

Abstract
While bacterial iterative type I polyketide synthases (iPKSs) are now known to participate in the biosynthesis of a small set of diverse natural products, the subsequent downstream modification of the resulting polyketide products remains poorly understood. Toward this goal, we report functional characterization of the putative orsellinic acid C2-O-methyltransferase (CalO6) involved in calicheamicin biosynthesis. This study suggests C2-O-methylation precedes C3-hydroxylation/methylation and C5-iodination and to require a coenzyme A- (CoA) or acyl carrier protein- (ACP) bound substrate.
Keywords: S-adenosylmethionine, iterative PKS, polyketide, acyl carrier protein, orsellinic acid, AdoMet, SAM
Bacterial iterative type I PKSs (iPKSs) are single polypeptides comprised of a series of fused distinctly functional domains that operate in a progressive, iterative fashion. These multifunctional enzymes assemble structurally diverse mono- or bicyclic- aromatic polyketides (Scheme 1)[1] from acyl Co-enzyme A (CoA) precursors where subsequent modification/utilization of the aromatic core has been proposed or demonstrated to be dependent upon a dedicated acyltransferase (AT) (chlorothricin,[2] pactamycin,[3] avilamycin,[4] tiacumicin B,[5] and calicheamicin[6] ) or via adenylated intermediates (maduropeptin,[7] polyketomycin,[8] azinomycin B,[9] and neocarzinostatin,[10]). A primary point of structural divergence of iPKS-derived aromatic polyketides derives from downstream ‘tailoring’ modifications (hydroxylation, epoxidation, acylation, glycosylation, transamination, halogenation and/or methylation) of the resulting polyketide scaffold. Published work to date suggests the fundamental timing and/or biochemical basis of these subsequent modifications to also be widely variant and poorly understood. For example, the napthoic acid O-methyltransferase (O-MT) NcsB1 in neocarzinostatin biosynthesis operates on the free napthoic acid moiety prior to adenylation[10c] while MdpB1-catalyzed C-methylation during the biosynthesis of maduropeptin utilizes a CoA-tethered 6-methylsalicylic acid substrate.[7b] In contrast, TiaM-catalyzed halogenation occurs as the last step in tiacumicin B maturation, after the iPKS-derived aromatic component has been attached to nearly mature natural product.[5] Thus, additional fundamental studies are needed to provide a general understanding of the factors that dictate the order and substrate preference of post-iPKS events.
Scheme 1.
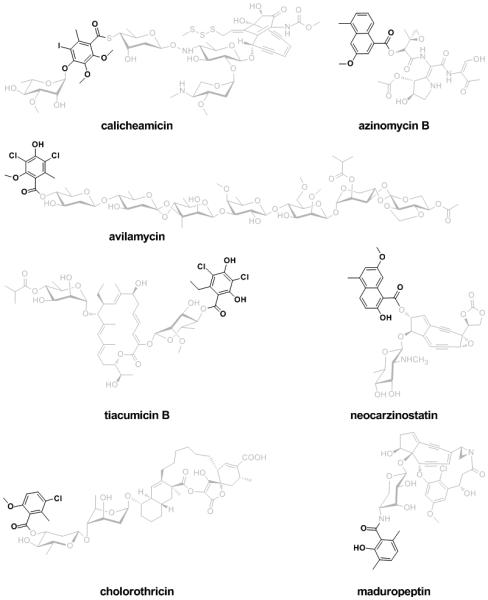
Representative natural products that contain aromatic polyketides moieties (highlighted in dark color) synthesized by bacterial iterative type I PKS.
A total of six enzymes (CalO1-CalO6) are postulated to participate in constructing the iPKS-derived modified orsellinic acid (OSA, 2,4-dihydroxy-6-methylbenzoic acid) component of the prototype naturally-occurring enediyne calicheamicin (Scheme 2). These include: CalO5, the iPKS OSA synthase; CalO2, the P450 hydroxylase; CalO3, the putative flavin-dependent halogenase; and CalO1/O6, putative AdoMet-dependent aromatic O-MTs. While CalO1 and CalO2 ligand-binding and crystallography studies implicated the requirement of thioester-conjugated (CoA- or ACP-) substrates for CalO1-catalyzed methylation and CalO2-catalyzed hydroxylation reactions,[11] additional biochemical characterization of these, or the other, OSA-related enzymes in this pathway has been restricted by a lack of suitable substrates or knowledge regarding the timing of each enzymatic step. Herein we report the biochemical characterization of the calicheamicin OSA C2-O-MT CalO6 using S-N-acetylcysteaminyl OSA (SNAC-OSA) as a substrate. This study confirms the regiospecificity of CalO6 and, in conjunction with the prior work on CalO1/O2, offers additional insights regarding the possible sequence of biosynthetic events of the key aromatic component of the calicheamicin aryltetrasaccharide.
Scheme 2.

The proposed pathway for OSA biosynthesis and modification en route to calicheamicin. The architecture and domain organization of the iPKS (CalO5) is illustrated at the top and the key CalO6-catalyzed C2-OH methylation reaction is highlighted within the box. Based upon the CalO6 biochemical studies described, ‘R’ is anticipated to be an ACP or CoA thioester.
For biochemical characterization, N-His6-CalO6 was overproduced in E. coli BL21(DE3) and purified by affinity chromatography. In initial attempts, the free acids 2,4-dihydroxy benzoic acid (6), salicylic acid (7), 5-iodo-OSA (8), OSA (9), 3,4-dihydroxy benzoic acid (10) or 5-iodosalicylic acid (6) failed to turnover in presence of CalO6. Based on prior ligand binding studies that revealed the P450 hydroxylase CalO2 to bind SNAC-linked substrate mimics,[11a] the corresponding SNAC-linked substrates (1-5, respectively; Figure 1A) were subsequently synthesized and assessed as substrates for CalO6. From this putative substrate set, only SNAC-OSA (4 in Figure 1A) resulted in methylated products in the presence of CalO6. Specifically, two products (Figure 1B, P1 and P2 with retention times of 15.3 min and 20.0 min, respectively) were observed, both confirmed as mono-methylated SNAC-OSA analogs by high-resolution mass spectrometry (HRMS m/z 306.07603 [M+Na]+; calcd for C13H17NO4S, 306.07705 [M+Na]+). For full characterization of products, the CalO6 reaction was scaled to provide ~900 μg (30%) and ~400 μg (13%) of P1 and P2, respectively, after purification. Subsequent NMR spectroscopy revealed P1 and P2 as SNAC-2-methoxy-OSA and SNAC-4-methoxy-OSA, respectively (Figure 1C). The inability of CalO6 to utilize SNAC-2,4-dihydroxy-benzoic acid (1 in Figure 1A) or SNAC-5-iodo-OSA (3 in Figure 1A), implicates CalO6 the first step among the putative OSA tailoring reactions en route to calicheamicin. As ligand-binding studies previously revealed iodination to be critical for CalO2 binding, we put forth the putative sequence of events as illustrated in Scheme 2.
Figure 1.
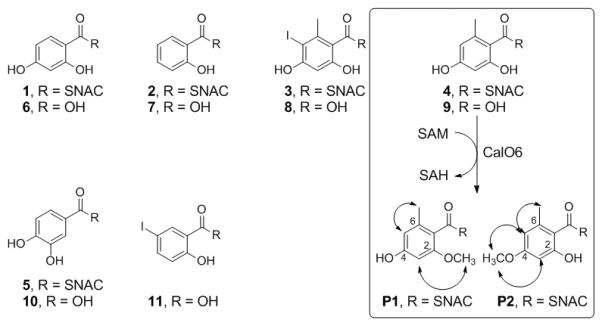

Range of substrates assessed in this study with the CalO6 substrate SNAC-OSA (4) and CalO6 reaction products P1 and P2 highlighted within the box. B. HPLC chromatogram of a representative CalO6 reactions with 4 at 37 °C for 1 h (i: SAM; ii: 4; iii-v: CalO6 reactions containing 8, 16 and 24 μg of enzyme, respectively). Peaks labeled with S, P1 and P2 are substrate, SNAC-OSA, SNAC-2-methoxy-OSA and SNAC-4-methoxy OSA, respectively and SAH and/or SAM degradation products are denoted by stars (*) C. Sections of the P1 and P2 NOESY spectra (mixing time = 300 ms) where arrows in A indicate key NOE connections.
The CalO6 steady-state kinetic parameters in the context of P1 and P2 formation were determined based upon product formation monitored by HPLC (Table 1). Importantly, compared to typical kinetic values reported for aromatic O-MTs,[10c, 7b] the CalO6 KM values reflected in this study are >10-fold higher while the corresponding kcat values are ~1000-fold lower, suggesting the SNAC conjugates to be rather poor mimics of the putative native (CoA- or ACP-) substrate conjugate. However, this analysis revealed a ~3-fold kinetic bias toward P1 formation, consistent with C2-OH methylation as the preferred CalO6 regiospecificity.
Table 1.
Kinetic parameters for CalO6 using variable SNAC-OSA (0.1 - 5 mM) and SAM (0.1 - 4 mM) in 25 mM Tris, 5 mM MgCl2, pH 8.0 at 35 °C.
| Product | Constant Substrate |
Varied Substrate |
kcat × 10−3
(min−1) |
KM (mM) |
kcat / KM × 10−3
(mM−1min−1) |
|---|---|---|---|---|---|
| P1 | SAM | SNAC-OSA | 1.03 ± 0.03 | 1.27 ± 0.08 | 0.81 |
| SNAC-OSA | SAM | 0.94 ± 0.03 | 0.30 ± 0.03 | 3.13 | |
|
| |||||
| P2 | SAM | SNAC-OSA | 0.33 ± 0.01 | 1.31 ± 0.11 | 0.25 |
| SNAC-OSA | SAM | 0.36 ± 0.01 | 0.56 ± 0.06 | 0.64 | |
|
| |||||
| Total | SAM | SNAC-OSA | 1.35 ± 0.03 | 1.27 ± 0.07 | 1.06 |
| SNAC-OSA | SAM | 1.29 ± 0.03 | 0.35 ± 0.04 | 3.69 | |
A CalO6 BLAST search revealed 39% sequence identity with the mitomycin 7-O-MT MmcR,[12] 37% with the neocarzinostatin O-MT NcsB1[10d] and 34% with the calicheamicin sugar 4’-O-MT, CalO1 [11b] (see supporting information Figure S1). To further explore the basis of CalO6 regiospecificity, a CalO6 homology model was constructed based upon MmcR using Swiss-Model (Figure 2).[13] In most MTs, general acid/base catalysis contributes to rate acceleration wherein an active-site His participates as a catalytic base, as exemplified by the rebeccamycin 4’-O-MT, RebM.[14] Sequence alignment (see supporting information Figure S1) implicates His252-Glu311 as the CalO6 catalytic diad and, consistent with this, the putative active-site base His252 is within hydrogen-bonding distance (3.1 Å) of Glu311 in the homology model. Manual docking of 4 and SAH to position the favored C2-OH within appropriate conformation for methylation (4 C2-OH-His252 side chain imidazole, 3.1 Å; 4 C2-OH- SAH thiol 3.1 Å) invokes an unfavorable distance for C4 methylation (4 C4-OH-His252 side chain imidazole, 6.6 Å; Figure 2). While this is consistent with a bias toward C2-O-methylation, the perceived suboptimal binding and slow turnover of 4 by CalO6 may allow for ‘slipping’ within the active and corresponding misalkylation of the substrate mimetic 4 (i.e., C4-O-methylation rather than C2-O-methylation).
Figure 2.

CalO6 homology model based upon mitomycin-7-O-methyltransferase, MmcR (PDB: 3GWZ) with the distances between the putative catalytic histidine (His252) and hydroxyl groups of C2- and C4- of SNAC-OSA indicated. In the stick model, SAH and SNAC-OSA are shaded dark, and CalO6 residues proposed to take part in the catalysis are in light grey.
In summary, we have characterized CalO6 as the calicheamicin OSA C2-O-MT. This work implicates a dependence upon a thioester-conjugated substrate, the nature of which, while anticipated to be either ACP-based or CoA-based, cannot be gleaned from the current study. Re-examination of the calicheamicin gene cluster reveals no apparent gene encoding for a dedicated ACP or CoA ligase, suggesting the ACP domain within the orsellinate synthase CalO5 (Scheme 2) may serve as the putative substrate carrier for some or all subsequent tailoring reactions. In conjunction with the ligand-binding studies conducted with CalO2, the current study suggests C2-O-methylation to be an initiating tailoring event. From a more global perspective, prior studies to elucidate the aryltetrasaccharide glycosylation sequence of events failed to demonstrate 4 C4-O-rhamnosylation.[15] While not confirmatory, when considered in the context of the current CalO6 study and previous CalO1/CalO2 work, the prior glycosylation studies are consistent with the currently favored hypothesis that orsellinate halogenation and O-methylation occur prior to aryltetrasaccharide assembly in the context of calicheamicin biosynthesis.
Supplementary Material
Acknowledgements
We thank the School of Pharmacy Analytical Instrumentation Centre (University of Wisconsin-Madison) for analytical support. This work was supported by NIH RO1 CA84374 (JST) and the National Center for Advancing Translational Sciences (UL1TR000117).
Footnotes
Supporting information for this article is available
References
- [1].(a) Zhang O, Pang B, Ding W, Liu W. ACS Catal. 2013;3:1439–1447. [Google Scholar]; (b) Van Lanen SG, Shen B. Curr. Opin. Drug Discovery Dev. 2008;11:186–195. [PubMed] [Google Scholar]; (c) Van Lanen SG, Shen B. Curr. Top. Med Chem. 2008;8:448–449. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen B. Curr. Opin. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- [2].a) Shao L, Qu XD, Jia XY, Zhao QF, Tian ZH, Wang M, Tang GL, Liu W. Biochem Bioph Res Co. 2006;345:133–139. doi: 10.1016/j.bbrc.2006.04.069. [DOI] [PubMed] [Google Scholar]; b) Jia XY, Tian ZH, Shao L, Qu XD, Zhao QF, Tang J, Tang GL, Liu W. Chem Biol. 2006;13:575–585. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- [3].Ito T, Roongsawang N, Shirasaka N, Lu WL, Flatt PM, Kasanah N, Miranda C, Mahmud T. Chembiochem. 2009;10:2253–2265. doi: 10.1002/cbic.200900339. [DOI] [PubMed] [Google Scholar]
- [4].Weitnauer G, Muhlenweg A, Trefzer A, Hoffmeister D, Sussmuth RD, Jung G, Welzel K, Vente A, Girreser U, Bechthold A. Chem Biol. 2001;8:569–581. doi: 10.1016/s1074-5521(01)00040-0. [DOI] [PubMed] [Google Scholar]
- [5].Xiao Y, Li SM, Niu SW, Ma LA, Zhang GT, Zhang HB, Zhang GY, Ju JH, Zhang CS. J Am Chem Soc. 2011;133:1092–1105. doi: 10.1021/ja109445q. [DOI] [PubMed] [Google Scholar]
- [6].Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang KX, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- [7].a) Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. J Am Chem Soc. 129;2007:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ling J, Horsman GP, Huang SX, Luo Y, Lin S, Shen B. J Am Chem Soc. 2010;132:12534–12536. doi: 10.1021/ja1050814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Daum M, Peintner I, Linnenbrink A, Frerich A, Weber M, Paululat T, Bechthold A. Chembiochem. 2009;10:1073–1083. doi: 10.1002/cbic.200800823. [DOI] [PubMed] [Google Scholar]
- [9].Zhao QF, He QL, Ding W, Tang MC, Kang QJ, Yu Y, Deng W, Zhang Q, Fang J, Tang GL, Liu W. Chem Biol. 2008;15:693–705. doi: 10.1016/j.chembiol.2008.05.021. W. [DOI] [PubMed] [Google Scholar]
- [10].a) Liu W, Nonaka K, Nie LP, Zhang J, Christenson SD, Bae J, Van Lanen SG, Zazopoulos E, Farnet CM, Yang CF, Shen B. Chem Biol. 2005;12:293–302. doi: 10.1016/j.chembiol.2004.12.013. [DOI] [PubMed] [Google Scholar]; b) Cooke HA, Zhang J, Griffin MA, Nonaka K, Van Lanen SG, Shen B, Bruner SD. J Am Chem Soc. 2007;129:7728–7729. doi: 10.1021/ja071886a. [DOI] [PubMed] [Google Scholar]; c) Luo Y, Lin S, Zhang J, Cooke HA, Bruner SD, Shen B. J. Biol Chem. 2008;283:14694–14702. doi: 10.1074/jbc.M802206200. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cooke HA, Guenther EL, Luo Y, Shen B, Bruner SD. Biochemistry. 2009;48:9590–9598. doi: 10.1021/bi901257q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Mccoy JG, Johnson HD, Singh S, Bingman CA, Lee IK, Thorson JS, Phillips GN. Acta Crystallogr D Biol Crystallogr. 2011;74:50–60. [Google Scholar]; b) Chang A, Singh S, Bingman CA, Thorson JS, Phillips GN. Proteins. 2009;67:197–203. doi: 10.1107/S090744491100360X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Singh S, Chang A, Goff RD, Bingman CA, Grüschow S, Sherman DH, Phillips GN, Thorson JS. >Proteins. 2011;79:2181–2188. doi: 10.1002/prot.23040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]; b) Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Peitsch MC. Bio/Technology. 1995;13:658–660. [Google Scholar]
- [14].Singh S, McCoy JG, Bingman CA, Phillips GN, Thorson JS. J Biol Chem. 2008;283:22628–22636. doi: 10.1074/jbc.M800503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.