2′,6′-Difluorostyryl)-2-(N-methylamino)pyridine (7b)
Yield 83%, mp 130–132 °C (from ethanol–water). 1H NMR (DMSO-d6): δ 8.12 (d, 1H, J = 2.4 Hz), 7.78 (dd, 1H, J1 = 8.6 Hz, J2 = 2.6 Hz), 7.32–7.24 (m, 1H), 7.23 (d, 1H, J = 16.8 Hz), 7.16–7.08 (m, 2H), 6.87–6.81 (m, 2H), 6.49 (d, 1H, J = 9.2 Hz), 2.80 (d, 3H, J = 4.8 Hz). 13C NMR (DMSO-d6): δ 159.95 (dd, J1 = 247.4 Hz, J = 8.4 Hz, two C), 159.37, 148.44, 133.03, 132.9 (t, J = 8.0 Hz), 127.97 (t, J = 10.6 Hz), 120.60, 114.61 (t, J = 15.9 Hz), 111.96 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 109.55, 108.31, 27.89. HRMS (ESI) calcd for C14H13F2N2 [MH+], 247.10413; found, 247.10318. Anal. Calcd for C14H12F2N2: C, 68.28; H, 4.91. Found: C, 68.12; H, 5.04.
(E)-5-(2′,6′-Difluorostyryl)-2-(N-ethylamino)pyridine (7c)
The general procedure was repeated using 2-(tert-butyloxycarbonyl)aminopyridine-5-carboxaldehyde and 2,6-difluorobenzyl diethyl phosphonate to afford (E)-2-(tert-butoxycarbonylamino)-5-(2′,6′-difluorostyryl)pyridine: yield 86%, mp 190–191 °C (from dichloromethane). 1H NMR (CDCl3): δ 9.16 (br s, 1H), 8.45 (d, 1H, J = 2.4 Hz), 8.04 (d, 1H, J = 8.8 Hz), 7.89 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4), 7.36 (d, 1H, J = 16.8 Hz), 7.12–7.20 (m, 1H), 7.08 (d, 1H, J = 16.8 Hz), 6.88–6.95 (m, 2H), 1.58 (s, 9H). 13C NMR (CDCl3): δ 160.95 (dd, J1 = 250.4 Hz, J = 7.6 Hz, two C), 152.61, 152.15, 146.88, 135.12, 131.10 (t, J = 8.6 Hz), 128.04 (t, J = 10.6 Hz), 115.02, 114.50 (t, J = 15.1 Hz), 112.28, 111.61 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 81.10, 28.38 (three C). HRMS (ESI) calcd for C18H19F2N2O2 [MH+], 333.14091; found, 333.13949. Anal. Calcd for C18H18F2N2O2: C, 65.05; H, 5.46. Found: C, 65.12; H, 5.59. To 615 mg of (E)-2-(tert-butoxycarbonylamino)-5-(2′,6′-difluorostyryl)pyridine (1.85 mmol) in 18 mL of anhydrous N,N-dimethylformamide at 0 °C was added 163 mg of 60% sodium hydride (4.07 mmol) in portions. The suspension was stirred for 20 min while maintaining the temperature below 5 °C, and 0.16 mL of ethyl iodide (2.04 mmol) was added dropwise. The mixture was stirred at 5 °C for 30 min and allowed to stir at 25 °C for 12 h. The reaction was quenched with water, extracted with dichloromethane, and washed successively with water, 0.1 M hydrochloric acid solution, saturated aqueous NaHCO3 solution, and brine, and dried over anhydrous MgSO4 to afford 633 mg (95%) of (E)-2-(N-(tert-butoxycarbonyl)-N-ethylamino)-5-(2′,6′-difluorostyryl)pyridine as a clear, colorless oil that was used in the next step without further purification. To 613 mg (1.7 mmol) of (E)-2-(N-(tert-butoxycarbonyl)-N-ethylamino)-5-(2′,6′-difluorostyryl)pyridine in 17 mL of dichloromethane was added 4.4 mL of trifluoroacetic acid (57.1 mmol). The mixture was stirred for 12 h at 25 °C. A precipitate was collected and dissolved in water, cooled to 0 °C, and neutralized with aqueous solution of Na2CO3. The product was dissolved in dichloromethane, washed with water and brine, and dried over anhydrous MgSO4. The product was purified by chromatography using 1:3 ethyl acetate–hexane (Rf = 0.23) to afford 398 mg (90%) of 7c as a white solid. mp 96–97 °C. 1H NMR (CDCl3): δ 8.16 (br s, 1H), 7.70 (d, 1H, J = 7.6 Hz), 7.31 (d, 1H, J = 16.8 Hz), 7.13–7.06 (m, 1H), 6.91–6.86 (m, 3H), 6.39 (d, 1H, J = 8.8 Hz), 4.73 (br s, 1H), 3.33 (m, 2H, J = 7.2 Hz), 1.26 (t, 3H, J = 7.2 Hz). 13C NMR (CDCl3): δ 160.79 (dd, J1 = 249.7 Hz, J = 7.6 Hz, two C), 158.48, 148.32, 134.06, 132.12 (t, J = 8.3 Hz), 127.10 (t, J = 10.6 Hz), 122.74, 115.11 (t, J = 15.1 Hz), 111.53, 111.47 (dd, J1 = 19.7 Hz, J2 = 6.8 Hz, two C), 106.56, 36.92, 14.82. HRMS (ESI) calcd for C15H15F2N2 [MH+], 261.11978; found, 261.11883. Anal. Calcd for C15H14F2N2: C, 69.22; H, 5.42. Found: C, 69.27; H, 5.43.
(E)-5-(2′-Chloro-6′-fluorostyryl)-2-(N,N-dimethylamino)pyridine (8a)
Yield 76%, mp 63–65 °C (from hexane). 1H NMR (DMSO-d6): δ 8.23 (d, 1H, J = 2.4 Hz), 7.87 (dd, 1H, J1 = 9.0 Hz, J2 = 2.6 Hz), 7.38–7.35 (m, 1H), 7.29–7.26 (m, 2H), 7.20 (d, 1H, J = 16.8 Hz), 6.97 (d, 1H, J = 16.8 Hz), 6.70 (d, 1H, J = 9.2 Hz), 3.07 (s, 6H). 13C NMR (DMSO-d6): δ 160.30 (d, J = 248.9 Hz), 158.75, 148.02, 133.81, 133.50 (d, J = 12.1 Hz), 132.97 (d, J = 6.0 Hz), 128.40 (d, J = 10.6 Hz), 125.87 (d, J = 3.1 Hz), 123.95 (d, J = 15.1 Hz), 120.30, 115.14 (d, J = 23.5 Hz), 114.25 (d, J = 2.3 Hz), 106.05, 37.64 (two C). HRMS (ESI) calcd for C15H15ClFN2 [MH+], 277.09023; found, 277.08944. Anal. Calcd for C15H14ClFN2: C, 65.10; H, 5.10. Found: C, 65.01; H, 5.21.
(E)-5-(2′-Chloro-6′-fluorostyryl)-2-(N-methylamino)pyridine (8b)
Yield 86%, mp 106–108 °C (from hexane). 1H NMR (DMSO-d6): δ 8.12 (d, 1H, J = 2.4 Hz), 7.76 (dd, 1H, J1 = 9.0 Hz, J2 = 2.6 Hz), 7.37–7.33 (m, 1H), 7.29–7.24 (m, 2H), 7.17 (d, 1H, J = 16.4 Hz), 6.91 (d, 1H, J = 16.4 Hz), 6.87 (q, 1H, J = 4.6 Hz), 6.51 (d, 1H, J = 8.8 Hz), 2.80 (d, 3H, J = 4.8 Hz). 13C NMR (DMSO-d6): δ 160.29 (d, J = 248.1 Hz), 159.44, 148.46, 133.80 (d, J = 11.4 Hz), 133.06, 132.93 (d, J = 5.3 Hz), 128.28 (d, J = 8.3 Hz), 125.86 (d, J = 3.0 Hz), 124.02 (d, J = 15.2 Hz), 120.43, 114.14 (d, J = 22.7 Hz), 113.68, 108.33, 27.90. HRMS (ESI) calcd for C14H13ClFN2 [MH+], 263.07458; found, 263.07375. Anal. Calcd for C14H12ClFN2: C, 64.01; H, 4.60. Found: C, 64.13; H, 4.63.
(E)-5-(2′,6′-Dichlorostyryl)-2-(N,N-dimethylamino)pyridine (9a)
Yield 78%, mp 54–56 °C (from hexane). 1H NMR (DMSO-d6): δ 8.22 (d, 1H, J = 2.8 Hz), 7.87 (dd, 1H, J1 = 8.8 Hz, J2 = 2.8 Hz), 7.51 (d, 2H, J = 8.0 Hz), 7.29–7.25 (m, 1H), 7.00 (d, 1H, J = 16.8 Hz), 6.92 (d, 1H, J = 16.8 Hz), 6.70 (d, 1H, J = 8.8 Hz), 3.07 (s, 6H). 13C NMR (DMSO-d6): δ 158.78, 147.82, 134.47, 134.19, 133.98, 133.44 (two C), 128.90 (two C), 128.69, 119.86, 117.80, 106.01, 37.66 (two C). HRMS (ESI) calcd for C15H15Cl2N2 [MH+], 293.06068; found, 293.05978. Anal. Calcd for C15H14Cl2N2: C, 61.45; H, 4.81. Found: C, 61.51; H, 4.96.
(E)-5-(2′,6′-Dichlorostyryl)-2-(N-methylamino)pyridine (9b)
Yield 88%, mp 129–131 °C (from hexane). 1H NMR (DMSO-d6): δ 8.11 (d, 1H, J = 2.0 Hz), 7.77 (dd, 1H, J1 = 9.0 Hz, J2 = 2.6 Hz), 7.50 (d, 2H, J = 8.0 Hz), 7.28–7.24 (m, 1H), 6.97 (d, 1H, J = 16.4 Hz), 6.88–6.83 (m, 2H), 6.50 (d, 1H, J = 8.4 Hz), 2.80 (d, 3H, J = 4.8 Hz). 13C NMR (DMSO-d6): δ 159.44 148.23, 134.52, 134.47, 133.43 (two C), 133.24, 128.89 (two C), 128.60, 119.97, 117.21, 108.22, 27.89. HRMS (ESI) calcd for C14H13Cl2N2 [MH+], 279.04503; found, 279.04430. Anal. Calcd for C14H12Cl2N2: C, 60.23; H, 4.33. Found: C, 60.35; H, 4.51.
(E)-5-(2′,6′-Difluorostyryl)-2-(N,N-dimethylamino)pyrimidine (10a)
Yield 85%, mp 134–136 °C (from hexane). 1H NMR (DMSO-d6): δ 8.65 (s, 2H), 7.35–7.28 (m, 1H), 7.20 (d, 1H, J = 17.2 Hz), 7.17–7.10 (m, 2H), 7.00 (d, 1H, J = 17.2 Hz), 3.16 (s, 6H). 13C NMR (DMSO-d6): δ 161.61, 160.43 (dd, J1 = 248.2 Hz, J = 7.6 Hz, two C), 156.55 (two C), 130.23 (t, J = 8.0 Hz), 128.99 (t, J = 10.6 Hz), 118.88, 114.65 (t, J = 15.2 Hz), 112.43 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 111.75, 37.17 (two C). HRMS (ESI) calcd for C14H14F2N3 [MH+], 262.11503; found, 262.11417. Anal. Calcd for C14H13F2N3: C, 64.36; H, 5.02. Found: C, 64.45; H, 5.03.
(E)-5-(2′,6′-Difluorostyryl)-2-(N-methylamino)pyrimidine (10b)
Yield 90%, mp 143–144 °C (from ethanol). 1H NMR (DMSO-d6): δ 8.60 (s, 2H), 7.42 (q, 1H, J = 4.6 Hz), 7.36–7.28 (m, 1H), 7.21–7.11 (m, 3H), 6.99 (d, 1H, J = 16.8 Hz), 2.83 (d, 3H, J = 4.8 Hz). 13C NMR (DMSO-d6): δ 162.19, 160.00 (dd, J1 = 248.2 Hz, J = 7.6 Hz, two C), 156.38 (two C), 129.97 (t, J = 7.6 Hz), 128.50 (t, J = 10.6 Hz), 119.08, 114.26 (t, J = 15.2 Hz), 112.02 (dd, J1 = 19.4 Hz, J2 = 6.4 Hz, two C), 111.02, 27.93. HRMS (ESI) calcd for C13H12F2N3 [MH+], 248.09938; found, 248.09838. Anal. Calcd for C13H11F2N3: C, 63.15; H, 4.48. Found: C, 63.14; H, 4.55.
(E)-5-(2′,6′-Difluorostyryl)-2-(N-ethylamino)pyrimidine (10c)
Yield 89%, mp 147–149 °C (from ethanol). 1H NMR (DMSO-d6): δ 8.58 (s, 2H), 7.48 (t, 1H, J = 5.6 Hz), 7.35–7.28 (m, 1H), 7.20–7.10 (m, 3H), 6.98 (d, 1H, J = 16.8 Hz), 3.56–3.29 (m, 2H), 1.12 (t, 3H, J = 7.4 Hz). 13C NMR (DMSO-d6): δ 161.62, 160.00 (dd, J1 = 248.2 Hz, J = 7.6 Hz, two C), 156.40 (two C), 129.97 (t, J = 7.6 Hz), 128.51 (t, J = 10.6 Hz), 119.08, 114.27 (t, J = 15.6 Hz), 112.00 (dd, J1 = 19.4 Hz, J2 = 6.1 Hz, two C), 110.94, 35.50, 14.65. HRMS (ESI) calcd for C14H14F2N3 [MH+], 262.11503; found, 262.11424. Anal. Calcd for C14H13F2N3: C, 64.36; H, 5.02. Found: C, 64.54; H, 4.96.
(E)-5-(2′-Chloro-6′-fluorostyryl)-2-(N,N-dimethylamino)pyrimidine (11a)
Yield 74%, mp 105–106 °C (from hexane). 1H NMR (DMSO-d6): δ 8.64 (s, 2H), 7.38–7.25 (m, 3H), 7.14 (d, 1H, J = 16.8 Hz), 7.42 (d, 1H, J = 16.8 Hz), 3.16 (s, 6H). 13C NMR (DMSO-d6): δ 161.22, 160.32 (d, J = 248.9 Hz), 156.13 (two C), 133.12 (d, J = 5.3 Hz), 130.73 (d, J = 11.3 Hz), 128.83 (d, J = 8.4 Hz), 125.91 (d, J = 1.5 Hz), 123.67 (d, J = 14.5 Hz), 118.29, 115.36, 115.17 (d, J = 22.7 Hz), 36.73 (two C). HRMS (ESI) calcd for C14H14ClFN3 [MH+], 278.08548; found, 278.08458. Anal. Calcd for C14H13ClFN3: C, 60.55; H, 4.72. Found: C, 60.66; H, 4.79.
(E)-5-(2′-Chloro-6′-fluorostyryl)-2-(N-methylamino)pyrimidine (11b)
Yield 78%, mp 163–165 °C (from ethanol). 1H NMR (DMSO-d6): δ 8.58 (s, 2H), 7.43 (q, 1H, J = 4.6 Hz), 7.38–7.36 (m, 1H), 7.33–7.25 (m, 2H), 7.12 (d, 1H, J = 16.8 Hz), 7.04 (d, 1H, J = 16.8 Hz), 2.83 (d, 3H, J = 4.8 Hz). 13C NMR (DMSO-d6): δ 162.30, 160.38 (d, J = 248.9 Hz), 156.45 (two C), 133.18 (d, J = 5.3 Hz), 130.97 (d, J = 11.4 Hz), 128.86 (d, J = 9.9 Hz), 125.97 (d, J = 3.1 Hz), 123.79 (d, J = 14.4 Hz), 118.98, 115.36, 115.13, 27.99. HRMS (ESI) calcd for C13H12ClFN3 [MH+], 264.06983; found, 264.06918. Anal. Calcd for C13H11ClFN3: C, 59.21; H, 4.20. Found: C, 59.19; H, 4.34.
(E)-5-(2′,6′-Dichlorostyryl)-2-(N,N-dimethylamino)pyrimidine (12a)
Yield 82%, mp 103–104 °C (from hexane). 1H NMR (DMSO-d6): δ 8.64 (s, 2H), 7.52 (d, 2H, J = 8.0 Hz), 7.31–7.27 (m, 1H), 7.04 (d, 1H, J = 16.8 Hz), 6.94 (d, 1H, J = 16.8 Hz), 3.16 (s, 6H). 13C NMR (DMSO-d6): δ 161.28, 156.09 (two C), 134.20, 133.49 (two C), 131.42, 129.03, 128.90 (two C), 118.92, 117.84, 36.76 (two C). HRMS (ESI) calcd for C14H14Cl2N3 [MH+], 294.05593; found, 294.05516. Anal. Calcd for C14H13Cl2N3: C, 57.16; H, 4.45. Found: C, 57.26; H, 4.59.
(E)-5-(2′,6′-Dichlorostyryl)-2-(N-methylamino)pyrimidine (12b)
Yield 91%, mp 191–193 °C (from ethanol). 1H NMR (DMSO-d6): δ 8.58 (s, 2H), 7.52 (d, 2H, J = 8.0 Hz), 7.42 (q, 1H, J = 4.8 Hz), 7.31–7.27 (m, 1H), 7.02 (d, 1H, J = 16.8 Hz), 6.91 (d, 1H, J = 16.8 Hz), 2.83 (d, 3H, J = 5.2 Hz). 13C NMR (DMSO-d6): δ 162.29, 156.34 (two C), 134.26, 133.50 (two C), 131.57, 128.99, 128.88 (two C), 118.62, 118.46, 27.93. HRMS (ESI) calcd for C13H12Cl2N3 [MH+], 280.04028; found, 280.03979. Anal. Calcd for C13H11Cl2N3: C, 55.73; H, 3.96. Found: C, 55.77; H, 4.07.
Biological Studies
MAT2A Inhibition Assay
l-Methionine (50 μM) and ATP (50 μM) were incubated with purified His-tagged MAT2A holoenzyme (3 μg) in 0.3 mL of reaction buffer (50 mM Tris pH8.0, 50 mM KCl, 10 mL MgCl2) at room temperature for 25 min. The Pi released from the reaction was measured with SensoLyte Malachite Green (MG) phosphate assay kit (AnaSpec, 71103). The absorbance was measured at 635 nm on a microplate reader (Spectra MR, DYNEX Technologies). For the inhibition assay, MAT2A holoenzyme was incubated with FIDAS agents at room temperature for 10 min and then mixed with l-methionine and ATP in 0.3 mL of reaction buffer.
Cell Proliferation Studies
LS174T colon cancer cells were plated into 12-well plates (3 × 104 cells/mL). On the next day, the cells were treated with FIDAS agents. Effects of FIDAS analogues on cell proliferation were analyzed using Cell Viability Analyzer (Beckman Coulter, Vi-Cell XR).
hERG Binding Studies
The HEK-293 cell line stably expressing the hERG potassium channel (accession no. U04270), referred to as hERG-HEK cells, were received at passage 11 (P11) from Millipore (CYL3006, lot 2, Billerica, MA). [3H]-Dofetilide (specific activity of 80 Ci/mmol; labeled on the N-methyl group) was obtained from American Radiolabeled Chemicals (St. Louis, MO). Other chemicals and solvents were obtained from Sigma-Aldrich (Milwaukee, WI) with exceptions of polyethylenimine (PEI), which was obtained from Fluka/Sigma-Aldrich (St. Louis, MO), and minimium essential medium (MEM) with GlutaMAX and phenol red, MEM nonessential amino acids solution (NEAA, 100×), G418 disulfate salt solution, fetal bovine serum (FBS), 0.05% Trypsin-EDTA 1× with phenol red, and Hank’s balanced salt solution (HBSS), which were obtained from Life Technologies (Carlsbad, CA).
hERG-HEK Cell Culture
hERG-HEK cells were cultured according to the protocol provided by Millipore. Cells were maintained in MEM (with glutamax and phenol red) supplemented with 10% FBS, 1% NEAA. and 400 μg/mL Geneticin, and incubated at 37 °C in a humidified atmosphere with 5% CO2. Frozen aliquots of cells were transferred into T-75 cm2 flasks and allowed to adhere for 4–8 h. The medium was replaced every 2 days. Passages were carried out at least three times at 6 day intervals after thawing. Cells were dissociated with trypsin/EDTA and seeded into new 150 × 25 mm dishes at (2–3) × 106 cells per dish and placed at 30 °C, 5% CO2, for 40–48 h prior to membrane preparation. Membrane preparation occurred 6 days after the last passage (passage 20).
Membrane Preparation
Cell membrane preparation was based on previous methods.19−22 Cells were rinsed twice with HBSS at 37 °C and collected by scraping the dishes in ∼20 mL of ice-cold 0.32 M sucrose and homogenized on ice with a Teflon pestle using a Maximal Digital homogenizer (Fisher Scientific, Pittsburgh, PA) at ∼280 rpm for 30 s. Homogenates were centrifuged at 300g and 800g for 4 min each at 4 °C. Pellets were resuspended in 9 mL of ice-cold Milli-Q water, and osmolarity was restored by addition of 1 mL of 500 mM Tris buffer (pH 7.4) followed by suspension and centrifugation at 20 000g for 30 min at 4 °C. Pellets were homogenized in 2 mL assay buffer (50 mM Tris, 10 mM KCl, and 1 mM MgCl2, 4 °C), and aliquots of cell membrane suspensions were stored at −80 °C and thawed the day of the [3H]-dofetilide binding assay. Protein content was determined prior to the assay using a Bradford protein assay with bovine albumin as the standard.
[3H]-Dofetilide Binding Assay
[3H]-Dofetilide binding assays using hERG-HEK293 cell membranes were based on previous methods.19 Assays determining concentration–response were conducted in duplicate, and three independent assays were performed for each analogue evaluated. Cell membrane suspension (5 μg) was added to duplicate tubes containing assay buffer, 25 μL of a single concentration of FIDAS agent (concentration range of 10 nM to 100 μM for each experiment), and 25 μL of [3H]-dofetilide (5 nM, final concentration) for an assay volume of 250 μL. Binding occurred for 60 min at 25 °C and was terminated by rapid filtration through Whatman GF/B filters, which were presoaked in 0.25% PEI overnight, using a Brandel cell/membrane harvester (M-48; Brandel Inc., Gaithersburg, MD). Filters were washed three times with ∼1 mL of ice-cold assay buffer. Radioactivity was determined by liquid scintillation spectrometry using the Tri-Carb 2100-TR liquid scintillation analyzer (PerkinElmer Life and Analytical Sciences).
Data Analysis
Compound concentrations that produced 50% inhibition (IC50) in the biological studies were determined from the concentration–response curves via the nonlinear regression one-site competition-fitting program (Prism 5.04; GraphPad Software Inc., San Diego, CA).
Acknowledgments
C.L. and D.S.W. were supported by grant nos. R21 CA139359 and CA172379 from the NIH. L.P.D. and J.R.N. were supported by grant nos. U01 DA013519, UL1TR000117, and T32 DA016176 from the NIH. D.S.W. was supported by the Office of the Dean of the College of Medicine and by NIH grant no. P20 RR020171 from the National Institute of General Medical Sciences to L. Hersh, PI. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NIGMS. The authors thank Craig Vander Kooi for helpful discussions regarding the fluorescence assays.
Glossary
Abbreviations Used
- ATP
adenosine triphosphate
- MAT2A
catalytic subunit of methionine S-adenosyltransferase-2
- CRC
colorectal cancer
- FIDAS
difluorinated N,N′-dialkylaminostilbenes
- DMSO
dimethyl sulfoxide
- ESI
electrospray ionization
- FBS
fetal bovine serum
- HBSS
Hank’s balanced salt solution
- hERG-HEK
HEK-293 cell membranes stably expressing the hERG channel
- hERG
human ether-à-go-go-related protein
- IND
investigational new drug
- LC
liver cancer
- MEM
minimum essential medium
- NMR
nuclear magnetic resonance
- NEAA
nonessential amino acids
- PEI
polyethylenimine
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- SAR
structure–activity relationship
Supporting Information Available
Examples of IC50 determination for the cell proliferation assay and effects of FIDAS agent on colon cancer organoids. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): In accord with University policy, Wen Zhang, Chunming Liu, David S. Watt, and Vitaliy M. Sviripa have disclosed this work to the University’s Intellectual Property Committee that has sought patent protection and that has given a private firm, Liu-Watt, LLC, in which the four principals have partial ownership, an option to license this technology.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhang W.; Sviripa V.; Kril L. M.; Chen X.; Yu T.; Shi J.; Rychahou P.; Evers B. M.; Watt D. S.; Liu C. Fluorinated N,N-dialkylaminostilbenes for Wnt pathway inhibition and colon cancer repression. J. Med. Chem. 2011, 54, 1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Sviripa V.; Chen X.; Shi J.; Yu T.; Hamza A.; Ward N. S.; Kril L. M.; Vander Kooi C. W.; Zhan C.-G.; Evers B. M.; Watt D. S.; Liu C. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem. Biol. 2013, 8, 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt D. S.; Liu C.; Sviripa V.; Zhang W.. Stilbene analogs and methods of treating cancer. Patent US2012/0196874, August 2, 2012.
- Paul S.; Mizuno C. S.; Lee H. J.; Zheng X.; Chajkowisk S.; Rimoldi J. M.; Conney A.; Suh N.; Rimando A. M. In vitro and in vivo studies on stilbene analogs as potential treatment agents for colon cancer. Eur. J. Med. Chem. 2010, 45, 3702–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini F.; Sello G. Natural stilbenes and analogues as antineoplastic agents. Stud. Nat. Prod. Chem. 2008, 34, 77–127. [Google Scholar]
- Maccario C.; Savio M.; Ferraro D.; Bianchi L.; Pizzala R.; Pretali L.; Forti L.; Stivala L. A. The resveratrol analog 4,4′-dihydroxy-trans-stilbene suppresses transformation in normal mouse fibroblasts and inhibits proliferation and invasion of human breast cancer cells. Carcinogenesis 2012, 33, 2172–2180. [DOI] [PubMed] [Google Scholar]
- Mikuła-Pietrasik J.; Sosińska P.; Wierzchowski M.; Piwocka K.; Książek K. Synthetic resveratrol analogue, 3,3′,4,4′,5,5′-hexahydroxy-trans-stilbene, accelerates senescence in peritoneal mesothelium and promotes senescence-dependent growth of gastrointestinal cancers. Int. J. Mol. Sci. 2013, 14, 22483–22498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae Y. S.; Kim J. G.; Jung H. J.; Yang J. D.; Jung J. H.; Aiyar S. E.; Kim S.; Park H. Anticancer effect of (E)-2-hydroxy-3′,4′,5′-trimethoxystilbene on breast cancer cells by mitochondrial depolarization. Cancer Chemother. Pharmacol. 2011, 68, 349–358. [DOI] [PubMed] [Google Scholar]
- Kelkel M.; Jacob C.; Dicato M.; Diederich M. Potential of the dietary antioxidants resveratrol and curcumin in prevention and treatment of hematologic malignancies. Molecules 2010, 15, 7035–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discovery Today 2010, 15, 757–765. [DOI] [PubMed] [Google Scholar]
- Cai J.; Sun W. M.; Hwang J. J.; Stain S. C.; Lu S. C. Changes in S-adenosylmethionine synthetase in human liver cancer: molecular characterization and significance. Hepatology 1996, 24, 1090–1097. [DOI] [PubMed] [Google Scholar]
- Chen H.; Xia M.; Lin M.; Yang H.; Kuhlenkamp J.; Li T.; Sodir N. M.; Chen Y. H.; Josef-Lenz H.; Laird P. W.; Clarke S.; Mato J. M.; Lu S. C. Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells. Gastroenterology 2007, 133, 207–218. [DOI] [PubMed] [Google Scholar]
- Ito K.; Ikeda S.; Kojima N.; Miura M.; Shimizu-Saito K.; Yamaguchi I.; Katsuyama I.; Sanada K.; Iwai T.; Senoo H.; Horikawa S. Correlation between the expression of methionine adenosyltransferase and the stages of human colorectal carcinoma. Surg. Today 2000, 30, 706–710. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Chen J.; Liu L.; Zhang J.; Wang D.; Ma L.; He Y.; Liu Y.; Liu Z.; Wu J. The X protein of hepatitis B virus inhibits apoptosis in hepatoma cells through enhancing the methionine adenosyltransferase 2A gene expression and reducing S-adenosylmethionine production. J. Biol. Chem. 2011, 286, 17168–17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Elliott P.. Principles and Practice of Clinical Cardiovascular Genetics; Oxford University Press: New York, 2010. [Google Scholar]
- Shafqat N.; Muniz J. R. C.; Pilka E. S.; Papagrigoriou E.; von Delft F.; Oppermann U.; Yue W. W. Insight into S-adenosylmethionine biosynthesis from the crystal structures of the human methionine adenosyltransferase catalytic and regulatory subunits. Biochem. J. 2013, 452, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanoff B. E.; Reitz A. B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar]
- Hageman H. A. The von Braun cyanogen bromide reaction. Org. React. 1953, 7, 198–262. [Google Scholar]
- A Practical Guide to Assay Development and High-Throughput Screening in Drug Discovery; Chen T., Ed.; CRC Press: Boca Raton, FL, 2010. [Google Scholar]
- Biswas T.; Resto-Roldán E.; Sawyer S. K.; Artsimovitch I.; Tsodikov O. V. A novel non-radioactive primase-pyrophosphatase activity assay and its application to the discovery of inhibitors of Mycobacterium tuberculosis primase DnaG. Nucleic Acids Res. 2013, 41, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengrass P. M.; Stewart M.; Wood C. M.. Affinity-assay for the human ERG potassium channel. Patent WO2003021271 A2, March 13, 2003.
- Jo S.-H.; Youm J. B.; Lee C. O.; Earm Y. E.; Ho W.-K. Blockade of the HERG human cardiac K+ channel by the antidepressant drug amitryptyline. Br. J. Pharmacol. 2000, 129, 1474–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern W. S.; Carlsson L.; Davis A. S.; Lynch W. G.; MacKenzie I.; Palethorpe S.; Siegl P. K.; Strang I.; Sullivan A. T.; Wallis R.; Camm A. J.; Hammond T. G. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.