

Abstract
Complex biological processes such as inflammation, cell death, migration, proliferation, and the release of biologically active molecules can be used as outcomes in phenotypic assays during early stages of drug discovery. Although target-based approaches have been widely used over the past decades, a disproportionate number of first-in-class drugs have been identified using phenotypic screening. This review details phenotypic assays based on inhibition of microglial activation and their utility in primary and secondary screening, target validation, and pathway elucidation. The role of microglia, both in normal as well as in pathological conditions such as chronic neurodegenerative diseases, is reviewed. Methodologies to assess microglia activation in vitro are discussed in detail, and classes of therapeutic drugs known to decrease the proinflammatory and cytotoxic responses of activated microglia are appraised, including inhibitors of glutaminase, cystine/glutamate antiporter, nuclear factor κB, and mitogen-activated protein kinases.
Keywords: cytokines, reactive oxygen species, nitric oxide, glutamate, screening, cell-based assays
Introduction
Advances in molecular biology and genetics in the 1980s, in addition to the publication of the human genome sequence in 2001, led the pharmaceutical industry to consider every functional protein as a potential therapeutic drug target. Moreover, unwanted side effects to drugs have been attributed to nonspecific interactions between drugs and proteins or systems other than the anticipated target. In that respect, for more than 20 years, drug discovery efforts have been focused on the rational tailoring of high-affinity drugs capable of selectively and specifically inhibiting a particular target known to play a role in disease pathology.1,2
Target-based drug discovery led to the production of many of the drugs currently on the market.3 This approach is most appropriate when there is a clear understanding of the molecular basis of a disease and the structure, function, and regulation of the therapeutic target. However, present levels of productivity in the pharmaceutical industry are suboptimal, and it has been suggested that productivity could increase if a shift from solely target-based assay approaches to inclusion of phenotypic assays (PAs) occurs in the early stages of compound screening.4
In support of this concept, from 1999 to 2008, most new molecular entities approved by the Food and Drug Administration (FDA) were identified using PAs.3 Considering that the number of pharmaceutical programs that used PAs as primary screening assays was much smaller than those employing target-based assays, the implications are that the former strategy is significantly more successful at producing new drugs.3,5,6 PA-based approaches have been particularly effective in challenging therapeutic areas such as the central nervous system (CNS). Seven of the eight small-molecule new molecular entities for CNS pathologies approved between 1999 and 2008 were discovered using PAs.3 However, these two drug discovery approaches have unique advantages and disadvantages; therefore, case-by-case evaluation of each new target or disease is necessary.
Pharmaceutical companies such as Lilly, Novartis, and GlaxoSmithKline have championed the renaissance of phenotypic screening. Examples of some of the recently approved CNS first-in-class drugs discovered using PAs include Chantix (varenicline), a drug prescribed for smoking cessation; the antipsychotic Abilify (aripiprazole); Namenda (memantine), prescribed for dementia; and the antiepileptic Inovelon (rufinamide).3 Out of the four examples mentioned, memantine was identified via in vitro cell-based PAs, varenicline was identified using both cell-based and in vivo PAs, and rufinamide's anticonvulsive and aripirazole's antipsychotic properties were tested directly in animal models (in vivo PAs). Therefore, both in vitro and in vivo PAs have been fundamental for the discovery and development of successful CNS drugs.
Williams and Enna7,8 argued in favor of phenotypic screening for CNS therapeutics based on the high attrition rate for small molecules identified using a target-centric drug development process. These authors point out that historically, much of the successful CNS drug development has been an empirical and in many cases serendipitous process. For example, drugs such as diazepam (anxiety), chlorpromazine (schizophrenia), and iproniazide (depression) were all initially developed for other conditions. It was later found, in patients, that these drugs were able to control the conditions for which they are currently used.
The discovery and development of drugs for complex neurodegenerative diseases such as multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), human immunodeficiency virus 1 (HIV-1)–associated neurocognitive disorders (HAND), Parkinson disease (PD), and Alzheimer disease (AD) could benefit from the use of PAs. The inflammation and neurodegeneration seen in these diseases have been linked to the uncontrolled chronic activation of microglial cells9–16; therefore, use of small molecules to block the deleterious effects of these cells could be therapeutic. This article will review both the role of activated microglia in neurodegeneration and the current methodologies to monitor its inhibition for the identification of novel neurotherapeutics.
Microglial Cells and Their Activation
Microglial cells, as the brain-resident macrophages, are one of the key players in the immunity of the CNS. They are distributed throughout the parenchyma and account for approximately 10% to 20% of the total glial cell population in the brain.17,18 These cells were first described in 1932 by Pío del Rio Hortega.19 Their origin, characteristics, and role under both physiological and pathological conditions have been tirelessly debated since their discovery. However, the general consensus is that microglial cells are derived from primitive yolk sac macrophages that migrate to the brain and become resident cells during early embryo development (E10 to E19 in rodents and between the first and second trimester in humans).20,21
Under normal physiological conditions in a healthy mature brain, microglial cells display surveillance behavior and morphology, exhibiting low cell body mobility and an extensive display of fine processes that are constantly scanning their surroundings.22,23 These cells are sensitive to changes of the brain microenvironment, and as soon as an alteration of homeostasis occurs, activation is triggered. This causes changes in cell shape, behavior, and gene and protein expression, leading to the release of an array of factors that include cytokines (interleukin [IL]–1β, tumor necrosis factor [TNF]–α, IL-6), chemokines (MCP-1), excitatory neurotransmitters (glutamate), complement factors, prostaglandins, and reactive oxygen and nitrogen species (ROS and RNS, respectively). Moreover, activated cells adopt amoeboid morphology, their cellular processes disappear, and their capacity to move, proliferate, and phagocytose increase. Expression levels of membrane receptors and ability to present antigens increase upon activation as well (for an in-depth review of microglia physiology, refer to Kettenmann et al.24).
An acute response of activated microglial cells allows the clearance of the stimulus that originally caused the activation and the repair of the resulting tissue damage. This constitutes a beneficial activation or healthy response designed to effectively reestablish brain homeostasis.25–29 However, when the insults, stresses, or infections cannot be controlled, a prolonged, self-propagating chronic microglial response occurs. Both experimental and epidemiological evidence suggests that this type of response is associated with neurodegenerative diseases, as described in detail below.15,30–32
Activation of microglial cells can be triggered by pathological conditions in vivo or by stimulation with proteins, cytokines, and chemical agents both in vivo and in vitro (Table 1). The most widely used method of triggering microglia activation is the treatment with lipopolysaccharide (LPS), a Gram-negative bacterial immunostimulant that triggers a cascade of proinflammatory events that mimic pathological responses.17,33 Microglial cells, like macrophages and other cells that belong to the innate immunity response, express Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD) proteins (NLRs), and C-type lectin receptors (CLRs) (Fig. 1). These receptors give cells the ability to recognize activating stimuli such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).34 PAMPs capable of inducing microglia activation include bacterial, fungal, parasitic, and viral molecules such as α- and β-glucan, O-linked mannan, viral RNA and DNA, flagellin, chitin, and microbial cell wall components. DAMPs include molecules not normally found in healthy CNS, such as blood-clotting factors, RNA and DNA released by necrotic cells, phosphatidylserine externalized on apoptotic cells, immunoglobulin-antigen complexes, opsonizing complement, and abnormally folded proteins or aggregates.33,35
Table 1.
Examples of Treatments and Conditions That Induce Microglial Cell Activation In Vitro and In Vivo.
| Stimuli | Response induced | Reference |
|---|---|---|
| Pathological condition | ||
| Mechanical nerve injury and neuronal damage | Phagocytosis, proliferation | 123 |
| Hypoxia | ROS | 124, 125 |
| Ischemia | ROS | 126 |
| Biological toxins | ||
| LPS | Cytokines, chemokines, ROS, RNS, glutamate, Ca2+ mobilization | 98 |
| Dopamine quinine | TNF-α, glutamate, prostaglandin E2 | 127 |
| ATP | Ca2+ influx, cytokines, ROS | 128–130 |
| α-Synuclein | Cytokines, chemokines, ROS | 131, 132 |
| Amyloid-β | Cytokines, chemokines, NO, ROS, glutamate, Ca2+ mobilization | 10 |
| Tat | Cytokines, chemokines, glutamate, RNS, iNOS, Ca2+ elevation | 133–136 |
| gp120 | Cytokines, chemokines, glutamate, Ca2+ mobilization | 134,142, 143 |
| tPA (tissue plasminogen activator), thrombin, fibrinogen | Proliferation, NO, TNF-α, IL-6, IL-12, Ca2+ mobilization | 12, 144, 145 |
| Chemical toxins | ||
| MPTP, rotenone, paraquat | ROS, TNF-α | 146–148 |
| Alcohol | ROS | 149 |
| Air pollution | ROS | 150, 151 |
| Phorbol ester (PMA) | ROS | 152 |
ATP, adenosine triphosphate; IFN, interferon; IL, interleukin; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NO, nitric oxide; RNS, reactive nitrogen species; ROS, reactive oxygen species; TNF-α, tumor necrosis factor–α.
Figure 1.
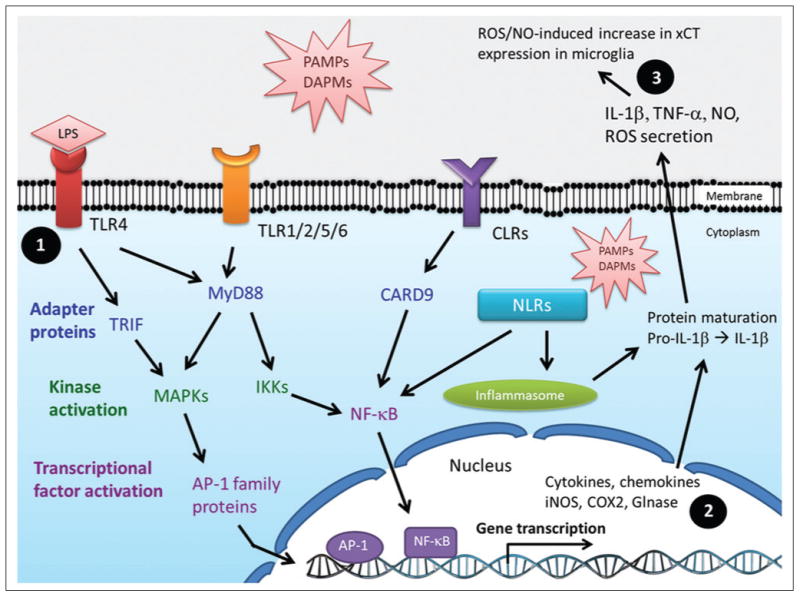
Microglial cell activation signaling pathways. Pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPs, respectively) are recognized by membrane receptors such as Toll-like receptors (TLR1–9), C-type lectin receptors (CLRs), and cytoplasmic sensors like nucleotide-binding oligomerization domain (NOD)–like receptors (NRLs). PAMPs and DAMPs include RNA, DNA, proteins, bacterial and fungal cell wall components, and microbial antigens such as lipopolysaccharides, lipoproteins, peptidoglycan, glycolipids, α- and β-mannan, β-glucan, fucose, and glycosylphosphatidylinositol. (1) Ligand binding to TLRs triggers events dependent on myeloid differentiation primary response protein 88 (MyD88) and TIR domain-containing adaptor protein inducing interferon β (TRIF). MyD88 activates nuclear factor–κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, inducing transcriptional activation of proinflammatory genes such as interleukin (IL)–1 β, tumor necrosis factor–α (TNF-α), IL-6, inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX2). (2) Glutaminase (Glnase) levels and activity have been shown to be increased during inflammatory responses in a NF-κB–dependent manner. (3) Reactive oxygen species (ROS)/nitric oxide (NO) released by activated microglia induce increased expression levels and activity of cystine/glutamate antiporter (xCT) on nearby microglial cells.
There are at least 13 different TLR family members in humans, 9 of which (TLR1–9) have been reported in microglial cells.36 In general, ligand binding to TLRs leads to the activation of signaling pathways that involve the cytoplasmic adapter proteins MyD88 and/or TRIF. These events are followed by the activation of multiple kinases such as IRAK1/4, IKKs, and MAPKs and subsequent activation of transcriptional factors nuclear factor–κB (NF-κB) and AP-1. Ultimately, expression of an array of proinflammatory and neurotoxic genes occurs, and levels of cytokines, chemokines, and enzymes such as cyclooxygenase 2 (COX2), inducible nitric oxide synthase (iNOS),37 and glutaminase38 increase (Fig. 1). Despite the diversity of stimuli that activate microglial cells (Table 1), there is a convergence of signaling pathways that leads to common steps and similar responses downstream (Fig. 1). Induction of microglial cell activation by LPS treatment is considered a broad overarching approach due to the simultaneous triggering of responses dependent on the two main TLR signaling pathways, TRIF and MyD88. From the biological processes evoked in activated microglia, perhaps cell proliferation and the release of biologically active molecules such as cytokines, chemokines, glutamate, ROS, and RNS are the most amenable to in vitro monitoring and manipulation.
Microglia Activation in Neurodegenerative Disorders
As the brain's resident macrophage, microglial cells play a key role in protection against exogenous and endogenous insults. Disruption of brain homeostasis caused by physiological or pathological conditions (Table 1) induces microglial cell activation and the release of cytokines, chemokines, and toxins that lead to inflammation. This appears to be a common process in a number of acute and chronic neurodegenerative diseases such as stroke,39 AD,30 PD,14 MS,40 ALS,41 Huntington disease,15 and HAND,16 inducing or exacerbating neuronal damage. Postmortem analyses of human tissues and animal models of slow progressing diseases such as ALS, PD, and AD have shown neuronal loss concomitantly with increased levels of activated microglia in areas of the brain with lesions.9,11
A well-documented example of the role of microglial cells in neurodegenerative diseases is represented by HAND. HIV-1 crosses the blood-brain barrier (BBB) and infects CD4+ microglial cells, inducing their activation; triggering the release of NO, cytokines, chemokines, and glutamate; upregulating the expression of membrane receptors; and increasing antigen presentation. Infected cells permit virus replication and release of viral particles and toxins such as Tat and gp120 (Table 1), which in turn induce activation of uninfected microglial cells. One of the mechanisms that contributes to neuronal cell death is the overstimulation of NMDA receptors by extracellular glutamate released by activated microglial cells, leading to increased intracellular Ca2+ levels and apoptotic cell death. Furthermore, proinflammatory cytokines and oxidative factors released by HIV-1–infected microglial cells decrease the capacity of oligodendrocytes and astrocytes to uptake extracellular glutamate, thereby altering their neurosupportive/protective role. All these proinflammatory conditions and cytotoxic events exacerbate the neurodegeneration and cognitive decline seen in HIV-infected patients.13,18,24,42
Activated microglial also play a role in ALS pathology. Increased release of NO and superoxide (ROS) from microglia and neuronal cell death are seen in approximately 20% of the patients with the familial form of the disease. These individuals have mutations in the superoxide dismutase 1 (SOD1) gene, which appear to be associated with microglial cell activation mediated by the MyD88 pathway.43–45 Microglial cells are also implicated in AD. Aβ plaques, one of the hallmarks of the disease, have been shown to be surrounded and infiltrated by activated microglia and astrocytes. Moreover, analysis of both animal models and postmortem brains of AD patients shows upregulation of TLR2, 4, and 7, as well as TLR pathway-related genes in microglial cells.45 Interestingly, a genetic polymorphism of TLR4 (Asp299Gly) that weakens the response to receptor stimulation in microglial cells is associated with a 2.7-fold reduction in the susceptibility to late-onset AD.46 Finally, a loss-of-function mutation of TLR4 inhibits Aβ-induced microglial cell activation and reduces levels of released proinflammatory factors NO, IL-6, and TNF-α.47
Interestingly, patients on chronic nonsteroidal anti-inflammatory drugs (NSAIDs) have a lower incidence of AD and show improved performance in cognitive function tests.48 Moreover, a delayed onset of AD has been documented for twins and siblings on anti-inflammatory drugs.49,50 Therefore, both clinical and epidemiological evidence suggest that anti-inflammatory drugs can delay the onset of neurodegenerative disorders. However, it is important to mention two points of debate. First, not all the anti-inflammatory clinical trials have resulted in protection against disease such as AD and ALS.51–60 And second, evidence seems to indicate that early rather than late anti-inflammatory intervention in terms of disease progression might be crucial for therapeutic efficacy.61 These two points highlight the complexity of neurotherapeutics and the challenges that lie ahead in drug discovery. In summary, there is abundant literature supporting the role of activated microglial cells in neurodegenerative diseases; therefore, a reasonable therapeutic approach could involve the neutralization of the harmful proinflammatory responses of microglia.
Assessing Microglial Activation
The most frequently used methods for in vitro activation of microglial cells are shown in Table 1. Treatment with LPS is by far the most employed activation strategy, and despite the complexity of the signaling processes (Fig. 1), the responses that LPS and other stimuli trigger are reasonably well understood. This has allowed the establishment of methodologies to monitor microglial activation both in vitro and in vivo.18 Table 2 compiles the most widely used in vitro microglial cell activation assays.
Table 2.
Commonly Used Methods to Monitor Microglia Activation.
| Method | Response Detected | Reference |
|---|---|---|
| ELISA, ELISPOT | Production and release of TNF-α, IL-1β, IL-6, CCL6, MCP-1 | 62, 153 |
| Western blots, in-cell Western, FACS, and immunocytochemistry | Expression iNOS, COX2, CD86, IL-1β, IL-6 | 62, 76, 154, 155 |
| Colorimetric assays | Release of NO | 76 |
| Fluorescence and luminescence assays | Release of glutamate, ROS, and RNS; calcium mobilization; and cell viability | 63, 156–160 |
| Quantitative RT-PCR | Changes in levels of activation marker mRNAs (iNOS, COX2, CD86, IL-1β, TNF-α, CD11b) | 155 |
| HPLC | Glutamate release | 156, 161, 162 |
| Electrophysiological measurements | Alterations of microglial membrane potential | 163, 164 |
| Mass spectrometry | Glutamate levels and in vivo cell proliferation | 66, 161 |
COX2, cyclooxygenase 2; ELISA, enzyme-linked immunosorbent assay; ELISPOT, enzylme-linked immunosorbent spot; FACS, fluorescence-activated cell sorting; HPLC, high-performance liquid chromatography; IL, interleukin; iNOS, inducible nitric oxide synthase; mRNA, messenger RNA; NO, nitric oxide; RNS, reactive nitrogen species; ROS, reactive oxygen species; RT-PCR, reverse transcription PCR; TNF-α, tumor necrosis factor–α.
The most common way of monitoring microglial cell activation is the determination of changes in extracellular levels of cytokines, chemokines, or nitric oxide (NO). There is a broad selection of commercially available cytokine and chemokine enzyme-linked immunosorbent assay (ELISA) kits rendering the detection of TNF-α, IL-6, IL-1β, and MCP-1 a simple, reproducible, and relatively inexpensive process.62 The diazotization assay (Griess reaction) is one of the quickest, most widely used, and inexpensive methods to estimate extracellular NO levels in biological samples. This assay measures NO2− (nitrite) derived from NO upon reaction with O2. In general, levels of NO2− correlate well with NO levels. For a detailed review on alternative strategies to detect this metabolite, refer to Hetrick and Shoenfisch.63
One aspect of microglial cell activation that is often undervalued is the change in proliferation rate, an important mechanism of the cellular response implicated in either exacerbation or amelioration of neurodegeneration.64,65 Shankaran et al.66 developed an elegant mass spectrometry– based method to determine in vivo microglial proliferation rates. By giving 2H2 O to mice treated with or without LPS, the authors were able to label the DNA of proliferating cells. Brains were harvested at different time points, and microglial cells were isolated by cell sorting. DNA purification followed by enzymatic hydrolysis, purine derivatization, and mass spectrometry analysis allowed determination of deuterium incorporation into DNA as a function of time, allowing estimation of the percentage of new microglial cells generated as a consequence of LPS-induced activation.
Several of the methods mentioned in Table 2 are routinely used in basic research studies of microglial cell activation; however, PA development can equally benefit from these methodologies. For example, quantitative reverse transcription PCR (qRT-PCR) for high-throughput screening (HTS) has been used to determine the effects of small molecules on messenger RNA (mRNA) levels of a component in a pathway of interest. Due to lower reagent costs, the use of liquid-handling systems, and the miniaturization of reactions, such HTS assays are now cost-effective, rapid, and robust.67 Since microglial cell activation induces changes in gene expression of proteins such as COX2, iNOS, IL-1R, SOCS3, and CD86, qRT-PCR could be an efficient way to determine if small molecules block activation responses.68 Ultimately, it is likely that the number of strategies to monitor activated microglial cell phenotypes and functions (Table 2) will only continue to grow, making these assays more accessible, inexpensive, robust, and HTS adaptable.
Microglia Activation Assays
Primary Phenotypic Screening
Use of cell-based assays to identify small-molecule inhibitors of microglial cell activation permits simultaneous probing of multiple targets without bias (Fig. 1). For example, a decrease in the levels of a toxin released by activated microglia could be due to a variety of events such as direct inhibition of the enzyme responsible for toxin production, inhibition of the toxin release pathway, increase in enzyme or toxin degradation, inhibition of enzyme transcription, or translation, blockade of posttranslational modifications, or protein-protein interactions required for enzyme activity.69–74 It is also possible that compound efficacy could be achieved only when multiple pathways or targets are engaged simultaneously.75 Hence, the power of PAs in primary screenings is underscored in the case of complex and not yet fully understood biological processes and diseases. Figure 2 depicts the basic steps involved in conducting microglial activation PAs.
Figure 2.
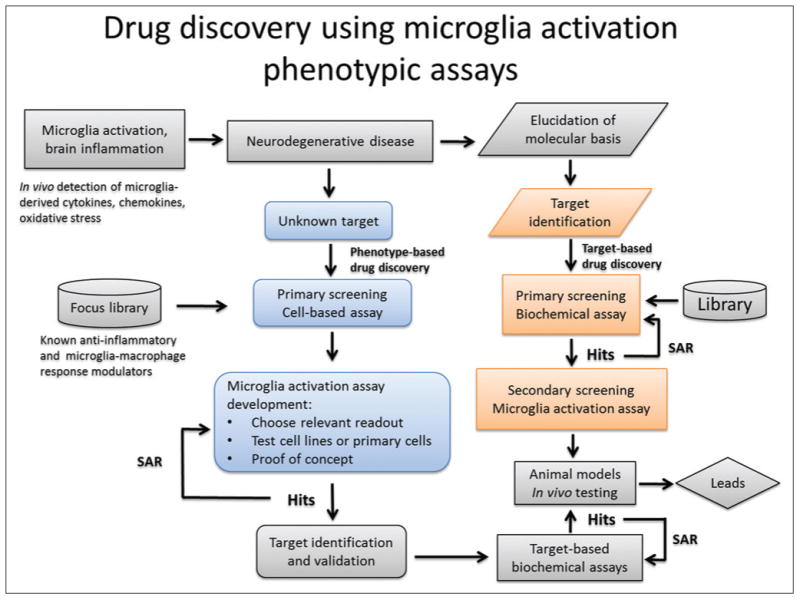
Schematic representation of work flow for primary and secondary phenotypic assay screening strategies based on microglial activation. SAR, structure-activity relationship.
Both immortalized cell lines and primary microglia have been used in PAs.76,77 The most widely used rodent cell lines are BV-2, C8-B4, N9, N11, various EOC lines, highly aggressive proliferating immortalized (HAPI) cell line, and MG5.62,78,79 Nagai et al.80 generated and described the only human immortalized microglial cell line (HMO6) with a phenotype closely related to primary microglia. A human cell line would be preferred for neurotherapeutic studies; however, thus far, only a few groups have used these cells, as reflected by the sparse literature, and due to commercial licensing restrictions, no wide distribution of the cell line is possible.
All these cell lines retain different degrees of the morphological, phenotypical, and functional properties seen in freshly isolated primary microglia. The most popular of those cell lines, BV-2, was generated in 1990 by viral transformation with v-raf/v-myc of murine neonatal microglial cells.81 Perhaps due to their ease of growth, maintenance, and use, BV-2 are the preferred cells for in vitro assays. LPS stimulation of these cells causes release of cytokines, RNS, ROS, and glutamate with a similar but slightly reduced response as compared with primary microglia, particularly in the case of glutamate production.62 Nonetheless, there is abundant literature demonstrating that these cells can be used to study activation induced by various stimuli using PAs such as cytokine ELISA or qRT-PCR and NO determination with Griess reagent.62,82 Figure 3A shows modulation of LPS-induced TNF-α levels in BV-2 cells by the flavonoids apigenin and fisetin.
Figure 3.
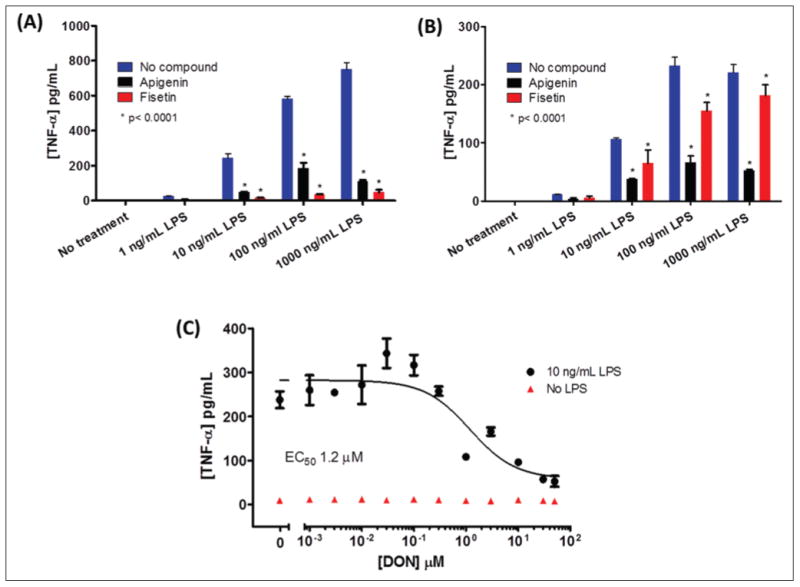
Modulation of tumor necrosis factor–α (TNF-α) release from microglial cell lines. (A) BV-2 cells and (B) C8-B4 cells (3 × 104 cells/well plated 16 h before experiment in poly D-Lys–coated 96-well plates) were treated with lipopolysaccharide (LPS) (1–1000 ng/mL) for 4 h in 2% fetal bovine serum (FBS) containing RPMI and Dulbecco's modified Eagle's medium, respectively. Media were removed from plates (25 μL) and used to determine levels of TNF-α released by the cells in the absence or presence of 20 μM microglia activation inhibitors apigenin and fisetin added to cells 30 min prior to LPS treatment. (C) Inhibition of microglial activation by the glutaminase inhibitor DON added to the cells 30 min prior to LPS treatment (10 ng/mL for 4 h). Levels of TNF-α were determined using an eBioscience (San Diego, CA) mouse TNF-α enzyme-linked immunosorbent assay kit (catalog number 88-7324-88) according to the manufacturer's instructions.
Another cell line that has been successfully used is the C8-B4. This is a spontaneously transformed mouse microglial cell line capable of producing cytokines, NO, and glutamate.83 Figure 3B,C shows how LPS-induced TNF-α release can also be modulated by apigenin and fisetin, as well as the glutaminase inhibitor 6-diazo-5-oxo-L-norleucine (DON). Glutaminase is an enzyme that produces glutamate by catalyzing the deamination reaction of glutamine; it is believed to play a role in HAND and MS38,84 (Fig. 1). Induction in expression of this enzyme has been linked to the MyD88 pathway and NF-κB transcriptional activity.38,85,86 The rest of the microglial cell lines mentioned have not been well characterized, although production of various cytokines and/or NO has been confirmed for most of them.87–90
Experimental evidence shows that compared with cell lines, primary microglial cells more closely resemble both the phenotype and the stimulus responses of microglial cells in vivo.79 The simplest and most inexpensive method of primary microglial isolation (>95% purity) consists of establishing a confluent mixed glial culture from the brains of neonate rodents. Isolation of the microglia can be accomplished by gentle shaking of the flask containing the cells and collecting the detached cells.91 Levels of extracellular glutamate released by mouse or rat primary microglial cells can be determined using an assay that consists of two reactions, one catalyzed by glutamate oxidase and the following one by horseradish peroxidase (HRP). In the presence of Amplex Red, these reactions generate the fluorescent product resorufin. Figure 4 shows how it is possible to modulate in vitro the levels of glutamate released from rat primary microglia using the flavonoids apigenin and fisetin, the tetracycline derivative minocycline (Fig. 4A), and a cystine/glutamate antiporter (xCT) inhibitor, erastin (Fig. 4B). It is believed that the xCT transporter plays a role in neurodegeneration by releasing excess glutamate in exchange for extracellular cystine, which is required to produce glutathione, an essential antioxidant molecule necessary to control activated microglial-induced oxidative stress42,92–94 (Fig. 1). Experimental evidence shows that NO, ROS, Aβ, LPS, and other treatments induce increased expression levels and activity of xCT.93,95,96
Figure 4.
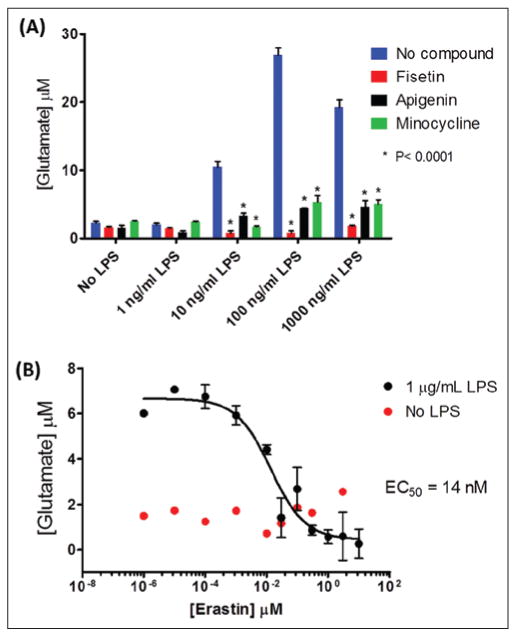
Modulation of lipopolysaccharide (LPS)–induced glutamate levels in rat primary microglia-conditioned media. (A) Extracellular levels of glutamate increase when rat primary microglial cells (3.5 × 104 cells/well plated in poly D-Lys-coated 96-well plates) were treated with 1 μg/mL LPS for 24 h in phenol red–free and serum-free Dulbecco's modified Eagle's medium containing 2 mM glutamine. The microglia activation inhibitors apigenin, fisetin, and minocycline (20 μM) reduced glutamate levels in activated microglia-conditioned media. (B) Inhibitory dose-response curve for LPS-induced glutamate production from rat primary microglia by the xCT inhibitor, erastin. Compounds were added to cells 30 min prior to LPS stimulation. Levels of glutamate in conditioned media were determined using a fluorescent assay that consisted of the oxidation of glutamate by glutamate oxidase (0.04 U/mL) to generate α-ketoglutarate, ammonia, and hydrogen peroxide, coupled to a horseradish peroxidase (0.125 U/mL) reaction in which the generated hydrogen peroxide reacts with 50 μM Amplex Red (Molecular Probes, Life Technologies, Grand Island, NY) to produce resorufin (excitation = 530 nm, emission fluorescence = 590 nm).
Recently, it has been possible to produce microglial cells from stem cells using a modified neuronal differentiation method.97–99 From a pathophysiological point of view, these microglial cells are more relevant than immortalized cell lines due to their similarity to freshly isolated primary microglia, essentially displaying undistinguishable morphology, functions, and cellular markers. In addition, a virtually unlimited supply of these cells could be generated by in vitro proliferation, eliminating the need to euthanize large numbers of animals and making achievable the HTS of thousands of compounds. Despite advances in the development of protocols for stem cell–derived microglial cell generation, access to these cells, technology, and expertise is still limited.
The mass spectrometry–based method developed by Shankaran et al.66 to determine microglial cell proliferation rates previously described has been used as a primary screen PA to test a panel of approved drugs with pleiotropic activities in the search of compounds capable of inhibiting in vivo LPS-induced microglial cell proliferation and neuroinflammation. This strategy allowed the identification of isotretinoin as an antiproliferative compound that was also shown to delay clinical symptoms in the experimental autoimmune encephalomyelitis (EAE) model.
To date, efforts to find compounds capable of inhibiting microglial cell activation, including the few HTS campaigns carried out so far, have yielded a set of drugs, natural products, and synthetic derivatives that exhibited anti-neuroinflammatory properties. These compounds have been identified using nitrite assays, cytokine ELISAs, and/or monitoring cytokine mRNA levels in microglial cell lines (BV-2 cells mostly) activated with stimuli such as LPS, Aβ, or necrotic neuronal cells. Some of the identified compounds include nonsteroidal anti-inflammatory drugs (NSAIDs),48 dexamethasone, pentoxifylline,100 tetracycline derivatives (minocycline),101,102 flavonoids (apigenin, fisetin, luteolin),103 extracts of ginger,104 tetrandrine from Stephania tetrandra (Menispermaceae plant) acting via NF-κB inhibition,105 3-amino-6-phenyl-pyridazine derivatives inhibitors of protein kinases,76,106 KT-15073 and KT-15087 causing NF-κB and MAPK inhibition,77 senkyunolide A and Z-ligustilide from Ligusticum chuanxiong (traditional Chinese herbal medicine),107 cannabinoids,108 and triamcinolone acetonide and amcinonide.109 Once a compound has been identified in a primary screen as an inhibitor of microglial cell activation, it is imperative to show if this inhibition is accompanied by neuroprotective effects. Assays with microglial/neuronal cell co-cultures have shown the neuroprotective properties of KT-15073, KT-15087, apigenin, luteolin, and minocycline. Furthermore, animal models and clinical trials have shown efficacy and tolerability for NSAIDs, pentoxifylline, and minocycline.56,77,101–103,110–114
Secondary Screening in Target-Based Drug Discovery
PAs can also play a critical role in target-based drug discovery as confirmatory functional assays for compounds identified by primary screening. If a target is known to be involved in the activation of microglia, a phenotypic assay such as those described above would allow testing the effects of the compounds in a biological context (Fig. 2).
As an example of target-based primary screening, Benicchi et al.115 described a novel homogeneous time-resolved fluorescence assay to identify small-molecule inhibitors of the TWEAK-Fn14 protein-protein interaction, which is involved in activation of the NF-κB pathway in models of microglia-dependent CNS inflammation. Even though it has not yet been done, this target-based primary assay could be followed by a secondary microglial activation PA to assess the functional efficacy of identified hit compounds.
PAs can also be used to determine the effects of modulation of microglia activation on other cells in the brain by assaying co-cultures of microglial and other cells such as neurons, astrocytes, and oligodendrocytes. For example, to assess if neuronal protection occurs when activation of microglial cells is inhibited, the viability of neurons could be monitored with assays such as lactate dehydrogenase activity, caspase activity, or MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) in microglia-neuron co-cultures.116
When experimental and/or epidemiological evidence supports the role of a target in disease pathology, validation of this target can be carried out using PAs with either chemical or genetic probes. Manipulation of the target using small molecules (chemical probes) or genetic approaches such as protein overexpression, gene knockouts, and RNAi (RNA interference) can be used to induce changes in target function or expression levels inducing a loss-of-function or a gain-of-function phenotype. The phenotypic outcome can then be monitored, validating the target's role in the functional assay system.
Similarly, chemical and genetic approaches are useful in pathway elucidation and unraveling the molecular mechanisms of a disease. An interesting example that illustrates the use of microglial cell activation assays as a tool in pathway building was published by Burguillos et al.117 In this work, activation of BV-2 cells by LPS was shown by RT-PCR and Western blots to induce increases in expression of iNOS and IKK-β and to produce ROS. Moreover, activation of the key apoptosis executioners, caspase 3 and 7, in the absence of poly(ADP-ribose) polymerase (PARP) cleavage and cell death was required for microglial activation. Downregulation of caspase 3/7 and caspase 8 expression by gene silencing with small interference RNA (siRNA) and inhibition of their enzymatic activity using chemical probes demonstrated reduction of microglial cell activation as monitored via ROS production and iNOS expression. Similarly, the activity of protein kinase C-δ (PKC-δ) and its cleavage by caspase 3/7 were required for microglial activation. Finally, both LPS and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) treatment of rats and postmortem analysis of brains of AD and PD patients showed microglial activation and caspase reactivity in regions of the brain with lesions. Based on these results, it could be of interest to conduct target-based screening for inhibitors of caspase 8, 3/7, or PKC-δ to control microglia-dependent chronic inflammation in the brain.
Once an active compound has been identified by phenotypic screening, it is important to find its molecular target so that lead optimization can be directed (Fig. 2). Some of the common methods used for target identification include (1) affinity purification of the target with beads coupled to the compound, (2) the use of expression libraries (phage display or bacterial libraries) to identify the protein(s) capable of interacting with the compound, and (3) protein microarrays of full-length expressed recombinant proteins to screen those capable of binding to the compound. For the use of protein microarrays in target identification, readers can refer to the work of MacBeath and Schreiber,118 in which the authors test three well-known small molecule– target interactions: streptavidin-biotin, anti-digoxigen anti-body–digoxigen, and FKBP12-AP1497. A thorough review of these and other strategies to accomplish target identification was done by Hart.119 Last, it is important to point out that target identification is not strictly required for structure-activity relationship (SAR) studies and that elucidation of a compound's mechanism of action is not necessary for FDA approval of a drug.3,5,120–122
Challenges and Perspective
Swinney and Anthony3 looked at the first-in-class new molecular entities approved by the FDA between 1999 and 2008 and found that a shift in drug discovery strategies has occurred from target to phenotype based. In that period, 28 (37%) of the 75 approved drugs were discovered using PAs, in contrast to 17 (23%) discovered using target-based approaches. Moreover, of the 8 CNS small-molecule drugs approved, 7 were discovered using PAs, highlighting the success of this approach in perhaps one of the most challenging therapeutic areas.
Although PAs can have significant utility in drug discovery, it is important to note their limitations. First is the inherent variability of using living cellular systems. Although this can be somewhat minimized by carefully controlling experimental conditions, the assay variability will likely be more significant relative to a biochemical assay with purified proteins. The second concern is that modulation of a response in a PA could be due to the ability of a compound to simultaneously block the activity of multiple targets, making SAR studies for compound optimization challenging.
However, on balance, PAs can aid the early drug discovery process, focusing on compounds that have functional implications in a pathologically relevant biological system. PAs can precede HTS as a means to validate therapeutic targets or to test the capacity of a library of compounds to inhibit a specific biological process. Moreover, PAs can also follow HTS as a tool to confirm lead compounds that are active in a functional cellular assay (Fig. 2). Improved success rates in drug discovery might be imminently achievable if diversification of screening strategies is implemented.
The use of activated microglia PAs has demonstrated that inhibition of targets such as glutaminase, xCT, NF-κB, and MAPKs101 is correlated with decreased release of toxic factors from activated microglia. Identification of safe and efficacious compounds capable of inhibiting microglial activation, as well as protecting neuronal cells, may provide a novel therapeutic approach for some of the most prevalent neurodegenerative diseases.
Acknowledgments
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research is supported by the National Institute of Mental Health (NIMH), Center for Novel Therapeutics for HIV-Associated Cognitive Disorders (P30 MH075673-06) and the Johns Hopkins Brain Science Institute.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Imming P, Sinning C, Meyer A. Drugs, Their Targets and the Nature and Number of Drug Targets. Nat Rev Drug Discov. 2006;5:821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay MA. Target Discovery. Nat Rev Drug Discov. 2003;2:831–838. doi: 10.1038/nrd1202. [DOI] [PubMed] [Google Scholar]
- 3.Swinney DC, Anthony J. How Were New Medicines Discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 4.Paul SM, Mytelka DS, Dunwiddie CT, et al. How to Improve R&D Productivity: The Pharmaceutical Industry's Grand Challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 5.Lee JA, Uhlik MT, Moxham CM, et al. Modern Phenotypic Drug Discovery Is a Viable, Neoclassic Pharma Strategy. J Med Chem. 2012;55:4527–4538. doi: 10.1021/jm201649s. [DOI] [PubMed] [Google Scholar]
- 6.Macarron R, Banks MN, Bojanic D, et al. Impact of High-Throughput Screening in Biomedical Research. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 7.Enna SJ, Williams M. Challenges in the Search for Drugs to Treat Central Nervous System Disorders. J Pharmacol Exp Ther. 2009;329:404–411. doi: 10.1124/jpet.108.143420. [DOI] [PubMed] [Google Scholar]
- 8.Williams M, Enna SJ. Prospects for Neurodegenerative and Psychiatric Disorder Drug Discovery. Expert Opin Drug Discov. 2011;6:457–463. doi: 10.1517/17460441.2011.562497. [DOI] [PubMed] [Google Scholar]
- 9.Polazzi E, Monti B. Microglia and Neuroprotection: From In Vitro Studies to Therapeutic Applications. Prog Neurobiol. 2010;92:293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Bianca VD, Dusi S, Bianchini E, et al. Beta-Amyloid Activates the O-2 Forming NADPH Oxidase in Microglia, Monocytes, and Neutrophils: A Possible Inflammatory Mechanism of Neuronal Damage in Alzheimer's Disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 11.Boillee S, Yamanaka K, Lobsiger CS, et al. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 12.Lee DY, Oh YJ, Jin BK. Thrombin-Activated Microglia Contribute to Death of Dopaminergic Neurons in Rat Mesencephalic Cultures: Dual Roles of Mitogen-Activated Protein Kinase Signaling Pathways. Glia. 2005;51:98–110. doi: 10.1002/glia.20190. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Eugenin EA, Berman JW, et al. The Neurology of AIDS. 3rd Oxford University Press; New York: 2012. [Google Scholar]
- 14.McGeer PL, Itagaki S, Boyes BE, et al. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson's and Alzheimer's Disease Brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 15.Moller T. Neuroinflammation in Huntington's Disease. J Neural Transm. 2010;117:1001–1008. doi: 10.1007/s00702-010-0430-7. [DOI] [PubMed] [Google Scholar]
- 16.Zindler E, Zipp F. Neuronal Injury in Chronic CNS Inflammation. Best Pract Res Clin Anaesthesiol. 2010;24:551–562. doi: 10.1016/j.bpa.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Lynch MA. The Multifaceted Profile of Activated Microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 18.Luo XG, Chen SD. The Changing Phenotype of Microglia from Homeostasis to Disease. Transl Neurodegener. 2012;1:9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.del Rio-Hortega P. Microglia in: Cytology and Cellular Pathology of the Nervous System. Paul B Hoeber; New York: 1932. p. 1267. [Google Scholar]
- 20.Ginhoux F, Greter M, Leboeuf M, et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monier A, Adle-Biassette H, Delezoide AL, et al. Entry and Distribution of Microglial Cells in Human Embryonic and Fetal Cerebral Cortex. J Neuropathol Exp Neurol. 2007;66:372–382. doi: 10.1097/nen.0b013e3180517b46. [DOI] [PubMed] [Google Scholar]
- 22.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 23.Davalos D, Grutzendler J, Yang G, et al. ATP Mediates Rapid Microglial Response to Local Brain Injury In Vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 24.Kettenmann H, Hanisch UK, Noda M, et al. Physiology of Microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 25.Imai F, Suzuki H, Oda J, et al. Neuroprotective Effect of Exogenous Microglia in Global Brain Ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- 26.Kim YJ, Park HJ, Lee G, et al. Neuroprotective Effects of Human Mesenchymal Stem Cells on Dopaminergic Neurons through Anti-Inflammatory Action. Glia. 2009;57:13–23. doi: 10.1002/glia.20731. [DOI] [PubMed] [Google Scholar]
- 27.Lalancette-Hebert M, Gowing G, Simard A, et al. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambertsen KL, Clausen BH, Babcock AA, et al. Microglia Protect Neurons against Ischemia by Synthesis of Tumor Necrosis Factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann J, Sauerzweig S, Ronicke R, et al. Microglia Cells Protect Neurons by Direct Engulfment of Invading Neutrophil Granulocytes: A New Mechanism of CNS Immune Privilege. J Neurosci. 2008;28:5965–5975. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao HM, Hong JS. Why Neurodegenerative Diseases Are Progressive: Uncontrolled Inflammation Drives Disease Progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pocock JM, Liddle AC, Hooper C, et al. Activated Microglia in Alzheimer's Disease and Stroke. Ernst Schering Res Found Workshop. 2002:105–132. doi: 10.1007/978-3-662-05073-6_7. [DOI] [PubMed] [Google Scholar]
- 32.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's Disease: Its Role in Neuronal Death and Implications for Therapeutic Intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanisch UK. Proteins in Microglial Activation—Inputs and Outputs by Subsets. Curr Protein Pept Sci. 2013;14:3–15. doi: 10.2174/1389203711314010003. [DOI] [PubMed] [Google Scholar]
- 34.Colton CA. Heterogeneity of Microglial Activation in the Innate Immune Response in the Brain. J Neuroimmune Pharmacol. 2009;4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Block ML, Zecca L, Hong JS. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 36.Saijo K, Glass CK. Microglial Cell Origin and Phenotypes in Health and Disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 37.Turrin NP, Rivest S. Molecular and Cellular Immune Mediators of Neuroprotection. Mol Neurobiol. 2006;34:221–242. doi: 10.1385/MN:34:3:221. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Zhao L, Jia B, et al. Glutaminase Dysregulation in HIV-1–Infected Human Microglia Mediates Neurotoxicity: Relevant to HIV-1–Associated Neurocognitive Disorders. J Neurosci. 2011;31:15195–15204. doi: 10.1523/JNEUROSCI.2051-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morioka T, Kalehua AN, Streit WJ. Characterization of Microglial Reaction after Middle Cerebral Artery Occlusion in Rat Brain. J Comp Neurol. 1993;327:123–132. doi: 10.1002/cne.903270110. [DOI] [PubMed] [Google Scholar]
- 40.Ziehn MO, Avedisian AA, Tiwari-Woodruff S, et al. Hippocampal CA1 Atrophy and Synaptic Loss during Experimental Autoimmune Encephalomyelitis, EAE. Lab Invest. 2010;90:774–786. doi: 10.1038/labinvest.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banati RB, Gehrmann J, Kellner M, et al. Antibodies against Microglia/Brain Macrophages in the Cerebrospinal Fluid of a Patient with Acute Amyotrophic Lateral Sclerosis and Presenile Dementia. Clin Neuropathol. 1995;14:197–200. [PubMed] [Google Scholar]
- 42.Potter MC, Figuera-Losada M, Rojas C, et al. Targeting the Glutamatergic System for the Treatment of HIV-Associated Neurocognitive Disorders. J Neuroimmune Pharmacol. 2013;8:594–607. doi: 10.1007/s11481-013-9442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang J, Rivest S. MyD88-Deficient Bone Marrow Cells Accelerate Onset and Reduce Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. J Cell Biol. 2007;179:1219–1230. doi: 10.1083/jcb.200705046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 45.Trudler D, Farfara D, Frenkel D. Toll-Like Receptors Expression and Signaling in Glia Cells in Neuro-Amyloidogenic Diseases: Towards Future Therapeutic Application. Mediators Inflamm. 2010;2010:497987. doi: 10.1155/2010/497987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minoretti P, Gazzaruso C, Vito CD, et al. Effect of the Functional Toll-Like Receptor 4 Asp299Gly Polymorphism on Susceptibility to Late-Onset Alzheimer's Disease. Neurosci Lett. 2006;391:147–149. doi: 10.1016/j.neulet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 47.Walter S, Letiembre M, Liu Y, et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer's Disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 48.Alafuzoff I, Overmyer M, Helisalmi S, et al. Lower Counts of Astroglia and Activated Microglia in Patients with Alzheimer's Disease with Regular Use of Non-Steroidal Anti-Inflammatory Drugs. J Alzheimers Dis. 2000;2:37–46. doi: 10.3233/jad-2000-2105. [DOI] [PubMed] [Google Scholar]
- 49.Breitner JC, Welsh KA, Helms MJ, et al. Delayed Onset of Alzheimer's Disease with Nonsteroidal Anti-Inflammatory and Histamine H2 Blocking Drugs. Neurobiol Aging. 1995;16:523–530. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- 50.Zandi PP, Anthony JC, Hayden KM, et al. Reduced Incidence of AD with NSAID but Not H2 Receptor Antagonists: The Cache County Study. Neurology. 2002;59:880–886. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
- 51.Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. Trial of Celecoxib in Amyotrophic Lateral Sclerosis. Ann Neurol. 2006;60:22–31. doi: 10.1002/ana.20903. [DOI] [PubMed] [Google Scholar]
- 52.Fourrier A, Letenneur L, Begaud B, et al. Nonsteroidal Antiinflammatory Drug Use and Cognitive Function in the Elderly: Inconclusive Results from a Population-Based Cohort Study. J Clin Epidemiol. 1996;49:1201. doi: 10.1016/0895-4356(96)00202-8. [DOI] [PubMed] [Google Scholar]
- 53.Gordon PH, Moore DH, Miller RG, et al. Efficacy of Minocycline in Patients with Amyotrophic Lateral Sclerosis: A Phase III Randomised Trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- 54.Henderson AS, Jorm AF, Christensen H, et al. Aspirin, Anti-Inflammatory Drugs and Risk of Dementia. Int J Geriatr Psychiatry. 1997;12:926–930. [PubMed] [Google Scholar]
- 55.Jonker C, Comijs HC, Smit JH. Does Aspirin or Other NSAIDs Reduce the Risk of Cognitive Decline in Elderly Persons? Results from a Population-Based Study. Neurobiol Aging. 2003;24:583–588. doi: 10.1016/s0197-4580(02)00188-4. [DOI] [PubMed] [Google Scholar]
- 56.Levin BJ, Thompson G, Mitsumoto LH, et al. Pentoxifylline in ALS: A Double-Blind, Randomized, Multicenter, Placebo-Controlled Trial. Neurology. 2006;66:1786–1787. doi: 10.1212/01.wnl.0000230561.21579.ad. [DOI] [PubMed] [Google Scholar]
- 57.May FE, Moore MT, Stewart RB, et al. Lack of Association of Nonsteroidal Anti-Inflammatory Drug Use and Cognitive Decline in the Elderly. Gerontology. 1992;38:275–279. doi: 10.1159/000213340. [DOI] [PubMed] [Google Scholar]
- 58.Nilsson SE, Johansson B, Takkinen S, et al. Does Aspirin Protect against Alzheimer's Dementia? A Study in a Swedish Population-Based Sample Aged > or =80 Years. Eur J Clin Pharmacol. 2003;59:313–319. doi: 10.1007/s00228-003-0618-y. [DOI] [PubMed] [Google Scholar]
- 59.Saag KG, Rubenstein LM, Chrischilles EA, et al. Nonsteroidal Antiinflammatory Drugs and Cognitive Decline in the Elderly. J Rheumatol. 1995;22:2142–2147. [PubMed] [Google Scholar]
- 60.Scharf S, Mander A, Ugoni A, et al. A Double-Blind, Placebo-Controlled Trial of Diclofenac/Misoprostol in Alzheimer's Disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 61.Tabas I, Glass CK. Anti-Inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horvath RJ, Nutile-McMenemy N, Alkaitis MS, et al. Differential Migration, LPS-Induced Cytokine, Chemokine, and NO Expression in Immortalized BV-2 and HAPI Cell Lines and Primary Microglial Cultures. J Neurochem. 2008;107:557–569. doi: 10.1111/j.1471-4159.2008.05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hetrick EM, Schoenfisch MH. Analytical Chemistry of Nitric Oxide. Annu Rev Anal Chem (Palo Alto Calif) 2009;2:409–433. doi: 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gowing G, Philips T, Van Wijmeersch B, et al. Ablation of Proliferating Microglia Does Not Affect Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Caused by Mutant Superoxide Dismutase. J Neurosci. 2008;28:10234–10244. doi: 10.1523/JNEUROSCI.3494-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tikka T, Fiebich BL, Goldsteins G, et al. Minocycline, a Tetracycline Derivative, Is Neuroprotective against Excitotoxicity by Inhibiting Activation and Proliferation of Microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shankaran M, Marino ME, Busch R, et al. Measurement of Brain Microglial Proliferation Rates In Vivo in Response to Neuroinflammatory Stimuli: Application to Drug Discovery. J Neurosci Res. 2007;85:2374–2384. doi: 10.1002/jnr.21389. [DOI] [PubMed] [Google Scholar]
- 67.Bittker JA. High-Throughput RT-PCR for Small-Molecule Screening Assays. Curr Protoc Chem Biol. 2012;4:49–63. doi: 10.1002/9780470559277.ch110204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colton CA, Mott RT, Sharpe H, et al. Expression Profiles for Macrophage Alternative Activation Genes in AD and in Mouse Models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhatia HS, Candelario-Jalil E, de Oliveira AC, et al. Mangiferin Inhibits Cyclooxygenase-2 Expression and Prostaglandin E2 Production in Activated Rat Microglial Cells. Arch Biochem Biophys. 2008;477:253–258. doi: 10.1016/j.abb.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 70.Chung HS, Kim SN, Jeong JH, et al. A Novel Synthetic Compound 4-Acetyl-3-methyl-6-(2-bromophenyl) pyrano[3,4-c]pyran-1,8-dione Inhibits the Production of Nitric Oxide and Proinflammatory Cytokines Via the NF-kappaB Pathway in Lipopolysaccharide-Activated Microglia Cells. Neurochem Res. 2013;38:807–814. doi: 10.1007/s11064-013-0983-6. [DOI] [PubMed] [Google Scholar]
- 71.Huo Y, Rangarajan P, Ling EA, et al. Dexamethasone Inhibits the Nox-Dependent ROS Production via Suppression of MKP-1–Dependent MAPK Pathways in Activated Microglia. BMC Neurosci. 2011;12:49. doi: 10.1186/1471-2202-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jana M, Pahan K. Gemfibrozil, a Lipid Lowering Drug, Inhibits the Activation of Primary Human Microglia via Peroxisome Proliferator–Activated Receptor Beta. Neurochem Res. 2012;37:1718–1729. doi: 10.1007/s11064-012-0781-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Terazawa R, Akimoto N, Kato T, et al. A Kavalactone Derivative Inhibits Lipopolysaccharide-Stimulated iNOS Induction and NO Production through Activation of Nrf2 Signaling in BV2 Microglial Cells. Pharmacol Res. 2013;71:34–43. doi: 10.1016/j.phrs.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 74.Rezai-Zadeh K, Ehrhart J, Bai Y, et al. Apigenin and Luteolin Modulate Microglial Activation via Inhibition of STAT1-Induced CD40 Expression. J Neuroinflammation. 2008;5:41. doi: 10.1186/1742-2094-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hellerstein MK. A Critique of the Molecular Target-Based Drug Discovery Paradigm Based on Principles of Metabolic Control: Advantages of Pathway-Based Discovery. Metab Eng. 2008;10:1–9. doi: 10.1016/j.ymben.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 76.Mirzoeva S, Sawkar A, Zasadzki M, et al. Discovery of a 3-Amino-6-phenyl-pyridazine Derivative as a New Synthetic Antineuroinflammatory Compound. J Med Chem. 2002;45:563–566. doi: 10.1021/jm015573g. [DOI] [PubMed] [Google Scholar]
- 77.Ock J, Hong SH, Suk K. Identification of KT-15073 as an Inhibitor of Lipopolysaccharide-Induced Microglial Activation. Biol Pharm Bull. 2010;33:461–467. doi: 10.1248/bpb.33.461. [DOI] [PubMed] [Google Scholar]
- 78.Ohsawa K, Imai Y, Nakajima K, et al. Generation and Characterization of a Microglial Cell Line, MG5, Derived from a p53-Deficient Mouse. Glia. 1997;21:285–298. [PubMed] [Google Scholar]
- 79.Stansley B, Post J, Hensley K. A Comparative Review of Cell Culture Systems for the Study of Microglial Biology in Alzheimer's Disease. J Neuroinflammation. 2012;9:115. doi: 10.1186/1742-2094-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagai A, Mishima S, Ishida Y, et al. Immortalized Human Microglial Cell Line: Phenotypic Expression. J Neurosci Res. 2005;81:342–348. doi: 10.1002/jnr.20478. [DOI] [PubMed] [Google Scholar]
- 81.Abd-El-Basset EM, Prashanth J, Ananth Lakshmi KV. Up-regulation of Cytoskeletal Proteins in Activated Microglia. Med Princ Pract. 2004;13:325–333. doi: 10.1159/000080469. [DOI] [PubMed] [Google Scholar]
- 82.Henn A, Lund S, Hedtjarn M, et al. The Suitability of BV2 Cells as Alternative Model System for Primary Microglia Cultures or for Animal Experiments Examining Brain Inflammation. ALTEX. 2009;26:83–94. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- 83.Alliot F, Marty MC, Cambier D, et al. A Spontaneously Immortalized Mouse Microglial Cell Line Expressing CD4. Brain Res Dev Brain Res. 1996;95:140–143. doi: 10.1016/0165-3806(96)00101-0. [DOI] [PubMed] [Google Scholar]
- 84.Werner P, Pitt D, Raine CS. Multiple Sclerosis: Altered Glutamate Homeostasis in Lesions Correlates with Oligodendrocyte and Axonal Damage. Ann Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- 85.Shijie J, Takeuchi H, Yawata I, et al. Blockade of Glutamate Release from Microglia Attenuates Experimental Autoimmune Encephalomyelitis in Mice. Tohoku J Exp Med. 2009;217:87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
- 86.Pais TF, Figueiredo C, Peixoto R, et al. Necrotic Neurons Enhance Microglial Neurotoxicity through Induction of Glutaminase by a MyD88-Dependent Pathway. J Neuroinflammation. 2008;5:43. doi: 10.1186/1742-2094-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Righi M, Mori L, De Libero G, et al. Monokine Production by Microglial Cell Clones. Eur J Immunol. 1989;19:1443–1448. doi: 10.1002/eji.1830190815. [DOI] [PubMed] [Google Scholar]
- 88.Walker WS, Gatewood J, Olivas E, et al. Mouse Microglial Cell Lines Differing in Constitutive and Interferon-Gamma–Inducible Antigen-Presenting Activities for Naive and Memory CD4+ and CD8+ T Cells. J Neuroimmunol. 1995;63:163–174. doi: 10.1016/0165-5728(95)00146-8. [DOI] [PubMed] [Google Scholar]
- 89.Hensley K, Christov A, Kamat S, et al. Proteomic Identification of Binding Partners for the Brain Metabolite Lanthionine Ketimine (LK) and Documentation of LK Effects on Microglia and Motoneuron Cell Cultures. J Neurosci. 2010;30:2979–2988. doi: 10.1523/JNEUROSCI.5247-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tocharus J, Khonthun C, Chongthammakun S, et al. Melatonin Attenuates Methamphetamine-Induced Overexpression of Proinflammatory Cytokines in Microglial Cell Lines. J Pineal Res. 2010;48:347–352. doi: 10.1111/j.1600-079X.2010.00761.x. [DOI] [PubMed] [Google Scholar]
- 91.Giulian D, Baker TJ. Characterization of Ameboid Microglia Isolated from Developing Mammalian Brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen CJ, Ou YC, Chang CY, et al. Glutamate Released by Japanese Encephalitis Virus-Infected Microglia Involves TNF-Alpha Signaling and Contributes to Neuronal Death. Glia. 2012;60:487–501. doi: 10.1002/glia.22282. [DOI] [PubMed] [Google Scholar]
- 93.Conrad M, Sato H. The Oxidative Stress–Inducible Cystine/Glutamate Antiporter, System x (c) (–): Cystine Supplier and Beyond. Amino Acids. 2012;42:231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 94.Gupta S, Knight AG, Gupta S, et al. HIV-Tat Elicits Microglial Glutamate Release: Role of NAPDH Oxidase and the Cystine-Glutamate Antiporter. Neurosci Lett. 2010;485:233–236. doi: 10.1016/j.neulet.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qin S, Colin C, Hinners I, et al. System Xc- and Apolipoprotein E Expressed by Microglia Have Opposite Effects on the Neurotoxicity of Amyloid-Beta Peptide 1–40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mori M. Regulation of Nitric Oxide Synthesis and Apoptosis by Arginase and Arginine Recycling. J Nutr. 2007;137:1616S–1620S. doi: 10.1093/jn/137.6.1616S. [DOI] [PubMed] [Google Scholar]
- 97.Beutner C, Roy K, Linnartz B, et al. Generation of Microglial Cells from Mouse Embryonic Stem Cells. Nat Protoc. 2010;5:1481–1494. doi: 10.1038/nprot.2010.90. [DOI] [PubMed] [Google Scholar]
- 98.Nakamura Y, Si QS, Kataoka K. Lipopolysaccharide-Induced Microglial Activation in Culture: Temporal Profiles of Morphological Change and Release of Cytokines and Nitric Oxide. Neurosci Res. 1999;35:95–100. doi: 10.1016/s0168-0102(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 99.Tsuchiya T, Park KC, Toyonaga S, et al. Characterization of Microglia Induced from Mouse Embryonic Stem Cells and Their Migration into the Brain Parenchyma. J Neuroimmunol. 2005;160:210–218. doi: 10.1016/j.jneuroim.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 100.Chao CC, Hu S, Close K, et al. Cytokine Release from Microglia: Differential Inhibition by Pentoxifylline and Dexamethasone. J Infect Dis. 1992;166:847–853. doi: 10.1093/infdis/166.4.847. [DOI] [PubMed] [Google Scholar]
- 101.Nikodemova M, Duncan ID, Watters JJ. Minocycline Exerts Inhibitory Effects on Multiple Mitogen-Activated Protein Kinases and IkappaBalpha Degradation in a Stimulus-Specific Manner in Microglia. J Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- 102.Orsucci D, Mancuso M, Filosto M, et al. Tetracyclines and Neuromuscular Disorders. Curr Neuropharmacol. 2012;10:134–138. doi: 10.2174/157015912800604498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kumazawa Y, Kawaguchi K, Takimoto H. Immunomodulating Effects of Flavonoids on Acute and Chronic Inflammatory Responses Caused by Tumor Necrosis Factor Alpha. Curr Pharm Des. 2006;12:4271–4279. doi: 10.2174/138161206778743565. [DOI] [PubMed] [Google Scholar]
- 104.Choi DK, Koppula S, Suk K. Inhibitors of Microglial Neurotoxicity: Focus on Natural Products. Molecules. 2011;16:1021–1043. doi: 10.3390/molecules16021021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.He FQ, Qiu BY, Li TK, et al. Tetrandrine Suppresses Amyloid-Beta–Induced Inflammatory Cytokines by Inhibiting NF-kappaB Pathway in Murine BV2 Microglial Cells. Int Immunopharmacol. 2011;11:1220–1225. doi: 10.1016/j.intimp.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 106.Watterson DM, Velentza AV, Zasadzki M, et al. Discovery of a New Class of Synthetic Protein Kinase Inhibitors That Suppress Selective Aspects of Glial Activation and Protect Against Beta-Amyloid Induced Injury: A Foundation for Future Medicinal Chemistry Efforts Focused on Targeting Alzheimer's Disease Progression. J Mol Neurosci. 2003;20:411–423. doi: 10.1385/JMN:20:3:411. [DOI] [PubMed] [Google Scholar]
- 107.Or TC, Yang CL, Law AH, et al. Isolation and Identification of Anti-Inflammatory Constituents from Ligusticum chuanxiong and Their Underlying Mechanisms of Action on Microglia. Neuropharmacology. 2011;60:823–831. doi: 10.1016/j.neuropharm.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 108.Martin-Moreno AM, Reigada D, Ramirez BG, et al. Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer's Disease. Mol Pharmacol. 2011;79:964–973. doi: 10.1124/mol.111.071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hong J, Kim BK, Lim H, et al. Identification and Characterization of Triamcinolone Acetonide, a Microglial-Activation Inhibitor. Immunopharmacol Immunotoxicol. 2012;34:912–918. doi: 10.3109/08923973.2012.671332. [DOI] [PubMed] [Google Scholar]
- 110.Ock J, Kim S, Suk K. Anti-Inflammatory Effects of a Fluorovinyloxyacetamide Compound KT-15087 in Microglia Cells. Pharmacol Res. 2009;59:414–422. doi: 10.1016/j.phrs.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 111.Orsucci D, Calsolaro V, Mancuso M, et al. Neuroprotective Effects of Tetracyclines: Molecular Targets, Animal Models and Human Disease. CNS Neurol Disord Drug Targets. 2009;8:222–231. doi: 10.2174/187152709788680689. [DOI] [PubMed] [Google Scholar]
- 112.Cukras CA, Petrou P, Chew EY, et al. Oral Minocycline for the Treatment of Diabetic Macular Edema (DME): Results of a Phase I/II Clinical Study. Invest Ophthalmol Vis Sci. 2012;53:3865–3874. doi: 10.1167/iovs.11-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Meininger V, Asselain B, Guillet P, et al. Pentoxifylline in ALS: A Double-Blind, Randomized, Multicenter, Placebo-Controlled Trial. Neurology. 2006;66:88–92. doi: 10.1212/01.wnl.0000191326.40772.62. [DOI] [PubMed] [Google Scholar]
- 114.Fan LW, Pang Y, Lin S, et al. Minocycline Attenuates Lipopolysaccharide-Induced White Matter Injury in the Neonatal Rat Brain. Neuroscience. 2005;133:159–168. doi: 10.1016/j.neuroscience.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 115.Benicchi T, Iozzi S, Svahn A, et al. A Homogeneous HTRF Assay for the Identification of Inhibitors of the TWEAK-Fn14 Protein Interaction. J Biomol Screen. 2012;17:933–945. doi: 10.1177/1087057112447873. [DOI] [PubMed] [Google Scholar]
- 116.Hoing S, Rudhard Y, Reinhardt P, et al. Discovery of Inhibitors of Microglial Neurotoxicity Acting through Multiple Mechanisms Using a Stem-Cell–Based Phenotypic Assay. Cell Stem Cell. 2012;11:620–632. doi: 10.1016/j.stem.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 117.Burguillos MA, Deierborg T, Kavanagh E, et al. Caspase Signalling Controls Microglia Activation and Neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- 118.MacBeath G, Schreiber SL. Printing Proteins as Microarrays for High-Throughput Function Determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 119.Hart CP. Finding the Target after Screening the Phenotype. Drug Discov Today. 2005;10:513–519. doi: 10.1016/S1359-6446(05)03415-X. [DOI] [PubMed] [Google Scholar]
- 120.Gough W, Hulkower KI, Lynch R, et al. A Quantitative, Facile, and High-Throughput Image-Based Cell Migration Method Is a Robust Alternative to the Scratch Assay. J Biomol Screen. 2011;16:155–163. doi: 10.1177/1087057110393340. [DOI] [PubMed] [Google Scholar]
- 121.Lee JA, Chu S, Willard FS, et al. Open Innovation for Phenotypic Drug Discovery: The PD2 Assay Panel. J Biomol Screen. 2011;16:588–602. doi: 10.1177/1087057111405379. [DOI] [PubMed] [Google Scholar]
- 122.Low J, Huang S, Blosser W, et al. High-Content Imaging Characterization of Cell Cycle Therapeutics through In Vitro and In Vivo Subpopulation Analysis. Mol Cancer Ther. 2008;7:2455–2463. doi: 10.1158/1535-7163.MCT-08-0328. [DOI] [PubMed] [Google Scholar]
- 123.Maeda M, Tsuda M, Tozaki-Saitoh H, et al. Nerve Injury–Activated Microglia Engulf Myelinated Axons in a P2Y12 Signaling-Dependent Manner in the Dorsal Horn. Glia. 2010;58:1838–1846. doi: 10.1002/glia.21053. [DOI] [PubMed] [Google Scholar]
- 124.Morigiwa K, Quan M, Murakami M, et al. P2 Purinoceptor Expression and Functional Changes of Hypoxia-Activated Cultured Rat Retinal Microglia. Neurosci Lett. 2000;282:153–156. doi: 10.1016/s0304-3940(00)00887-9. [DOI] [PubMed] [Google Scholar]
- 125.Park SY, Lee H, Hur J, et al. Hypoxia Induces Nitric Oxide Production in Mouse Microglia via p38 Mitogen-Activated Protein Kinase Pathway. Brain Res Mol Brain Res. 2002;107:9–16. doi: 10.1016/s0169-328x(02)00421-7. [DOI] [PubMed] [Google Scholar]
- 126.Hur J, Lee P, Kim MJ, et al. Ischemia-Activated Microglia Induces Neuronal Injury via Activation of gp91phox NADPH Oxidase. Biochem Biophys Res Commun. 2010;391:1526–1530. doi: 10.1016/j.bbrc.2009.12.114. [DOI] [PubMed] [Google Scholar]
- 127.Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine Quinones Activate Microglia and Induce a Neurotoxic Gene Expression Profile: Relationship to Methamphetamine-Induced Nerve Ending Damage. Ann N Y Acad Sci. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- 128.Chakfe Y, Seguin R, Antel JP, et al. ADP and AMP Induce Interleukin-1beta Release from Microglial Cells through Activation of ATP-Primed P2X7 Receptor Channels. J Neurosci. 2002;22:3061–3069. doi: 10.1523/JNEUROSCI.22-08-03061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brough D, Le Feuvre RA, Iwakura Y, et al. Purinergic (P2X7) Receptor Activation of Microglia Induces Cell Death via an Interleukin-1–Independent Mechanism. Mol Cell Neurosci. 2002;19:272–280. doi: 10.1006/mcne.2001.1054. [DOI] [PubMed] [Google Scholar]
- 130.Bartlett R, Yerbury JJ, Sluyter R. P2X7 Receptor Activation Induces Reactive Oxygen Species Formation and Cell Death in Murine EOC13 Microglia. Mediators Inflamm. 2013;2013:271813. doi: 10.1155/2013/271813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee EJ, Woo MS, Moon PG, et al. Alpha-Synuclein Activates Microglia by Inducing the Expressions of Matrix Metalloproteinases and the Subsequent Activation of Protease-Activated Receptor-1. J Immunol. 2010;185:615–623. doi: 10.4049/jimmunol.0903480. [DOI] [PubMed] [Google Scholar]
- 132.Su X, Maguire-Zeiss KA, Giuliano R, et al. Synuclein Activates Microglia in a Model of Parkinson's Disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Polazzi E, Levi G, Minghetti L. Human Immunodeficiency Virus Type 1 Tat Protein Stimulates Inducible Nitric Oxide Synthase Expression and Nitric Oxide Production in Microglial Cultures. J Neuropathol Exp Neurol. 1999;58:825–831. doi: 10.1097/00005072-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 134.D'Aversa TG, Eugenin EA, Berman JW. NeuroAIDS: Contributions of the Human Immunodeficiency Virus-1 Proteins Tat and gp120 as Well as CD40 to Microglial Activation. J Neurosci Res. 2005;81:436–446. doi: 10.1002/jnr.20486. [DOI] [PubMed] [Google Scholar]
- 135.D'Aversa TG, Yu KO, Berman JW. Expression of Chemokines by Human Fetal Microglia after Treatment with the Human Immunodeficiency Virus Type 1 Protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- 136.Sheng WS, Hu S, Hegg CC, et al. Activation of Human Microglial Cells by HIV-1 gp41 and Tat Proteins. Clin Immunol. 2000;96:243–251. doi: 10.1006/clim.2000.4905. [DOI] [PubMed] [Google Scholar]
- 137.Albright AV, Martin J, O'Connor M, et al. Interactions between HIV-1 gp120, Chemokines, and Cultured Adult Microglial Cells. J Neurovirol. 2001;7:196–207. doi: 10.1080/13550280152403245. [DOI] [PubMed] [Google Scholar]
- 138.Brooke SM, Sapolsky RM. Glucocorticoid Exacerbation of gp120 Neurotoxicity: Role of Microglia. Exp Neurol. 2002;177:151–158. doi: 10.1006/exnr.2002.7956. [DOI] [PubMed] [Google Scholar]
- 139.He BP, Wen W, Strong MJ. Activated Microglia (BV-2) Facilitation of TNF-Alpha–Mediated Motor Neuron Death In Vitro. J Neuroimmunol. 2002;128:31–38. doi: 10.1016/s0165-5728(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 140.Block ML, Hong JS. Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers with a Common Mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 141.Simi A, Tsakiri N, Wang P, et al. Interleukin-1 and Inflammatory Neurodegeneration. Biochem Soc Trans. 2007;35:1122–1126. doi: 10.1042/BST0351122. [DOI] [PubMed] [Google Scholar]
- 142.Schachtele SJ, Hu S, Little MR, et al. Herpes Simplex Virus Induces Neural Oxidative Damage via Microglial Cell Toll-Like Receptor-2. J Neuroinflammation. 2010;7:35. doi: 10.1186/1742-2094-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bobyn J, Mangano EN, Gandhi A, et al. Viral-Toxin Interactions and Parkinson's Disease: Poly(I:C) Priming Enhanced the Neurodegenerative Effects of Paraquat. J Neuroinflammation. 2012;9:86. doi: 10.1186/1742-2094-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Siao CJ, Tsirka SE. Tissue Plasminogen Activator Mediates Microglial Activation via Its Finger Domain through Annexin II. J Neurosci. 2002;22:3352–3358. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang CF, Li G, Ma R, et al. Thrombin-Induced Microglial Activation Contributes to the Degeneration of Nigral Dopaminergic Neurons In Vivo. Neurosci Bull. 2008;24:66–72. doi: 10.1007/s12264-008-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sawada H, Hishida R, Hirata Y, et al. Activated Microglia Affect the Nigro-Striatal Dopamine Neurons Differently in Neonatal and Aged Mice Treated with 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci Res. 2007;85:1752–1761. doi: 10.1002/jnr.21241. [DOI] [PubMed] [Google Scholar]
- 147.Miwa H, Kubo T, Morita S, et al. Oxidative Stress and Microglial Activation in Substantia Nigra following Striatal MPP+ Neuroreport. 2004;15:1039–1044. doi: 10.1097/00001756-200404290-00021. [DOI] [PubMed] [Google Scholar]
- 148.Wu XF, Block ML, Zhang W, et al. The Role of Microglia in Paraquat-Induced Dopaminergic Neurotoxicity. Antioxid Redox Signal. 2005;7:654–661. doi: 10.1089/ars.2005.7.654. [DOI] [PubMed] [Google Scholar]
- 149.Zhao YN, Wang F, Fan YX, et al. Activated Microglia Are Implicated in Cognitive Deficits, Neuronal Death, and Successful Recovery following Intermittent Ethanol Exposure. Behav Brain Res. 2013;236:270–282. doi: 10.1016/j.bbr.2012.08.052. [DOI] [PubMed] [Google Scholar]
- 150.Levesque S, Taetzsch T, Lull ME, et al. Diesel Exhaust Activates and Primes Microglia: Air Pollution, Neuroinflammation, and Regulation of Dopaminergic Neurotoxicity. Environ Health Perspect. 2011;119:1149–1155. doi: 10.1289/ehp.1002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Genc S, Zadeoglulari Z, Fuss SH, et al. The Adverse Effects of Air Pollution on the Nervous System. J Toxicol. 2012;2012:782462. doi: 10.1155/2012/782462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hashioka S, Han YH, Fujii S, et al. Phospholipids Modulate Superoxide and Nitric Oxide Production by Lipopolysaccharide and Phorbol 12-Myristate-13-Acetate-Activated Microglia. Neurochem Int. 2007;50:499–506. doi: 10.1016/j.neuint.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 153.Hagen J, Houchins JP, Kalyuzhny AE. ELISPOT Assay for Neuroscience Research: Studying TNFalpha Secretion from Microglial Cells. Methods Mol Biol. 2012;792:97–104. doi: 10.1007/978-1-61779-325-7_8. [DOI] [PubMed] [Google Scholar]
- 154.Perego C, Fumagalli S, De Simoni MG. Temporal Pattern of Expression and Colocalization of Microglia/Macrophage Phenotype Markers following Brain Ischemic Injury in Mice. J Neuroinflammation. 2011;8:174. doi: 10.1186/1742-2094-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chhor V, Le Charpentier T, Lebon S, et al. Characterization of Phenotype Markers and Neuronotoxic Potential of Polarised Primary Microglia In Vitro. Brain Behav Immun. 2013;32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barger SW, Basile AS. Activation of Microglia by Secreted Amyloid Precursor Protein Evokes Release of Glutamate by Cystine Exchange and Attenuates Synaptic Function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- 157.Zhou M, Diwu Z, Panchuk-Voloshina N, et al. A Stable Nonfluorescent Derivative of Resorufin for the Fluorometric Determination of Trace Hydrogen Peroxide: Applications in Detecting the Activity of Phagocyte NADPH Oxidase and Other Oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 158.Mohanty JG, Jaffe JS, Schulman ES, et al. A Highly Sensitive Fluorescent Micro-Assay of H2O2 Release from Activated Human Leukocytes Using a Dihydroxyphenoxazine Derivative. J Immunol Methods. 1997;202:133–141. doi: 10.1016/s0022-1759(96)00244-x. [DOI] [PubMed] [Google Scholar]
- 159.Tarpey MM, Wink DA, Grisham MB. Methods for Detection of Reactive Metabolites of Oxygen and Nitrogen: In Vitro and In Vivo Considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 160.Coelho-Santos V, Goncalves J, Fontes-Ribeiro C, et al. Prevention of Methamphetamine-Induced Microglial Cell Death by TNF-Alpha and IL-6 through Activation of the JAK-STAT Pathway. J Neuroinflammation. 2012;9:103. doi: 10.1186/1742-2094-9-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang Za, Y ES. Selective Detection of Neurotransmitters by Fluorescence and Chemiluminescence Imaging. Pure Appl Chem. 2001;73:1599–1611. [Google Scholar]
- 162.Zhao J, Lopez AL, Erichsen D, et al. Mitochondrial Glutaminase Enhances Extracellular Glutamate Production in HIV-1–Infected Macrophages: Linkage to HIV-1 Associated Dementia. J Neurochem. 2004;88:169–180. doi: 10.1046/j.1471-4159.2003.02146.x. [DOI] [PubMed] [Google Scholar]
- 163.Schilling T, Lehmann F, Ruckert B, et al. Physiological Mechanisms of Lysophosphatidylcholine-Induced De-ramification of Murine Microglia. J Physiol. 2004;557:105–120. doi: 10.1113/jphysiol.2004.060632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Farber K, Kettenmann H. Purinergic Signaling and Microglia. Pflugers Arch. 2006;452:615–621. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]