

Abstract
Over the past forty years, stable isotope analysis of bone (and tooth) collagen and hydroxyapatite has become a mainstay of archaeological and paleoanthropological reconstructions of paleodiet and paleoenvironment. Despite this method's frequent use across anthropological subdisciplines (and beyond), the present work represents the first attempt at gauging the effects of inter-laboratory variability engendered by differences in a) sample preparation, and b) analysis (instrumentation, working standards, and data calibration). Replicate analyses of a 14C-dated ancient human bone by twenty-one archaeological and paleoecological stable isotope laboratories revealed significant inter-laboratory isotopic variation for both collagen and carbonate. For bone collagen, we found a sizeable range of 1.8‰ for δ13Ccol and 1.9‰ for δ15Ncol among laboratories, but an interpretatively insignificant average pairwise difference of 0.2‰ and 0.4‰ for δ13Ccol and δ15Ncol respectively. For bone hydroxyapatite the observed range increased to a troublingly large 3.5‰ for δ13Cap and 6.7‰ for δ18Oap, with average pairwise differences of 0.6‰ for δ13Cap and a disquieting 2.0‰ for δ18Oap. In order to assess the effects of preparation versus analysis on isotopic variability among laboratories, a subset of the samples prepared by the participating laboratories were analyzed a second time on the same instrument. Based on this duplicate analysis, it was determined that roughly half of the isotopic variability among laboratories could be attributed to differences in sample preparation, with the other half resulting from differences in analysis (instrumentation, working standards, and data calibration). These findings have serious implications for choices made in the preparation and extraction of target biomolecules, the comparison of results obtained from different laboratories, and the interpretation of small differences in bone collagen and hydroxyapatite isotope values. To address the issues arising from inter-laboratory comparisons, we devise a novel measure we term the Minimum Meaningful Difference (MMD), and demonstrate its application.
Introduction
The past thirty years have witnessed an explosive increase in the ubiquity of stable isotope analysis of osseous remains in the fields of archaeology, paleoanthropology, and paleoecology (Figure 1). Indeed, stable isotope analysis of preserved osseous tissues has become a mainstay of paleodietary and paleoenvironmental reconstruction across anthropological subdisciplines. However, this growth in popularity has outpaced validation of the method's assumptions in at least one key area – the assessment of inter-laboratory variation. The present work aims to rectify this lacuna through experimental establishment of the degree and possible causes of inter-laboratory variation in stable isotope signatures of ancient bone collagen (col) and hydroxyapatite (ap).
Figure 1. Frequency of publication of articles on archaeological bone stable isotope analysis in American Journal of Physical Anthropology, Journal of Archaeological Science, and PLoS One.

Marked increase in frequency is evident.
The importance of stable isotopes for archaeology was first realized by Robert Hall in the late 1960s when he noted anomalously young radiocarbon dates produced by maize or any other species enriched in 13C [1], leading him to posit the utility of stable isotope analysis for the differentiation of archaeological browsers and grazers [2]. The first practical application of stable isotope analysis to the study of ancient human diet did not come until 1977 [3]. In this first study [3], and many thereafter [4]–[7], the main matter of concern was the timing of the introduction of maize agriculture, an event that is fairly obviously evidenced by a dramatic enrichment of consumers' collagen and hydroxyapatite δ13C signatures. Shortly after this first publication, DeNiro [8] established the fundamentals of δ13Ccol and δ15Ncol in controlled diet experiments with a variety of animals [9], [10]. He then used these two isotope systems of bone collagen to demonstrate a diachronic dietary shift among the prehistoric inhabitants of the Tehuacan Valley of Mexico. More or less contemporaneously, Tauber [11] used collagen carbon isotope values in his study of prehistoric and historic Danish fishers and farmers, Chisholm and colleagues [12] studied the exploitation of salmon by Northwest Coast Amerindians, Schoeninger and colleagues [13]–[15] demonstrated that both δ13Ccol and δ15Ncol values could be used to discriminate between habitual consumers of marine versus terrestrial foodstuffs, and Ambrose documented the importance of both diet and environment on collagen isotope values [16]–[19].
Paleoanthropological and paleoecological applications of stable isotope analysis have a history of comparable duration, although hydroxyapatite of bone, and more often dental enamel, has been the target osseous biomolecule. In the early 1980s, Sullivan and Krueger [20], [21] outlined the basics of stable isotope paleodietary reconstruction from biological apatites (δ13Cap), and realized the potential for the technique's application to specimens from “well back into the Pleistocene” [20∶335]. With Lee-Thorp and Van Der Merwe's [22] confirmation that δ13Cap values of dental enamel preserved biogenic signatures, the die for such work was cast (although see also [23]). Focusing on carbon isotopes in the inorganic fraction of bone (and more often tooth enamel [24]), various studies have pushed back the temporal horizon of stable isotope analysis well into the Miocene and earlier (see [25]–[29] for review of the pertinent paleoanthropological literature and recent examples).
In the four decades since these first applications, isotope analysis of human (and hominid) dental and skeletal remains has become commonplace. Indeed, Figure 1 demonstrates clearly the method's increasing popularity over the past thirteen years, as represented by the number of publications in three topical journals.
Curiously, however, while the pace of archaeological and anthropological applications of stable isotope analysis has increased, the validation of the technique's assumptions has lagged behind. There have been numerous studies of relevant methodological issues, including isotopic routing [30]–[36], controlled diet experiments [31], [37]–[40], variability among individual laboratory preparation techniques [41]–[44], the causes and consequences of diagenetic and taphonomic change [24], [45]–[58], and the importance of consistent data normalization and calibration procedures for inter-laboratory comparability [59], [60]. However, to the best of our knowledge, there has never been a controlled study assessing the amount of inter-laboratory variation or the degree to which inter-laboratory variation stems from differences in preparation/extraction methods versus difference in analytical instrumentation and data calibration. The present work is intended to remedy these obvious lacunae in our knowledge and assess the confidence in which comparisons of results from different laboratories might be held. This represents a crucial step in assessing just how (dis)similar the conclusions of two laboratories might be when analyzing the same source materials.
The results of this study suggest that, in general, isotopic data from bone collagen (δ13Ccol, δ15Ncol) derived from different laboratories are directly comparable. However, the direct comparison of isotopic data derived from bone hydroxyapatite (δ13Cap, δ18Oap) is more dangerous because variability engendered by differences in pretreatment, analysis, and standardization is of a far greater magnitude. To remedy this issue, we introduce what we have termed the Minimum Meaningful Difference (MMD) value, which serves as an empirically derived threshold by which the significance of values obtained in different laboratories might be judged. In the end, the results of this study have serious implications for choices made in the preparation and extraction of target biomolecules, the comparison of results obtained from different laboratories, and the interpretation of small differences in bone collagen and hydroxyapatite isotope values.
Methods and Materials
The fundamental premise of the present study is that the best assessment of inter-laboratory variability in stable isotope analysis would require replicate preparation and analysis of the same demonstrably ancient bone sample by a large number of participating laboratories. As such, the initial four major methodological components were: 1) identification and sub-sampling of a suitable ancient human bone sample, 2) verification of this bone's antiquity, 3) recruitment of a representative cohort of participating laboratories, and 4) construction of a rigorous survey and reporting regime by which both laboratory methods and results could be compiled in a manner that would facilitate subsequent statistical analysis. The goal of this work is to characterize the amount of variation present among laboratories rather than comment on “better” or “worse” preparation methods or analytical facilities.
In 2011, one of us (WJP) obtained a presumably ancient unprovenienced human femoral diaphysis from the Museo Gustavo Le Paige in San Pedro de Atacama, Chile. All necessary permits were obtained for the described study (Consejo de Monumentos Nacionales Ord. No. 3682/12, FONDECYT No. 1120376), which complied with all relevant regulations, and the field studies did not involve endangered or protected species. This specimen was judged to be appropriate based on its apparent excellent state of preservation (which is typical of intentionally buried ancient human bone from this hyperarid region of Chile), large size (>100 g), and likely ancient date.
The specimen was AMS 14C dated at the University of Arizona NSF-AMS facility following their established protocols for 14C dating of bone (acid-base-acid pretreatment, gelatinization, filtration, graphitization). The resulting AMS date for this specimen (laboratory #AA99865) is 1728±47 14C years before present (δ13C -17.3‰), This equates to a 2-sigma calibrated age range of 238–470 cal AD when calibrated using Calib 7.0 and the SHCAL13 southern hemisphere terrestrial curve [61], [62].
Subsequent to radiocarbon dating, the authors solicited forty-six archaeological and paleoecological isotope laboratories in order to assess their willingness to participate in this study. Interested laboratories were informed that they would be provided with sufficient sample material to prepare and analyze at least three collagen and three hydroxyapatite replicates (although it was understood that not all laboratories would be able to comply with a full set of both collagen and apatite measurements). While this study was intended to document variation in the analysis of both collagen and hydroxyapatite, participants were asked to perform both types of analysis only if this was routine for their laboratory. In addition to isotopic data (δ13C, δ15N, δ18O), participating laboratories would be expected to provide details on pretreatment and analytical methods, as well as sample preservation assessments (e.g., sample yield, elemental values, amino acid analysis, FTIR spectra, etc.). All potential participants were informed that while their participation in the project would be made known, laboratory attributions of individual results would be kept confidential and all publicly disseminated data would be presented using randomly generated designators. However, in order for each participating laboratory to be able to assess its results compared to those for other study participants, the respective PI's were informed that they would be provided, at the conclusion of the study, with a full complement of the study's results with their data indicated.
Of the forty-six solicited laboratories, twenty-one (46%) ultimately committed to participate (Table 1). When laboratories provided reasons for not participating, they most often cited factors such as cost and time, although other laboratories declined on the basis that they were no longer performing such analyses. Based on the number of participating institutions, we used a handheld Dremel rotary tool equipped with a diamond cutoff wheel to divide the femur into 112 pieces, each weighing approximately 0.75 g. This large number of samples allowed each laboratory to receive five separate individual samples drawn at random from the overall assemblage of 112 pieces, thereby randomizing intra-bone variability and controlling for any random error engendered by differences in sample pretreatment within each laboratory.
Table 1. Participating institutions and laboratory PIs.
| Institution | Laboratory PI |
| Arizona State University | Knudson |
| California State University, Chico | Bartelink |
| Free University, Amsterdam | Laffoon |
| Max Planck Institute | Richards |
| Northern Arizona University | Kellner |
| Notre Dame University | Schurr |
| Oxford University | Hedges |
| University of California, San Diego | Schoeninger |
| University of California, Santa Cruz | Koch |
| University of Cincinnati | Crowley |
| University of Florida | Krigbaum |
| University of Idaho | Kohn |
| University of Illinois, Chicago | Pestle |
| University of Illinois, Urbana | Ambrose/Fort |
| University of Miami | Pestle |
| University of Munich | Grupe |
| University of Rochester | Higgins |
| University of South Florida | Tykot |
| University of Tübingen | Bocherens |
| University of Utah | Cerling |
| University of Wyoming | Martínez del Rio |
In addition to receiving five bone samples, each participating laboratory was provided with an instruction letter requesting that they prepare all five replicate samples using the same standard laboratory method and four standardized survey forms (Figures S1–S4) to use for recording (as appropriate): collagen preparation methods, collagen results, hydroxyapatite preparation methods, and hydroxyapatite results. The use of such standardized forms was intended to maximize comparability of laboratory protocols and to streamline statistical analysis. Some of the participating laboratories did not follow instructions to process all 5 samples or to do so with identical pretreatment.
Summaries of the collagen and hydroxyapatite protocols for each laboratory are provided (using anonymous identifiers) in Tables S1 and S2 (Supporting Information). Twenty of the twenty-one participating laboratories performed collagen extractions, with one laboratory, Laboratory D, performing two different kinds of extractions. Sixteen of the twenty-one laboratories extracted and analyzed hydroxyapatite, with one laboratory, Laboratory N, performing two different kinds of extractions. It should be immediately evident that while there are some broad similarities in sample preparation across laboratories (for example, twenty of the twenty-one collagen preparations (95%) were performed using hydrochloric acid (HCl) as the demineralizing agent), the variation in particle size, reagent concentrations, treatment times, temperature, etc., is substantial. The number and diversity of variables makes identification of particular causes of variability challenging (see discussion below), however we were able to identify protocols that overall yield more (or less) similar results.
To control for at least one potential source of variability, isotopic analysis, we reanalyzed as many samples as possible on one instrument. Eighteen of the twenty laboratories that performed collagen extractions (90%), and eleven of the sixteen laboratories that extracted hydroxyapatite (69%), returned aliquots of prepared material for reanalysis. Three aliquots of collagen and hydroxyapatite (when available) were selected from each laboratory's returned samples for reanalysis. Collagen samples were reanalyzed at the UC-Davis Stable Isotope Facility using a PDZ Europa ANCA-GSL elemental analyzer interfaced to a PDZ Europa 20–20 isotope ratio mass spectrometer (Sercon Ltd., Cheshire, UK). Elemental concentration was standardized by reference to Glutamic Acid, and stable isotope composition was standardized by reference to bovine liver, nylon, and USGS-41 Glutamic Acid. Hydroxyapatite samples were re-analyzed in the Stable Isotope Geochemistry Stable Isotope Laboratory at the Rosenstiel School of Marine and Atmospheric Sciences at the University of Miami using a Kiel-IV Carbonate Device (Thermo-Electron, Bremen, Germany) coupled to a Thermo-Finnigan DeltaPlus (Thermo-Electron, Bremen, Germany), and standardized in reference to NBS-19 (TS-Limestone). These duplicate analyses allowed us to independently assess the degree to which isotopic variability resulted from pretreatment versus analysis.
In addition to the use of a battery of well-established statistical analyses (z-score calculation, t-test, ANOVA, Levene's test for equality of variance, Pearson's bivariate correlation, all performed using SPSS v.20 [IBM, New York, USA]), we also used heat maps to visually identify which pretreatment protocols clustered together (i.e., produced similar results). Heat maps visually display data patterns by assigning a gradation of color to numerical values. The heat maps depict the difference in the values obtained by each pair of labs (yellow means no difference, with increasingly red values getting more different). Both axes were clustered using average linkage hierarchical clustering, with Euclidean distance as the distance metric. Heat maps were generated using the Genesis software package developed by Alexander Sturn and Rene Snajder (available freely at http://genome.tugraz.at/genesisclient/genesisclient_description.shtml). Significance for all analyses was set at α = 0.05.
Finally, we developed a novel metric for the evaluation of inter-laboratory variation, the Minimum Meaningful Difference (MMD). The intent of this metric is to establish a means by which to evaluate isotopic results obtained from different laboratories, or when comparing newly obtained results to previously published values in the literature. Our hope is that these values will be treated as an experimentally generated threshold value that one could quickly use when evaluating whether newly generated isotopic data are significantly more enriched or depleted than another laboratory's results or previously published isotopic data. This metric is far more meaningful than a simple t-test, for example, as it explicitly takes into account inter-laboratory variability.
The development of MMDs assumed that the values obtained in the course of the present study are representative of the possible isotope values that might be obtained from any laboratory currently performing such analysis. Minimum Meaningful Differences were calculated by adding the average pairwise inter-laboratory difference for each isotope system plus four times the average of the standard deviations obtained by each laboratory participating in the present study (we used four times the standard deviation for each laboratory in order to account for 95% of the laboratory error from both laboratories in each pairwise comparison). Using this value, a researcher can evaluate with ∼95% confidence the likelihood that a newly obtained isotope value is different from another value as a consequence of bona fide biogenic differences rather than laboratory pretreatment and analysis.
Results and Discussion
Collagen
Although the present work does not focus on the most-commonly employed indicators of collagen quality (collagen yield, weight %C, weight %N, and atomic C:N), we present these for comparability with other studies. Across all laboratories, the respective values for these metrics were: collagen yield = 16.4±7.9% (the large range of which is explained, at least in part, by the fact that some laboratories employed ultrafiltration whereas the majority did not), weight %C = 41.7±5.3%, weight %N = 15.2±1.9%, and atomic C:N ratio = 3.2±0.1. These data robustly confirm the excellent quality of preservation of the collagen in the selected specimen.
Across all laboratories, δ13Ccol values averaged −17.0±0.3‰ and had an overall range of 1.8‰ (Table 2, Figure 2, top). Of the ninety-six measured values, six were apparent outliers (note red cells in Figure 3, top): one from Laboratory L (z-score −2.1 [p = 0.02]) and all five from Laboratory Q (z-scores from 3.0 [p<0.01] to 4.1 [p<0.01]). Overall, the laboratories cluster into four distinct groups, with Laboratory Q as a clear outlier (Figure 2, top, Figure 3, top). Nitrogen isotope values averaged 9.0±0.3‰, with an overall range of 1.9‰ (Table 2, Figure 2, bottom). Of the ninety-six measured δ15Ncol values, seven were outliers (with z-scores greater than 2.0 [p = 0.02]): One from Laboratory B, two from Laboratory H, and four from Laboratory L (note red cells in Figure 3, bottom). Four major δ15Ncol groups emerged, with two clear outliers (Laboratories L and H) (Figure 3, bottom). A statistically significant, but overall weak, Pearson correlation (r = 0.26, p = 0.01) was observed between δ13Ccol and δ15Ncol values (Figure 4).
Table 2. Results of initial analysis of bone collagen.
| Lab | δ13Ccol-VPDB (‰) | δ15Ncol-AIR (‰) | ||||||||||||
| R1 | R2 | R3 | R4 | R5 | Mean | sd | R1 | R2 | R3 | R4 | R5 | Mean | sd | |
| A | −17.2 | −17.1 | −17.3 | −17.2 | −17.1 | −17.2 | 0.1 | 9.1 | 9.1 | 9.2 | 9.1 | 9.2 | 9.1 | 0.0 |
| B | −17.3 | −17.4 | −17.3 | −17.4 | −17.3 | −17.3 | 0.1 | 8.5 | 8.6 | 8.2 | 8.7 | 8.6 | 8.5 | 0.2 |
| C | −17.2 | −17.0 | −16.9 | −17.0 | −16.8 | −17.0 | 0.1 | 9.2 | 8.7 | 8.9 | 8.7 | 8.9 | 8.9 | 0.2 |
| D1 | −17.2 | −17.0 | −17.1 | −17.1 | −17.2 | −17.1 | 0.1 | 8.7 | 8.9 | 8.6 | 8.8 | 9.0 | 8.8 | 0.1 |
| D2 | −17.2 | −17.1 | −17.2 | −17.2 | −17.4 | −17.2 | 0.1 | 8.6 | 8.6 | 8.6 | 8.8 | 9.1 | 8.7 | 0.2 |
| F | −17.0 | −17.0 | −17.1 | −16.9 | −17.0 | −17.0 | 0.1 | 9.1 | 9.1 | 9.3 | 9.3 | 8.8 | 9.1 | 0.2 |
| G | −17.1 | −16.6 | −17.0 | −17.2 | −16.9 | −16.9 | 0.2 | 9.0 | 9.0 | 9.0 | 9.2 | 9.0 | 9.1 | 0.1 |
| H | −17.4 | −16.8 | −16.8 | −17.1 | −17.1 | −17.0 | 0.2 | 9.5 | 9.6 | 9.7 | 9.9 | 9.4 | 9.6 | 0.2 |
| I | −17.0 | −17.0 | −17.2 | −17.0 | −17.0 | −17.0 | 0.1 | 9.3 | 9.4 | 9.3 | 9.3 | 9.2 | 9.3 | 0.1 |
| J | −17.2 | −17.4 | −17.4 | −17.3 | −17.2 | −17.3 | 0.1 | 8.9 | 8.9 | 8.8 | 9.0 | 8.8 | 8.9 | 0.1 |
| K | −16.9 | −17.1 | −17.0 | −17.1 | −17.0 | 0.1 | 8.9 | 9.0 | 8.8 | 8.7 | 8.9 | 0.1 | ||
| L | −17.0 | −17.2 | −17.7 | −17.2 | −17.1 | −17.2 | 0.2 | 8.3 | 8.0 | 8.2 | 8.1 | 8.2 | 8.2 | 0.1 |
| M | −16.9 | −16.8 | −16.9 | −17.0 | −17.1 | −16.9 | 0.1 | 8.9 | 9.1 | 8.9 | 9.2 | 9.3 | 9.1 | 0.2 |
| N | −17.0 | −17.0 | −17.0 | −17.0 | −17.1 | −17.0 | 0.1 | 8.7 | 8.6 | 8.5 | 8.4 | 8.9 | 8.6 | 0.2 |
| O | −17.2 | −17.0 | −17.1 | 0.1 | 9.0 | 9.4 | 9.2 | 0.2 | ||||||
| P | −17.2 | −17.2 | −17.4 | −17.3 | −17.5 | −17.3 | 0.1 | 9.2 | 9.3 | 9.4 | 9.4 | 9.3 | 9.3 | 0.1 |
| Q | −16.2 | −15.9 | −16.2 | −16.1 | −16.1 | −16.1 | 0.1 | 8.9 | 9.5 | 9.1 | 9.5 | 9.4 | 9.3 | 0.2 |
| R | −17.1 | −17.2 | −17.0 | −17.1 | −17.1 | −17.1 | 0.1 | 9.2 | 9.0 | 9.1 | 9.1 | 8.8 | 9.0 | 0.1 |
| S | −17.1 | −17.0 | −17.1 | −17.1 | −17.0 | −17.1 | 0.0 | 9.2 | 9.0 | 9.1 | 9.1 | 8.8 | 9.0 | 0.1 |
| U | −16.9 | −17.0 | −17.0 | −17.0 | −17.0 | −17.0 | 0.0 | 9.0 | 8.9 | 9.0 | 8.8 | 9.1 | 9.0 | 0.1 |
| Mean | −17.0 | Mean | 9.0 | |||||||||||
| sd | 0.3 | sd | 0.3 | |||||||||||
| Median | −17.1 | Median | 9.0 | |||||||||||
| Max | −15.9 | Max | 9.9 | |||||||||||
| Min | −17.7 | Min | 8.0 | |||||||||||
Figure 2. Distribution of initial δ13Ccol (top) and δ15Ncol (bottom) values by laboratory.

Dots represent individual analyses and solid horizontal lines represent the median values for all participating laboratories (−17.1‰ for δ13Ccol and 9.1‰ for δ15Ncol).
Figure 3. Heat maps of initial δ13Ccol (top) and δ15Ncol (bottom) values by laboratory.

Each entry in the matrix depicts the difference in values obtained between the given pair of laboratories. The key is provided at the top – large differences are red, and minimal differences are yellow. Rows and columns have been clustered in order to place similar laboratories near each other (clusters are indicated by the trees above and to the left of the heat maps) – see Methods.
Figure 4. Scatterplot of individual sample δ13Ccol and δ15Ncol values presented for each laboratory.

Outlying δ13Cco values of Laboratory Q are particularly evident.
Analysis of inter-laboratory variation indicates significant differences among laboratories for the two isotope systems of interest. For δ13Ccol, the average pairwise inter-laboratory difference was 0.2‰ (Table 3, above diagonal), and the values obtained by the various laboratories were found to be significantly different (ANOVA, F19,76 = 19.3, p<0.01). Significant differences remained even after outliers were removed (ANOVA, F18,71 = 5.4, p<0.01). The average pairwise inter-laboratory difference for δ15Ncol values was 0.4‰ (Table 3, below diagonal), with highly significant differences among laboratories (ANOVA, F19,76 = 19.3, p<0.01). Again, significant differences remained even after outliers were removed (ANOVA, F19,69 = 10.7, p<0.01).
Table 3. Difference matrix showing mean isotopic differences between laboratories for bone collagen results, δ13Ccol above diagonal, δ15Ncol beneath diagonal.
| Lab | A | B | C | D1 | D2 | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | U | ||
| δ13Ccol-VPDB (‰) | −17.2 | −17.3 | −17.0 | −17.1 | −17.2 | −17.0 | −16.9 | −17.0 | −17.0 | −17.3 | −17.0 | −17.2 | −16.9 | −17.0 | −17.1 | −17.3 | −16.1 | −17.1 | −17.1 | −17.0 | ||
| δ15Ncol-AIR (‰) | 9.1 | 8.5 | 8.9 | 8.8 | 8.7 | 9.1 | 9.1 | 9.6 | 9.3 | 8.9 | 8.9 | 8.2 | 9.1 | 8.6 | 9.2 | 9.3 | 9.3 | 9.0 | 9.0 | 9.0 | ||
| A | −17.2 | 9.1 | - | 0.1 | 0.2 | 0.1 | 0.0 | 0.2 | 0.2 | 0.2 | 0.2 | 0.1 | 0.2 | 0.0 | 0.3 | 0.2 | 0.1 | 0.1 | 1.1 | 0.1 | 0.1 | 0.2 |
| B | −17.3 | 8.5 | 0.6 | - | 0.4 | 0.2 | 0.1 | 0.3 | 0.4 | 0.3 | 0.3 | 0.0 | 0.3 | 0.1 | 0.4 | 0.3 | 0.2 | 0.0 | 1.2 | 0.2 | 0.3 | 0.4 |
| C | −17.0 | 8.9 | 0.2 | 0.4 | - | 0.1 | 0.2 | 0.0 | 0.0 | 0.1 | 0.1 | 0.3 | 0.0 | 0.2 | 0.0 | 0.0 | 0.1 | 0.3 | 0.9 | 0.1 | 0.1 | 0.0 |
| D1 | −17.1 | 8.8 | 0.3 | 0.3 | 0.1 | - | 0.1 | 0.1 | 0.2 | 0.1 | 0.1 | 0.2 | 0.1 | 0.1 | 0.2 | 0.1 | 0.0 | 0.2 | 1.0 | 0.0 | 0.1 | 0.1 |
| D2 | −17.2 | 8.7 | 0.4 | 0.2 | 0.1 | 0.1 | - | 0.2 | 0.3 | 0.2 | 0.2 | 0.1 | 0.2 | 0.0 | 0.3 | 0.2 | 0.1 | 0.1 | 1.1 | 0.1 | 0.1 | 0.2 |
| F | −17.0 | 9.1 | 0.0 | 0.6 | 0.2 | 0.3 | 0.4 | - | 0.1 | 0.0 | 0.0 | 0.3 | 0.0 | 0.2 | 0.1 | 0.0 | 0.1 | 0.3 | 0.9 | 0.1 | 0.1 | 0.0 |
| G | −16.9 | 9.1 | 0.1 | 0.5 | 0.2 | 0.3 | 0.3 | 0.1 | - | 0.1 | 0.1 | 0.4 | 0.1 | 0.3 | 0.0 | 0.0 | 0.2 | 0.4 | 0.8 | 0.2 | 0.1 | 0.0 |
| H | −17.0 | 9.6 | 0.5 | 1.1 | 0.7 | 0.8 | 0.9 | 0.5 | 0.6 | - | 0.0 | 0.3 | 0.0 | 0.2 | 0.1 | 0.0 | 0.1 | 0.3 | 0.9 | 0.1 | 0.0 | 0.1 |
| I | −17.0 | 9.3 | 0.2 | 0.8 | 0.4 | 0.5 | 0.6 | 0.2 | 0.2 | 0.3 | - | 0.3 | 0.0 | 0.2 | 0.1 | 0.0 | 0.1 | 0.3 | 0.9 | 0.1 | 0.0 | 0.1 |
| J | −17.3 | 8.9 | 0.2 | 0.4 | 0.0 | 0.1 | 0.1 | 0.2 | 0.2 | 0.7 | 0.4 | - | 0.3 | 0.1 | 0.4 | 0.3 | 0.2 | 0.0 | 1.2 | 0.2 | 0.2 | 0.3 |
| K | −17.0 | 8.9 | 0.3 | 0.3 | 0.0 | 0.0 | 0.1 | 0.3 | 0.2 | 0.8 | 0.5 | 0.0 | - | 0.2 | 0.1 | 0.0 | 0.1 | 0.3 | 0.9 | 0.1 | 0.0 | 0.0 |
| L | −17.2 | 8.2 | 0.9 | 0.4 | 0.7 | 0.6 | 0.6 | 1.0 | 0.9 | 1.5 | 1.1 | 0.7 | 0.7 | - | 0.3 | 0.2 | 0.1 | 0.1 | 1.1 | 0.1 | 0.2 | 0.2 |
| M | −16.9 | 9.1 | 0.0 | 0.6 | 0.2 | 0.3 | 0.3 | 0.0 | 0.0 | 0.5 | 0.2 | 0.2 | 0.2 | 0.9 | - | 0.1 | 0.2 | 0.4 | 0.8 | 0.2 | 0.1 | 0.0 |
| N | −17.0 | 8.6 | 0.5 | 0.1 | 0.3 | 0.2 | 0.1 | 0.5 | 0.5 | 1.0 | 0.7 | 0.3 | 0.3 | 0.4 | 0.5 | - | 0.1 | 0.3 | 0.9 | 0.1 | 0.1 | 0.0 |
| O | −17.1 | 9.2 | 0.1 | 0.7 | 0.3 | 0.4 | 0.5 | 0.1 | 0.1 | 0.4 | 0.1 | 0.3 | 0.4 | 1.0 | 0.1 | 0.6 | - | 0.2 | 1.0 | 0.0 | 0.0 | 0.1 |
| P | −17.3 | 9.3 | 0.2 | 0.8 | 0.4 | 0.5 | 0.6 | 0.2 | 0.2 | 0.3 | 0.0 | 0.4 | 0.5 | 1.1 | 0.2 | 0.7 | 0.1 | - | 1.2 | 0.2 | 0.2 | 0.3 |
| Q | −16.1 | 9.3 | 0.2 | 0.8 | 0.4 | 0.5 | 0.5 | 0.2 | 0.2 | 0.3 | 0.0 | 0.4 | 0.4 | 1.1 | 0.2 | 0.7 | 0.1 | 0.0 | - | 1.0 | 1.0 | 0.9 |
| R | −17.1 | 9.0 | 0.1 | 0.5 | 0.2 | 0.2 | 0.3 | 0.1 | 0.0 | 0.6 | 0.3 | 0.2 | 0.2 | 0.9 | 0.0 | 0.4 | 0.2 | 0.3 | 0.2 | - | 0.0 | 0.1 |
| S | −17.1 | 9.0 | 0.1 | 0.5 | 0.2 | 0.2 | 0.3 | 0.1 | 0.0 | 0.6 | 0.3 | 0.2 | 0.2 | 0.9 | 0.0 | 0.4 | 0.2 | 0.3 | 0.2 | 0.0 | - | 0.1 |
| U | −17.0 | 9.0 | 0.2 | 0.4 | 0.1 | 0.2 | 0.2 | 0.2 | 0.1 | 0.7 | 0.3 | 0.1 | 0.1 | 0.8 | 0.1 | 0.4 | 0.2 | 0.3 | 0.3 | 0.1 | 0.1 | - |
It is worthwhile noting that neither the choice of demineralizing agent (HCl versus EDTA) nor the decision of whether/how to remove humic acids (NaOH, KOH, or no treatment) engendered significant differences in the resulting isotopic signatures. The offset seen between samples demineralized using HCl versus EDTA was only 0.2‰ for δ13Ccol (t = 1.3, df = 94, p = 0.2) and 0.2‰ δ15Ncol (t = 1.5, df = 94, p = 0.2). No significant differences in δ13Ccol or δ15Ncol were observed as a result of humic acid removal reagents; no treatment, NaOH, and KOH produced indistinguishable δ13Ccol (ANOVA, F2,93 = 1.8, p = 0.2) and δ15Ncol (ANOVA, F2,93 = 0.6, p = 0.5) values. It is possible that the lack of appreciable differences in isotope values between laboratories that did and did not remove humic acids could be a consequence of the sample's low initial humic content.
Collagen reanalysis
Reanalysis of a subset of the collagen samples on the same instrument shifted both collagen isotope systems by approximately 0.1‰, with δ13Ccol decreasing from -17.0±0.3‰ to −17.1±0.1‰ (t = 1.4, df = 145, p = 0.3; Table 4, Figure 5, top), and δ15Ncol values increasing significantly (albeit not meaningfully) from 9.0±0.4‰ to 9.1±0.2‰ (t = 2.5, df = 145, p<0.01; Table 4, Figure 5, bottom). In both instances, reanalysis on the same instrument significantly reduced the variance for the measured samples. Standard deviation was significantly reduced for carbon (from 0.3‰ to 0.1‰; W = 8.4, df = 145, p<0.01) and nitrogen (0.4‰ to 0.2‰; W = 8.0, df = 145, p<0.01).
Table 4. Results of secondary reanalysis of bone collagen.
| Lab | δ13Ccol-VPDB (‰) | δ15Ncol-AIR (‰) | ||||||||
| R1 | R2 | R3 | Mean | sd | R1 | R2 | R3 | Mean | sd | |
| A | −16.9 | −17.0 | −17.1 | −17.0 | 0.1 | 9.0 | 9.2 | 9.3 | 9.2 | 0.2 |
| B | −17.1 | −17.3 | −17.2 | −17.2 | 0.1 | 9.2 | 9.0 | 8.9 | 9.0 | 0.2 |
| D1 | −17.1 | −17.1 | −17.1 | −17.1 | 0.0 | 8.9 | 8.9 | 8.6 | 8.8 | 0.2 |
| D2 | −17.3 | −17.1 | −17.1 | −17.2 | 0.1 | 9.0 | 8.8 | 8.9 | 8.9 | 0.1 |
| F | −17.0 | −17.2 | −17.2 | −17.1 | 0.1 | 8.6 | 8.9 | 9.0 | 8.8 | 0.2 |
| G | −17.2 | −17.2 | −17.2 | 0.0 | 9.4 | 9.3 | 9.4 | 0.1 | ||
| I | −17.2 | −17.1 | −17.0 | −17.1 | 0.1 | 9.1 | 9.2 | 9.0 | 9.1 | 0.1 |
| J | −17.1 | −17.1 | −17.0 | −17.1 | 0.1 | 9.2 | 9.2 | 9.2 | 9.2 | 0.0 |
| K | −17.0 | −17.0 | −17.1 | −17.0 | 0.1 | 9.1 | 9.1 | 9.4 | 9.2 | 0.2 |
| L | −16.9 | −16.8 | −17.3 | −17.0 | 0.3 | 9.4 | 9.5 | 9.3 | 9.4 | 0.1 |
| M | −17.2 | −17.0 | −17.2 | −17.1 | 0.1 | 9.6 | 9.0 | 9.3 | 9.3 | 0.3 |
| N | −17.0 | −17.0 | −17.0 | −17.0 | 0.0 | 9.3 | 8.9 | 8.7 | 9.0 | 0.3 |
| O | −17.1 | −17.2 | −17.2 | 0.1 | 9.2 | 8.7 | 9.0 | 0.4 | ||
| P | −17.3 | .17.2 | −17.0 | −17.2 | 0.2 | 8.9 | 9.0 | 8.9 | 8.9 | 0.1 |
| Q | −17.3 | −17.1 | −16.9 | −17.1 | 0.2 | 9.2 | 9.4 | 9.1 | 9.2 | 0.2 |
| R | −17.1 | −17.1 | −16.9 | −17.0 | 0.1 | 9.1 | 9.1 | 9.0 | 9.1 | 0.1 |
| S | −17.0 | −17.0 | −17.0 | −17.0 | 0.0 | 9.3 | 9.0 | 9.0 | 9.1 | 0.2 |
| U | −17.1 | −17.2 | −17.1 | −17.1 | 0.1 | 9.3 | 9.3 | 9.1 | 9.2 | 0.1 |
| Mean | −17.1 | Mean | 9.1 | |||||||
| sd | 0.1 | sd | 0.2 | |||||||
| Median | −17.1 | Median | 9.1 | |||||||
| Max | −16.8 | Max | 9.6 | |||||||
| Min | −17.3 | Min | 8.6 | |||||||
Figure 5. Boxplot comparison of δ13Ccol (top) and δ15Ncol (bottom) values between original analysis and reanalysis on a single instrument.
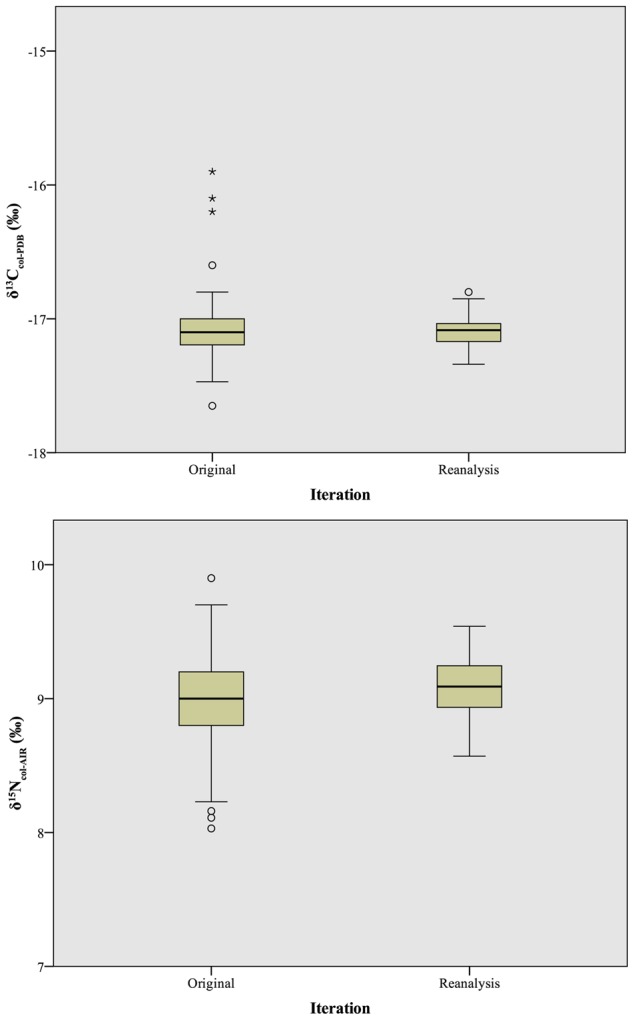
Box lines represent first quartile, second quartile (median), and third quartile; whiskers at 95% confidence intervals; dots represent weak outliers (more than 2 standard deviations from mean); asterisks represent strong outliers (more than 3 standard deviations from mean).
Reanalysis of the collagen samples affected the number and distribution of outliers. For δ13Ccol (Figure 5, top), three samples, one each from Laboratories A, B, and L, possessed z-scores between 2.1 (p = 0.02) and 2.5 (p<0.01). For δ15Ncol (Figures 5, bottom), a different set of three samples, one each from Laboratories D1, F, and M, had outlier z-scores between 2.0 (p = 0.02) and 2.3 (p = 0.01). Mean isotope values for laboratories that initially had uniformly outlying values (Laboratory Q for δ13Ccol and Laboratory L for δ15Ncol) were no longer aberrant after reanalysis. This strongly suggests that although collagen pretreatment methods are responsible for some of the observed isotopic differences among laboratories, differences in instrumentation or data calibration also drive a large amount of the observed variation in isotope values among laboratories: 69% (1.2‰ of the initially observed 1.8‰ range) for δ13Ccol and 48% (0.9‰ of the initially observed 1.9‰ range) for δ15Ncol [59], [60].
Establishing Minimum Meaningful Differences for Collagen
The Minimum Meaningful Difference (MMD) value, which takes into account both the average inter-laboratory difference and the typically observed intra-laboratory variability (in the form of the standard deviation of each laboratory's replicate measurements), was determined to be a modest 0.6‰ for δ13Ccol (Table 5). This means that a difference in isotope values obtained from two different analyses is likely to be bona fide if that difference exceeds the threshold value of 0.6‰. MMD for δ15Ncol (Table 5) was slightly higher (0.9‰). The relatively small magnitude of these values, as will be discussed below, provides substantial reassurance about the relative comparability of collagen isotope results obtained from different laboratories.
Table 5. Mean Measure of Difference values for four isotopic systems of interest.
| Isotopic system | Average pairwise difference (‰) | Average intra-laboratory standard deviation (‰) | MMD (‰) |
| δ13Cap | 0.6 | 0.15 | 1.2 |
| δ18Oap | 2 | 0.28 | 3.1 |
| δ13Ccol | 0.2 | 0.1 | 0.6 |
| δ15Ncol | 0.4 | 0.13 | 0.9 |
Hydroxyapatite
Across all laboratories, δ13Cap values averaged −11.7±0.6‰ and had an overall range of 3.5‰ (Table 6, Figure 6, top). Four measured values, all from the same laboratory (Laboratory R), were apparent outliers, with z-scores between 2.0 (p = 0.02) and 4.4 (p<0.01). Oxygen isotope values averaged −4.6±1.7‰, with an overall range of 6.7‰ (!) and no apparent outliers (Table 6, Figure 6, bottom). No significant Pearson correlation (r = −0.1, p = 0.2) was observed between δ13Cap and δ18Oap values (Figure 7).
Table 6. Results of initial analysis of bone hydroxyapatite.
| Lab | δ13Cap-VPDB (‰) | δ18Oap-VPDB (‰) | ||||||||||||
| R1 | R2 | R3 | R4 | R5 | Mean | sd | R1 | R2 | R3 | R4 | R5 | Mean | sd | |
| A | −11.5 | −11.4 | −11.5 | −11.7 | −11.5 | −11.5 | 0.1 | −2.5 | −2.7 | −2.7 | −2.7 | −2.6 | −2.6 | 0.1 |
| B | −11.4 | −11.7 | −11.5 | −11.8 | −11.5 | −11.6 | 0.1 | −6.4 | −6.6 | −7.7 | −8.0 | −6.8 | −7.1 | 0.6 |
| C | −12.0 | −12.1 | −11.8 | −12.0 | 0.1 | −3.3 | −2.9 | −3.1 | −3.1 | 0.2 | ||||
| D | −11.5 | −11.2 | −11.4 | −11.5 | −11.4 | −11.4 | 0.1 | −2.5 | −2.6 | −3.0 | −2.3 | −2.4 | −2.5 | 0.2 |
| F | −12.3 | −12.4 | −12.4 | −12.4 | −12.3 | −12.4 | 0.0 | −5.7 | −5.7 | −6.0 | −6.0 | −5.9 | −5.9 | 0.1 |
| G | −12.5 | −12.1 | −12.2 | −12.4 | −12.5 | −12.3 | 0.2 | −4.5 | −4.5 | −4.1 | −4.8 | −4.5 | −4.5 | 0.2 |
| H | −12.2 | −11.7 | −12.1 | −11.9 | −11.7 | −11.9 | 0.2 | −4.0 | −3.7 | −4.8 | −4.3 | −3.6 | −4.1 | 0.4 |
| I | −11.6 | −11.6 | −11.6 | −11.7 | −11.7 | −11.7 | 0.1 | −6.4 | −6.3 | −6.2 | −6.4 | −6.0 | −6.2 | 0.2 |
| K | −11.7 | −11.6 | −11.7 | −11.7 | −11.7 | −11.7 | 0.1 | −3.6 | −3.2 | −3.4 | −3.4 | −3.6 | −3.4 | 0.2 |
| N1 | −11.5 | −11.2 | −11.4 | 0.2 | −3.3 | −3.0 | −3.2 | 0.2 | ||||||
| N2 | −12.1 | −12.2 | −12.2 | −12.2 | 0.0 | −3.1 | −3.6 | −3.5 | −3.4 | 0.2 | ||||
| O | −11.3 | −11.3 | −11.3 | 0.0 | −6.7 | −6.6 | −6.7 | 0.0 | ||||||
| P | −11.8 | −11.9 | −12.1 | −11.9 | −11.9 | −11.9 | 0.1 | −3.4 | −3.4 | −3.8 | −3.6 | −3.5 | −3.5 | 0.2 |
| R | −10.7 | −10.3 | −9.1 | −10.5 | −10.1 | −10.1 | 0.6 | −7.3 | −7.7 | −6.3 | −7.8 | −7.7 | −7.4 | 0.6 |
| S | −12.5 | −12.3 | −12.6 | −12.6 | −12.2 | −12.4 | 0.2 | −6.2 | −6.4 | −6.5 | −6.1 | −5.8 | −6.2 | 0.3 |
| T | −11.8 | −11.8 | −11.5 | −11.2 | −11.5 | −11.6 | 0.2 | −4.0 | −3.1 | −2.3 | −3.5 | −1.3 | −2.8 | 0.9 |
| Mean | −11.7 | Mean | −4.6 | |||||||||||
| sd | 0.6 | sd | 1.7 | |||||||||||
| Median | −11.7 | Median | −4.0 | |||||||||||
| Max | −9.1 | Max | −1.3 | |||||||||||
| Min | −12.6 | Min | −8.0 | |||||||||||
Figure 6. Distribution of initial δ13Cap (top) and δ18Oap (bottom) values by laboratory.

Dots represent individual analyses and solid horizontal lines represent median values for all participating laboratories (−11.7‰ for δ13Cap and −4.0‰ for δ18Oap).
Figure 7. Scatterplot of individual δ13Cap and δ18Oap values presented for each laboratory.
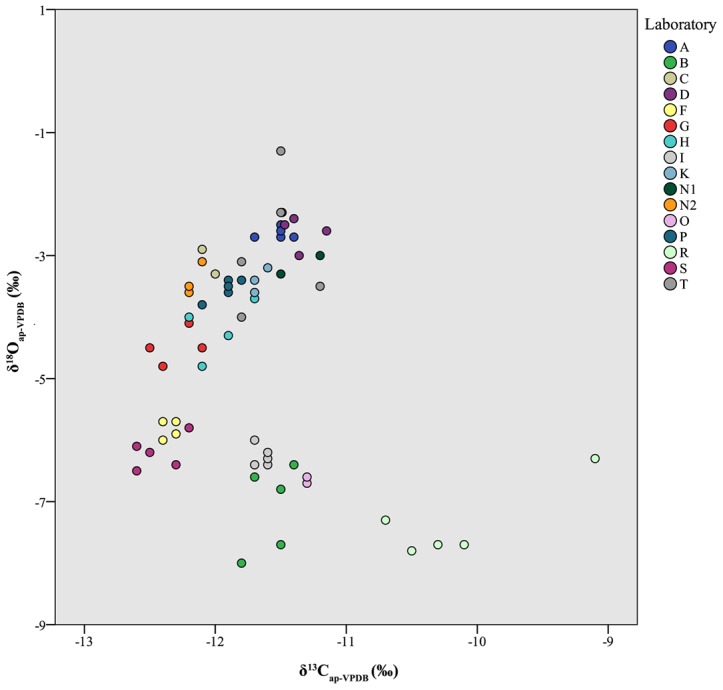
High variability of δ18Oap values and outlying values of Laboratory R are both apparent.
Analysis of inter-laboratory variation indicates significant differences among laboratories for the two isotope systems of interest (note red cells in Figure 8). For δ13Cap, the average pairwise inter-laboratory difference was 0.6‰ (Table 7, above diagonal). Mean δ13Cap values differed significantly among laboratories (ANOVA, F15,54 = 29.4, p<0.01), a finding that persisted following the removal of the four outlying values (ANOVA, F14,50 = 27.0, p<0.01). The average pairwise inter-laboratory difference for δ18Oap values was 2.0‰ (Table 7, below diagonal), with highly significant differences in distribution among laboratories (ANOVA, F15,54 = 66.9, p<0.01).
Figure 8. Heat maps of initial δ13Cap (top) and δ18Oap (bottom) values by laboratory.
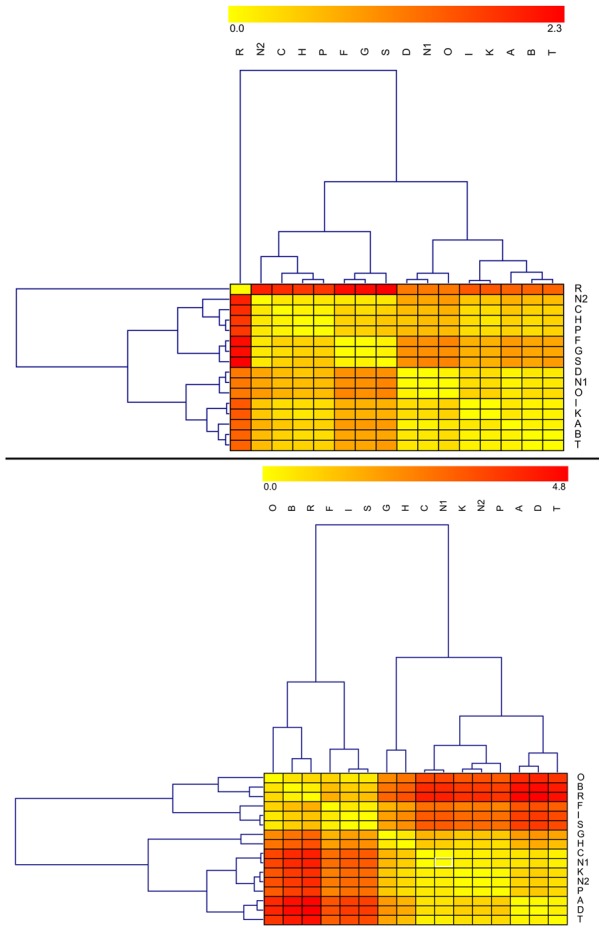
See Figure 3 legend for details.
Table 7. Difference matrix showing mean isotopic differences between laboratories for bone hydroxyapatite results; δ13Cap above diagonal, δ18Oap beneath diagonal.
| Lab | A | B | C | D | F | G | H | I | K | N1 | N2 | O | P | R | S | T | ||
| δ13Cap-VPDB (‰) | −11.5 | −11.6 | −12.0 | −11.4 | −12.4 | −12.3 | −11.9 | −11.7 | −11.7 | −11.4 | −12.2 | −11.3 | −11.9 | −10.1 | −12.4 | −11.6 | ||
| δ18Oap-VPDB (‰) | −2.6 | −7.1 | −3.1 | −2.5 | −5.9 | −4.5 | −4.1 | −6.2 | −3.4 | −3.2 | −3.4 | −6.7 | −3.5 | −7.4 | −6.2 | −2.8 | ||
| A | −11.5 | −2.6 | - | 0.1 | 0.5 | 0.1 | 0.9 | 0.9 | 0.4 | 0.2 | 0.2 | 0.1 | 0.7 | 0.2 | 0.4 | 1.4 | 0.9 | 0.1 |
| B | −11.6 | −7.1 | 4.5 | - | 0.4 | 0.2 | 0.8 | 0.8 | 0.3 | 0.1 | 0.1 | 0.2 | 0.6 | 0.3 | 0.3 | 1.4 | 0.8 | 0.0 |
| C | −12.0 | −3.1 | 0.5 | 4.0 | - | 0.6 | 0.4 | 0.3 | 0.1 | 0.3 | 0.3 | 0.6 | 0.2 | 0.7 | 0.1 | 1.9 | 0.4 | 0.4 |
| D | −11.4 | −2.5 | 0.1 | 4.6 | 0.6 | - | 1.0 | 1.0 | 0.5 | 0.3 | 0.3 | 0.0 | 0.8 | 0.1 | 0.6 | 1.2 | 1.0 | 0.2 |
| F | −12.4 | −5.9 | 3.2 | 1.2 | 2.7 | 3.3 | - | 0.0 | 0.4 | 0.7 | 0.7 | 1.0 | 0.2 | 1.1 | 0.4 | 2.2 | 0.1 | 0.8 |
| G | −12.3 | −4.5 | 1.9 | 2.6 | 1.4 | 2.0 | 1.4 | - | 0.4 | 0.7 | 0.7 | 1.0 | 0.2 | 1.0 | 0.4 | 2.2 | 0.1 | 0.8 |
| H | −11.9 | −4.1 | 1.4 | 3.0 | 1.0 | 1.6 | 1.8 | 0.4 | - | 0.3 | 0.2 | 0.6 | 0.2 | 0.6 | 0.0 | 1.8 | 0.5 | 0.4 |
| I | −11.7 | −6.2 | 3.6 | 0.9 | 3.1 | 3.7 | 0.4 | 1.8 | 2.2 | - | 0.0 | 0.3 | 0.5 | 0.4 | 0.3 | 1.5 | 0.7 | 0.1 |
| K | −11.7 | −3.4 | 0.8 | 3.7 | 0.3 | 0.9 | 2.4 | 1.0 | 0.6 | 2.8 | - | 0.3 | 0.5 | 0.4 | 0.3 | 1.5 | 0.7 | 0.1 |
| N1 | −11.4 | −3.2 | 0.5 | 4.0 | 0.0 | 0.6 | 2.7 | 1.3 | 0.9 | 3.1 | 0.3 | - | 0.8 | 0.0 | 0.6 | 1.2 | 1.1 | 0.2 |
| N2 | −12.2 | −3.4 | 0.8 | 3.7 | 0.3 | 0.9 | 2.5 | 1.1 | 0.7 | 2.8 | 0.0 | 0.3 | - | 0.9 | 0.2 | 2.0 | 0.2 | 0.6 |
| O | −11.3 | −6.7 | 4.0 | 0.5 | 3.5 | 4.1 | 0.8 | 2.2 | 2.6 | 0.4 | 3.2 | 3.5 | 3.3 | - | 0.6 | 1.2 | 1.1 | 0.3 |
| P | −11.9 | −3.5 | 0.9 | 3.6 | 0.4 | 1.0 | 2.3 | 1.0 | 0.6 | 2.7 | 0.1 | 0.4 | 0.1 | 3.1 | - | 1.8 | 0.5 | 0.4 |
| R | −10.1 | −7.4 | 4.7 | 0.3 | 4.2 | 4.8 | 1.5 | 2.9 | 3.3 | 1.1 | 3.9 | 4.2 | 4.0 | 0.7 | 3.8 | - | 2.3 | 1.4 |
| S | −12.4 | −6.2 | 3.6 | 0.9 | 3.1 | 3.7 | 0.3 | 1.7 | 2.1 | 0.0 | 2.8 | 3.1 | 2.8 | 0.4 | 2.7 | 1.2 | - | 0.9 |
| T | −11.6 | −2.8 | 0.2 | 4.3 | 0.3 | 0.3 | 3.0 | 1.7 | 1.2 | 3.4 | 0.6 | 0.3 | 0.6 | 3.8 | 0.7 | 4.5 | 3.4 | - |
One presumed driver of inter-laboratory variation in isotope values is the chosen method of organic removal, namely bleach (sodium hypochlorite, NaOCl) versus hydrogen peroxide (H2O2). Indeed, the isotope values of samples processed using hydrogen peroxide produced significantly enriched δ13Cap and δ18Oap values compared to those samples processed with bleach, with values of −11.8±0.7‰ versus −11.5±0.2‰ for δ13Cap (t = −2.9, df = 68, p<0.01; Figure 9, top), and −2.7±0.6‰ versus −5.1±1.6‰ for δ18Oap (t = −9.2, df = 68, p<0.01; Figure 9, bottom). Indeed, it is this preparation difference that helps to explain why there are two clear clusters (high-level branchings) in the heat maps of Figure 8. As the difference in mean δ18Oap values for samples processed by hydrogen peroxide versus bleach (2.4‰) is greater than the average pairwise difference in δ18Oap values between any two participating laboratories (2.0‰), it would appear that the choice of reagent used for organic removal is a prime driver in inter-laboratory variation in δ18Oap [41]. The same is not the case for δ13Cap, as the difference in means between oxidation methods (0.3‰) is less than the average pairwise inter-laboratory difference (0.6‰).
Figure 9. Boxplot comparison of δ13Cap (top) and δ18Oap (bottom) values obtained after oxidation with NaOCl versus H2O2.
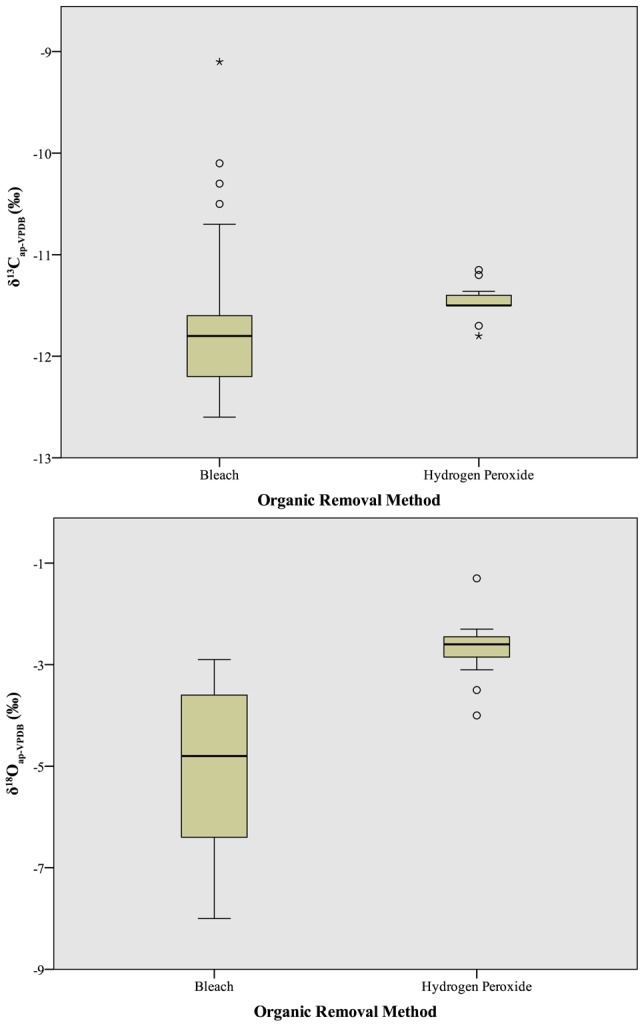
Box lines represent first quartile, second quartile (median), and third quartile; whiskers at 95% confidence intervals; dots represent weak outliers (more than 2 standard deviations from mean); asterisks represent strong outliers (more than 3 standard deviations from mean).
Differences in the labile carbonate removal technique (both concentration of acetic acid and the use of buffered versus un-buffered acetic) did not have a significant effect on δ13Cap or δ18Oap values. The offset seen between samples processed with 0.1–0.2 M versus 1.0 M acetic acid was only 0.03‰ for δ13Cap (t = −0.19, df = 68, p = 0.9) and 0.15‰ δ18Oap (t = −0.33, df = 68, p = 0.7). The differences between samples treated with buffered and un-buffered acetic acid were similarly small: 0.06‰ for δ13Cap (t = 0.37, df = 68, p = 0.7) and 0.2‰ for δ18Oap (t = −0.49, df = 68, p = 0.6). This result is somewhat unexpected, as previous studies [22], [42] have reported that acid strength has an impact on δ13Cap and δ18Oap values.
Hydroxyapatite reanalysis
Subsequent reanalysis of a subset of the hydroxyapatite samples on the same instrument revealed at least two interesting phenomena. First, reanalysis produced significantly enriched isotope results for carbon isotope values, which increased modestly from −11.7±0.6‰ to −11.2±0.5‰ (t = −4.0, df = 99, p<0.01; Table 8, Figure 10, top), as well as oxygen isotope values, which increased more dramatically from −4.6±1.7‰ to −3.4±0.9‰ (t = −4.6, df = 99, p<0.01; Table 8, Figure 10, bottom). Second, reanalysis on the same instrumentation significantly reduced the variance for δ18Oap, almost halving the standard deviation of the measured samples from 1.7‰ to 0.9‰ (W = 25.3, df = 99, p<0.01). While the variance for δ13Cap was also reduced during reanalysis, the observed difference (0.6‰ versus 0.5‰) was modest by comparison, and not significant (W = 0.01, df = 92, p = 0.92).
Table 8. Results of secondary reanalysis of bone hydroxyapatite.
| Lab | δ13Cap-VPDB (‰) | δ18Oap-VPDB (‰) | ||||||||
| R1 | R2 | R3 | Mean | sd | R1 | R2 | R3 | Mean | sd | |
| A | −11.4 | −11.8 | −11.4 | −11.5 | 0.2 | −2.3 | −3.5 | −2.3 | −2.7 | 0.7 |
| B | −10.9 | −10.9 | −11.3 | −11.0 | 0.2 | −4.3 | −4.8 | −4.1 | −4.4 | 0.3 |
| F | −11.6 | −11.5 | −11.6 | −11.6 | 0.1 | −3.0 | −2.5 | −3.0 | −2.9 | 0.3 |
| I | −11.6 | −11.0 | −11.4 | −11.3 | 0.3 | −4.7 | −4.0 | −4.4 | −4.4 | 0.3 |
| K | −11.7 | −11.5 | −11.6 | −11.6 | 0.1 | −2.7 | −2.2 | −2.3 | −2.4 | 0.2 |
| N2 | −11.0 | −11.3 | −11.2 | 0.2 | −3.2 | −3.0 | −3.1 | 0.1 | ||
| O | −10.0 | −10.3 | −10.2 | 0.1 | −3.7 | −4.1 | −3.9 | 0.3 | ||
| P | −11.8 | −12.0 | −11.2 | −11.7 | 0.4 | −3.7 | −3.7 | −3.3 | −3.6 | 0.2 |
| R | −10.8 | −11.2 | −11.5 | −11.2 | 0.4 | −4.5 | −4.7 | −4.5 | −4.5 | 0.1 |
| S | −11.6 | −10.7 | −11.7 | −11.4 | 0.5 | −3.3 | −2.7 | −4.1 | −3.4 | 0.7 |
| T | −10.3 | −10.4 | −10.9 | −10.6 | 0.3 | −1.7 | −1.8 | −2.4 | −2.0 | 0.4 |
| Mean | −11.2 | Mean | −3.4 | |||||||
| sd | 0.5 | sd | 0.9 | |||||||
| Median | −11.4 | Median | −3.3 | |||||||
| Max | −10.0 | Max | −1.7 | |||||||
| Min | −12.0 | Min | −4.8 | |||||||
Figure 10. Boxplot comparison of δ13Cap (top) and δ18Oap (bottom) values between original analysis and reanalysis on a single instrument.
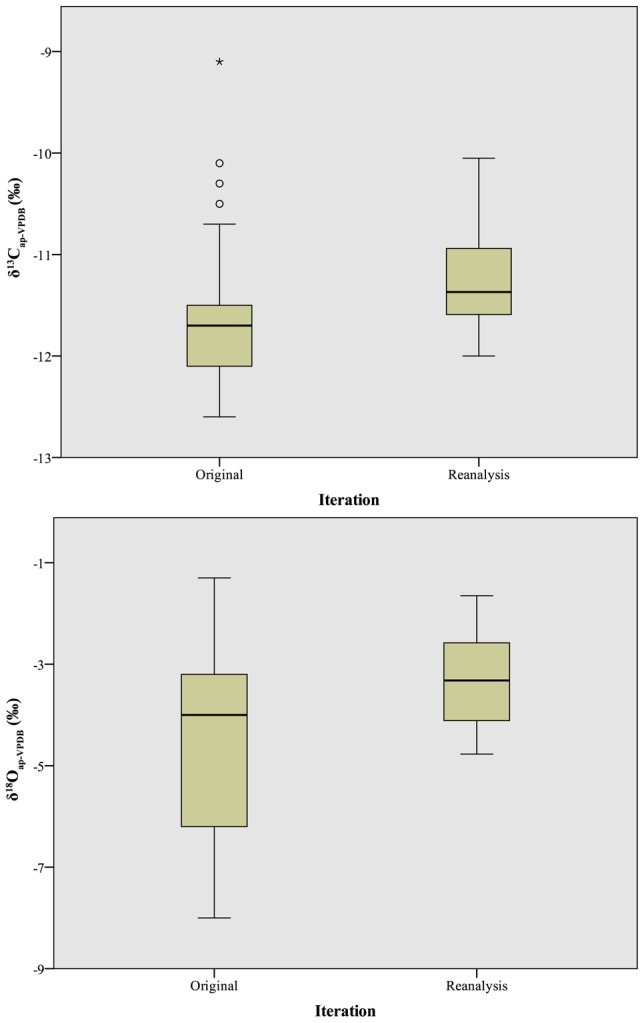
Box lines represent first quartile, second quartile (median), and third quartile; whiskers at 95% confidence intervals; dots represent weak outliers (more than 2 standard deviations from mean); asterisks represent strong outliers (more than 3 standard deviations from mean).
Two other phenomena of note were evident after reanalysis. First, the samples from Laboratory R, which had outlying δ13Cap values in the initial run, were not outliers in the reanalysis (z-scores between −0.5 [p = 0.3] and 0.9 [p = 0.18]). Indeed, there was only one outlier among the two isotope systems, a solitary samples from Laboratory O which had a δ13Cap value with a z-score of 2.4 (p<0.01). This finding suggests that Laboratory R's aberrant isotope values in the first round of analysis were the result of analytical instrumentation, working standards, or data calibration rather than a preparation step [59], [60]. Indeed, these factors, rather than pretreatment per se, would seem to drive a large portion of the observed variation in isotope values for hydroxyapatite: 44% (1.5‰ of the initially observed 3.5‰ range) for δ13Cap and 54% (3.6‰ of the initially observed 6.7‰ range) for δ18Oap. These results echo the recent findings of Carter and Fry [59] who demonstrated that differences in data calibration and correction can lead to substantial isotopic differences among laboratories.
Second, while reanalysis on the same instrument reduced the difference in δ13Cap values between samples oxidized using bleach versus hydrogen peroxide (<0.3‰, t = −1.1, df = 29, p = 0.3), a significant difference in δ18Oap values remained (1.3‰, t = −3.6, df = 29, p<0.01). Therefore, although instrumentation drove some of the isotopic variation among laboratories, differences in preparation (particularly oxidation/organic removal) were also responsible for observed differences in isotopic values obtained from different laboratories, a finding that is in agreement with previous studies [41], [43].
Establishing Meaningful Minimum Differences for Hydroxyapatite
As noted above, the average inter-laboratory pairwise differences for δ13Cap and δ18Oap were 0.6‰ and 2.0‰ respectively. The Minimum Meaningful Difference (MMD) value for δ13Cap was determined to be 1.2‰ (Table 5). This value suggests that a difference in isotopic signatures obtained from two different analyses is only likely to be bona fide when that difference exceeds the threshold value of 1.2‰. MMD for δ18Oap (Table 5) was much larger (3.1‰). This latter value is of particular concern, as it is greater than the difference in bona fide δ18O values that might be expected to result from biological or environmental differences (e.g., residency, residency or paleoclimate), as is more fully elucidated below.
Conclusions
The present study began with the goals of: 1) quantifying inter-laboratory variability in stable isotope analysis of bone collagen and hydroxyapatite, and 2) tracing the likely causes of this observed variability. For bone collagen, we found statistically significant inter-laboratory variation for both carbon and nitrogen isotope values among laboratories. However, the average pairwise difference between any two participating laboratories was only 0.2‰ for δ13Ccol and 0.4‰ for δ15Ncol. These values are of such a small magnitude as to not be cause for great concern. As to causality, neither of the most obvious differences in pretreatment between participating laboratories (demineralizing reagent or humic acid removal) had a significant effect on the resulting isotope values.
Subsequent reanalysis of a subset of samples on the same instrument indicates that the prime driver of inter-laboratory variation in collagen stable isotope analysis is differences in analytical instrumentation and/or standardization rather than pretreatment (accounting for 48–69% of the observed initial inter-laboratory variation). Finally, the Minimum Meaningful Difference (MMD) value establishes a threshold by which results obtained from two laboratories might be evaluated. Differences exceeding 0.6‰ for δ13Ccol and 0.9‰ for δ15Ncol have a high likelihood of being of biological origin rather than an artifact of pretreatment or analysis. In sum, it would appear that the results of stable isotope analysis of bone collagen from one laboratory can be compared (if cautiously) with results obtained elsewhere. Overall, inter-laboratory variability in collagen isotopes would not appear to be of paramount concern.
For bone hydroxyapatite, the results of the present study are somewhat less reassuring. Inter-laboratory variability for both δ13Cap and δ18Oap was significant, and while the average pairwise difference between any two participating laboratories was only 0.6‰ for δ13Cap, for δ18Oap, that value rose to 2.0‰, a difference which could easily change interpretations of past residency or paleomobility. It is unlikely that anything more than a small portion of this variability is the result of differential diagenesis [43], as variability within laboratories (each of which received a randomized set of bone samples) was significantly less than inter-laboratory variability. Instead, as previously suggested [41], differences in oxidation treatment (NaOCl versus H2O2) appear to be a prime driver of δ18Oap variability, but not for δ13Cap. However, differences in the method for removing labile carbonate (acid concentration and buffering agent) do not have a significant effect on either isotope system, counter previous suggestions [22], [42] to the contrary. Perhaps such difference were not observed because all laboratories that used strong acid used buffered acid or very short treatment times.
As with collagen, the subsequent reanalysis of a subset of samples on the same instrument indicates that differences in analytical instrumentation and/or standardization (rather than pretreatment per se) were a prime driver of inter-laboratory variation in hydroxyapatite stable isotope values (accounting for 44–54% of the observed initial inter-laboratory variation). Efforts to unify data correction among laboratories would likely decrease this variability [59], [60]. The Minimum Meaningful Difference (MMD) values suggest that results obtained from two laboratories have a high likelihood of being bona fide rather than an artifact of different pretreatment or analytical methods only if they exceed 1.2‰ for δ13Cap and 3.1‰ for δ18Oap. The magnitude of these MDD values, particularly for δ18Oap, might call into question the attribution of biological significance oftentimes given to different δ13Cap and δ18Oap values obtained in different laboratories. In sum, it would appear that inter-laboratory variability could be a significant concern for hydroxyapatite. Analytical results from different laboratories might not be directly comparable, particularly in the case of δ18Oap.
Three final points merit consideration. First, it should be noted that while the present study addresses the wisdom of (over)claiming the significance of dissimilar results from different laboratories, even small differences (if replicable) obtained in one laboratory can still be considered reliable. Second, the bone hydroxyapatite results presented here may not be directly applicable to comparisons of enamel hydroxyapatite, a tissue thought to be far more resistant to diagenesis [43]. However, the results presented here may be used as a cautionary starting point for enamel comparisons. Third, and finally, the results presented here ought to be thought of in terms of providing a minimum estimate of potential variability that could be generated among laboratories. The extremely high quality of preservation of the selected ancient bone specimen might lead us to underestimate possible inter-laboratory variation in preparation methods. We would expect to see larger isotopic differences among laboratories if they prepare and analyze a poorly preserved sample with lower collagen yield, greater post-mortem humic contamination, or more non lattice-bound carbonates.
Supporting Information
Sample hydroxyapatite extraction protocol form.
(EPS)
Sample hydroxyapatite result form.
(EPS)
Sample collagen extraction protocol form.
(EPS)
Sample collagen result form.
(EPS)
Summary of the collagen pretreatment methods for each participating laboratory.
(XLSX)
Summary of the hydroxyapatite pretreatment methods for each participating laboratory.
(XLSX)
Acknowledgments
The authors wish to thank the PIs, laboratory managers, and staff of the twenty-one participating laboratories. It is no overstatement to say that without them, this study would not have been possible. We also wish to thank Christina Torres-Rouff and Mark Hubbe of the Instituto de Estudios de Investigaciones Arqueológicas and Museo R.P. Le Paige for providing the sample on which this work was based. Finally, we wish to acknowledge Joy Matthews of the UC-Davis Stable Isotope Facility and Peter Swart and Monica Arienzo of the Stable Isotope Geochemistry Stable Isotope Laboratory at the Rosenstiel School of Marine and Atmospheric Sciences at the University of Miami for their aid in the reanalysis of the various laboratories' samples.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors have no support or funding to report.
References
- 1. Hall RL (1967) Those Late Corn Dates: Isotopic Fractionation as a Source of Error in Carbon-14 Dates. Michigan Archaeologist 13: 171–180. [Google Scholar]
- 2. Hall RL (1967) Current Research: Northern Mississippi Valley. American Antiquity 32: 570–572. [Google Scholar]
- 3. Vogel JC, Van Der Merwe NJ (1977) Isotopic Evidence for Early Maize Cultivation in New York State. American Antiquity 42: 238–242. [Google Scholar]
- 4. Bender MM, Baerreis DA, Steventon RL (1981) Further Light on Carbon Isotopes and Hopewell Agriculture. American Antiquity 46: 346–353. [Google Scholar]
- 5. Burleigh R, Brothwell D (1978) Studies on American dogs, 1. Carbon isotopes in relation to maize in the diet of domestic dogs from early Peru and Ecuador. Journal of Archaeological Science 5: 355–362. [Google Scholar]
- 6. van der Merwe NJ, Roosevelt AC, Vogel JC (1981) Isotopic evidence for prehistoric subsistence change at Parmana, Venezuela. Nature 292: 536–538. [Google Scholar]
- 7. van der Merwe NJ, Vogel JC (1978) 13C content of human collagen as a measure of prehistoric diet in woodland North America. Nature 276: 815–816. [DOI] [PubMed] [Google Scholar]
- 8. DeNiro MJ (1985) Postmortem preservation and alteration of in vivo bone collagen isotope ratios in relation to palaeodietary reconstruction. Nature 317: 806–809. [Google Scholar]
- 9. DeNiro MJ, Epstein S (1978) Influence of Diet on the Distribution of Carbon Isotopes in Animals. Geochimica et Chosmochimica Acta 42: 495–506. [Google Scholar]
- 10. DeNiro MJ, Epstein S (1981) Influence of Diet on the Distribution of Nitrogen Isotopes in Animals. Geochimica et Chosmochimica Acta 45: 341–351. [Google Scholar]
- 11. Tauber H (1981) 13C evidence for dietary habits of prehistoric man in Denmark. Nature 292: 332–333. [DOI] [PubMed] [Google Scholar]
- 12. Chisholm BS, Nelson DE, Schwarcz HP (1982) Stable-Carbon Isotope Ratios as a Measure of Marine Versus Terrestrial Protein in Ancient Diets. Science 216: 1131–1132. [DOI] [PubMed] [Google Scholar]
- 13. Schoeninger MJ (1985) Trophic Level Effects on 15N/14N and 13C/12C Ratios in Bone Collagen and Strontium Levels in Bone Mineral. Journal of human evolution 14: 515–525. [Google Scholar]
- 14. Schoeninger MJ, DeNiro MJ (1984) Nitrogen and Carbon Isotopic Composition of Bone Collagen from Marine and Terrestrial Animals. Geochimica et Cosmochimica Acta 48: 625–639. [Google Scholar]
- 15. Schoeninger MJ, DeNiro MJ, Tauber H (1983) Stable Nitrogen Isotope Ratios of Bone Collagen Reflect Marine and Terrestrial Components of Prehistoric Human Diet. Science 220: 1381–1383. [DOI] [PubMed] [Google Scholar]
- 16. Ambrose SH, DeNiro MJ (1986) The isotopic ecology of East African mammals. Oecologia 69: 395–406. [DOI] [PubMed] [Google Scholar]
- 17. Ambrose SH, DeNiro MJ (1986) Reconstruction of African human diet using bone collagen carbon and nitrogen isotope ratios. Nature 319: 321–324. [Google Scholar]
- 18. Ambrose SH, DeNiro MJ (1989) Climate and habitat reconstruction using stable carbon and nitrogen isotope ratios of collagen in prehistoric herbivore teeth from Kenya. Quaternary Research 31: 407–422. [Google Scholar]
- 19. Ambrose SH (1991) Effects of diet, climate and physiology on nitrogen isotope abundances in terrestrial foodwebs. Journal of Archaeological Science 18: 293–317. [Google Scholar]
- 20. Sullivan CH, Krueger HW (1981) Carbon isotope analysis of separate chemical phases in modern and fossil bone. Nature 292: 333–335. [DOI] [PubMed] [Google Scholar]
- 21. Sullivan CH, Krueger HW (1983) Carbon isotope ratios of bone apatite and animal diet reconstruction. Nature 301: 177–177. [DOI] [PubMed] [Google Scholar]
- 22. Lee-Thorp JA, van der Merwe NJ (1987) Carbon isotope analysis of fossil bone apatite. South African Journal of Science 83: 712–715. [Google Scholar]
- 23.Lee-Thorp JA (1989) Stable Carbon Isotopes in Deep Time: The Diets of Fossil Fauna and Hominids. Cape Town, South Africa: University of Cape Town.
- 24.LeGeros R (1991) Calcium Phosphates in Oral Biology and Medicine. Paris: Karger. [PubMed]
- 25. Lee-Thorp JA (2008) On Isotopes and Old Bones. Archaeometry 50: 925–950. [Google Scholar]
- 26.Sponheimer M, Lee-Thorp JA (2007) Hominin Paleodiets: The Contribution of Stable Isotopes. In: Henke W, Tattersall I, editors.Handbook of Paleoanthropology.Berlin: Springer. pp. 555–585. [Google Scholar]
- 27. Sponheimer M, Codron D, Passey BH, de Ruiter DJ, Cerling TE, et al. (2009) Using carbon isotopes to track dietary change in modern, historical, and ancient primates. American Journal of Physical Anthropology 140: 661–670. [DOI] [PubMed] [Google Scholar]
- 28. White TD, Ambrose SH, Suwa G, Su DF, DeGusta D, et al. (2009) Macrovertebrate paleontology and the Pliocene habitat of Ardipithecus ramidus. Science 326: 87–93. [PubMed] [Google Scholar]
- 29. Secord R, Wing SL, Chew A (2008) Stable isotopes in early Eocene mammals as indicators of forest canopy structure and resource partitioning. Paleobiology 34: 87–93. [Google Scholar]
- 30.Ambrose SH (1993) Isotopic Analysis of Paleodiets: Methodological and Interpretive Considerations. In: Sanford MK, editor.Investigation of Ancient Human Tissue Chemical Analyses in Anthropology.Langhorne, PA: Gordon and Breach. pp. 59–130. [Google Scholar]
- 31.Ambrose SH, Norr L (1993) Experimental Evidence for the Relationship of the Carbon Isotope Ratios of Whole Diet and Dietary Protein to those of Bone Collagen and Carbonate. In: Lambert JB, Grupe G, editors.Prehistoric Human Bone: Archaeology at the Molecular Level.New York: Springer-Verlag. pp. 1–37. [Google Scholar]
- 32. Froehle AW, Kellner CM, Schoeninger MJ (2012) Multivariate carbon and nitrogen stable isotope model for the reconstruction of prehistoric human die. American Journal of Physical Anthropology 147: 352–369. [DOI] [PubMed] [Google Scholar]
- 33. Jim S, Jones V, Ambrose SH, Evershed RP (2007) Quantifying dietary macronutrient sources of carbon for bone collagen biosynthesis using natural abundance stable carbon isotope analysis. British Journal of Nutrition 95: 1055–1062. [DOI] [PubMed] [Google Scholar]
- 34. Kellner CM, Schoeninger MJ (2007) A Simple Carbon Isotope Model for Reconstructing Prehistoric Human Diet. American Journal of Physical Anthropology 133: 1112–1127. [DOI] [PubMed] [Google Scholar]
- 35. Schwarcz HP (1991) Some Theoretical Aspects of Isotope Paleodiet Studies. Journal of Archaeological Science 18: 261–275. [Google Scholar]
- 36. Schwarcz HP, Schoeninger MJ (1991) Stable Isotope Analyses in Human Nutritional Ecology. Yearbook of Physical Anthropology 34: 283–321. [Google Scholar]
- 37.Ambrose SH (2000) Controlled Diet and Climate Experiments on Nitrogen Isotope Ratios of Rats. In: Ambrose SH, Katzenberg MA, editors.Biogeochemical Approaches to Paleodietary Analysis.New York: Kluwer Academic/Plenum Publishers. pp. 243–259. [Google Scholar]
- 38. Howland MR, Corr LT, Young SMM, Jones V, Jim S, et al. (2003) Expression of the dietary isotope signal in the compound-specific 13C values of pig bone lipids and amino acids. International Journal of Osteoarchaeology 13: 54–65. [Google Scholar]
- 39. Sponheimer M, Robinson T, Ayliffe L, Roeder B, Hammer J, et al. (2003) Nitrogen Isotopes in Mammalian Herbivores: Hair δ15N Values from a Controlled Feeding Study. International Journal of Osteoarchaeology 13: 80–87. [Google Scholar]
- 40. Tieszen LL, Boutton TW, Tesdahl KG, Slade NA (1983) Fractionation and turnover of stable carbon isotopes in animal tissues: Implications for δ13C analysis of diet. Oecologia 57: 32–37. [DOI] [PubMed] [Google Scholar]
- 41.Crowley BE, Wheatley PV (In review) To bleach or not to bleach? Comparing treatment methods for isolating biogenic carbonate. Chemical Geology.
- 42. Garvie-Lok SJ, Varney TL, Katzenberg MA (2004) Preparation of bone carbonate for stable isotope analysis: the effects of treatment time and acid concentration. Journal of Archaeological Science 31: 763–776. [Google Scholar]
- 43. Koch PL, Tuross N, Fogel ML (1997) The Effects of Sample Treatment and Diagenesis on the Isotopic Integrity of Carbonate in Biogenic Hydroxylapatite. Journal of Archaeological Science 24: 417–429. [Google Scholar]
- 44. Pestle WJ (2010) Chemical, elemental, and isotopic effects of acid concentration and treatment duration on ancient bone collagen: an exploratory study. Journal of Archaeological Science 37: 3124–3128. [Google Scholar]
- 45. King CL, Tayles N, Gordon KC (2011) Re-examining the chemical evaluation of diagenesis in human bone apatite. Journal of Archaeological Science 38: 2222–2230. [Google Scholar]
- 46. Lee-Thorp JA, Sponheimer M (2003) Three Case Studies Used to Reassess the Reliability of Fossil Bone and Enamel Isotope Signals for Paleodietary Studies. Journal of Anthropological Archaeology 22: 208–216. [Google Scholar]
- 47. Lee-Thorp JA, van der Merwe NJ (1991) Aspects of the chemistry of modern and fossil biological apatites. Journal of Archaeological Science 18: 343–354. [Google Scholar]
- 48. Nielsen-Marsh CM, Hedges REM (2000) Patterns of Diagenesis in Bone I: The Effects of Site Environments. Journal of Archaeological Science 27: 1139–1150. [Google Scholar]
- 49. Nielsen-Marsh CM, Hedges REM (2000) Patterns of Diagenesis in Bone II: Effects of Acetic Acid Treatment and the Removal of Daigenetic CO3 2− . Journal of Archaeological Science 27: 1151–1159. [Google Scholar]
- 50. Pestle WJ, Colvard M (2012) Bone Collagen Preservation in the Tropics: A Case Study from Ancient Puerto Rico. Journal of Archaeological Science 39: 2079–2090. [Google Scholar]
- 51. Shemesh A (1990) Crystallinity and Diagenesis of Sedimentary Apatites. Geochimica et Chosmochimica Acta 52: 2433–2438. [Google Scholar]
- 52. Sponheimer M, Lee-Thorp JA (1999) Alteration of Enamel Carbonate Environments During Fossilization. Journal of Archaeological Science 26: 143–150. [Google Scholar]
- 53. Trueman CN, Behrensmeyer AK, Tuross N, Weiner S (2004) Mineralogical and compositional changes in bones exposed on soil surfaces in Amboseli National Park, Kenya: diagenetic mechanisms and the role of sediment pore fluids. Journal of Archaeological Science 31: 721–739. [Google Scholar]
- 54. Trueman CN, Privat KL, Field J (2008) Why do crystallinity values fail to predict the extent of diagenetic alteration of bone mineral? Palaeogeography, Palaeoclimatology, Palaeoecology 266: 160–167. [Google Scholar]
- 55. Weiner S, Bar-Yosef O (1990) States of Preservation of Bones from Prehistoric Sites in the Near East: A Survey. Journal of Archaeological Science 17: 187–196. [Google Scholar]
- 56. Wright LE, Schwarcz HP (1996) Infrared and Isotopic Evidence for Diagenesis of Bone Apatite at Dos Pilas, Guatemala: Palaeodietary Implications. Journal of Archaeological Science 23: 933–944. [Google Scholar]
- 57. Zazzo A, Lecyer C, Marioti A (2004) Experimentally controlled carbon and oxygen isotope exchange between bioapatites and water under inorganic and microbially mediated conditions. Geochimica et Chosmochimica Acta 68: 1–12. [Google Scholar]
- 58. Zazzo A, Lecyer C, Sheppard SMF, Grandjean P, Marioti A (2004) Diagenesis and the reconstruction of paleoenvironments: A method to restore original δ18O values of carbonate and phosphate from fossil tooth enamel. Geochimica et Chosmochimica Acta 68: 2245–2258. [Google Scholar]
- 59. Carter JF, Fry B (2013) Ensuring the reliability of stable isotope ratio data—beyond the principle of identical treatment. Analytical and Bioanalytical Chemistry 405: 2799–2814. [DOI] [PubMed] [Google Scholar]
- 60. Skrzypek G (2013) Normalization procedures and reference material selection in stable HCNOS isotope analyses: an overview. Analytical and Bioanalytical Chemistry 405: 2815–2823. [DOI] [PubMed] [Google Scholar]
- 61.Stuiver M, Reimer PJ, Reimer RW (2013) CALIB 7.0.
- 62. Hogg AG, Hua Q, Blackwell PG, Niu M, Buck CE, et al. (2013) SHCal13 Southern Hemisphere Calibration, 0–50,000 Years cal BP. Radiocarbon 55: 1889–1903. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample hydroxyapatite extraction protocol form.
(EPS)
Sample hydroxyapatite result form.
(EPS)
Sample collagen extraction protocol form.
(EPS)
Sample collagen result form.
(EPS)
Summary of the collagen pretreatment methods for each participating laboratory.
(XLSX)
Summary of the hydroxyapatite pretreatment methods for each participating laboratory.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.