

Abstract
Cardiac muscle is unique because it contracts ceaselessly throughout the life and is highly resistant to fatigue. The marvelous nature of the cardiac muscle is attributed to its matrix that maintains structural and functional integrity and provides ambient micro-environment required for mechanical, cellular and molecular activities in the heart. Cardiac matrix dictates the endothelium-myocyte (E-M) coupling and contractility of cardiomyocytes. The Matrix metalloproteinases (MMPs) and their tissue inhibitor of metalloproteinases (TIMPs) regulate matrix degradation that determines cardiac fibrosis and myocardial performance. We have shown that MMP-9 regulates differential expression of micro RNAs (miRNAs), calcium cycling and contractility of cardiomyocytes. The differential expression of miRNAs is associated with angiogenesis, hypertrophy and fibrosis in the heart. MMP-9, which is involved in the degradation of cardiac matrix and induction of fibrosis, is also implicated in inhibition of survival and differentiation of cardiac stem cells (CSC). Cardiac matrix is distinct because it renders mechanical properties and provides a framework essential for differentiation of cardiac progenitor cells (CPC) into specific lineage. Cardiac matrix regulates myocyte contractility by E-M coupling and calcium transients and also directs miRNAs required for precise regulation of continuous and synchronized beating of cardiomyocytes that is indispensible for survival. Alteration in the matrix homeostasis due to induction of MMPs, altered expression of specific miRNAs or impaired signaling for contractility of cardiomyocytes leads to catastrophic effects. This review describes the mechanisms by which cardiac matrix regulates myocardial performance and suggests future directions for the development of treatment strategies in cardiovascular diseases.
Keywords: Heart, VEGF, MMP, TIMP, miRNA, stem cell, angiogenesis, cardiovascular diseases
Introduction
Although heart is a unique and dynamic organ functioning ceaselessly throughout life, it is highly vulnerable to disease pathology. Cardiovascular disease (CVD) is the leading cause of morbidity and mortality across the world. Although majority of research emphasize on CVD, less attention has been paid to the sophisticated regulatory mechanisms and micro-environment provided by cardiac matrix. Cardiac matrix is crucial for synchronized beating of cardiomyocytes that maintain the untiring contraction-relaxation cycle of the heart. Recently, it was discovered that elasticity of matrix plays a pivotal role in lineage specification [1] and self-renewal [2] of stem cells. The heart has self-renewing capacity due to endogenous cardiac stem cells [3]. It was reported that during cardiac stem cell therapy, paracrine effect causing inhibition of cardiac fibrosis, apoptosis and enhanced contractility [4], could be a possible factor mediated by matrix modulation. Another report using a cocktail of prosurvival miRNA-21, -24 and -221 was shown to enhance the engraftment and viability of transplanted cardiac progenitor cells [5] corroborating the fact that synergism of miRNA and stem cell could be a better therapeutic approach in heart failure [6]. We have demonstrated that targeted deletion of MMP-9 induces miRNAs that are down regulated in failing hearts and improves contractility and calcium handling by up regulating SERCA2 in cardiomyocytes [7]. Additionally, induction of MMP-9 and attenuation of TIMP-4 contribute to cardiac fibrosis in diabetic hearts whereas ablation of MMP-9 decreases cardiac fibrosis and increases cardiac stem cells (CPCs) in the heart [8, 9]. However, the cross-talk between MMP-9, miRNA and stem cells is unclear. The understanding of complex interactions among MMPs, miRNA and CPC in the milieu of cardiac matrix can be exploited for regeneration and improvement of myocardial contractility. In this review, the plausible mechanism of structural and functional remodeling of cardiac matrix in the context of heart failure and future therapeutic approaches is elaborated.
Matrix metalloproteinases as key players in cardiac matrix remodeling
MMPs are zinc containing calcium-dependent endopeptidases which are released as inactive zymogens in a latent form [10, 11] and are activated by auto-proteolysis, serine proteases, or other activated MMPs [10]. Pathological cardiac remodeling can be triggered by pressure (hypertension) or volume overload, hyperhomocysteinemia, and/or activation of renin-angiotensin-aldosterone system mediated oxidative/redox stress that alters the levels of different MMPs and TIMPs and signaling molecules leading to heart failure (Figure 1). Cardiac remodeling includes degradation of extracellular matrix (ECM), myocyte hypertrophy, impaired angiogenesis, collateralization, changes in receptor signaling cascade, fibrosis, autophagy, apoptosis, impaired differentiation and survival of CSC, fetal gene reprogramming, differential expressions of miRNAs and epigenetic modifications. Although MMPs are involved in cardiovascular remodeling, they have a distinct spatial and temporal role. The temporal activation of MMP and TIMP has been elucidated in myocardial infarction. While MMP-2 induction was observed on day1 and peaked at two weeks post-MI, TIMP-4 induction and activation remained the same from day1 [12]. Similarly, MMP-9 but not MMP-2 was activated in end-stage heart failure [13]. The spatial translocation of MMP-9 into mitochondria is associated with arrhythmia and cardiomyocyte contractility dysfunction [14, 15]. MMP-2 is constitutively expressed [16-18], whereas MMP-9 is inducible and instigates pathophysiological remodeling [7, 9, 19]. Although transgenic expression of MMP-2 impairs myocardial contractility [20], the disruption of myocardial filament by MMP-2 may not be true. The induction of MMP-2 may have an effect on other MMPs. For example in diabetics, MMP-2 is attenuated but MMP-9 shows robust activation[9] resulting in contractile dysfunction [7]. MMP-2 is reported to be up regulated in human aortic atherosclerotic lesions [21]. In heart failure, the level of MMP-2 increases during compensatory stage but during de-compensatory stage MMP-9 supersedes the levels of MMP-2 resulting in failure [16]. Recently, the existence of a novel intracellular MMP-2 isoform was reported in the mitochondria. This isoform is (65 kDA) induced by oxidative stress and has been shown to play an important role in promoting cardiac hypertrophy, apoptosis and systolic failure [22]. Despite the pathological role described above, further studies are required to confirm these findings. The vascular MMP versus cardiac MMP is interesting in the sense that the remodeling outcome may be compensatory in cardiac matrix while it is detrimental in the arterial pathology. Accumulation of collagen and loss of elastin the arterial wall correlate with decreased arterial compliance in hypertension and aging. Enhanced extracellular matrix remodeling in the arterial wall affects the course of disease pathology in atherosclerosis [23]. Plaque disruption due to vascular remodeling mostly results in thrombotic occlusion [24].
Figure 1.
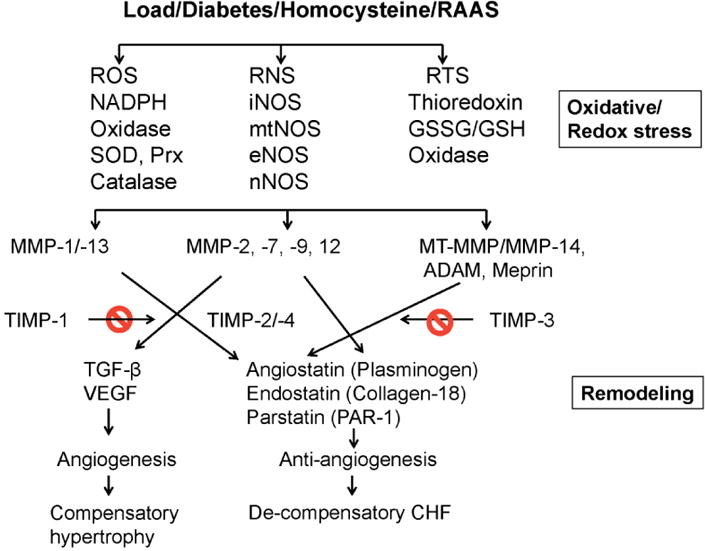
Pressure and/volume overload, diabetes, homocysteine and renin-angiotensin- aldosterone system (RAAS) engendered oxidative stress that activates latent MMPs and imbalances MMP/TIMP axis (remodeling). This generates angiogenic and anti-angiogenic factors, leading to compensatory to de-compensatory congestive heart failure (CHF).
MMPs are inhibited by tissue inhibitor of metalloproteinases (TIMPs). There are four types of TIMPs: TIMP-1, -2, -3 and -4. TIMPs are implicated in cardiac fibrosis [25], angiogenesis [26, 27], and apoptosis [28, 29]. Cardiac enriched TIMP-4 plays an important role in matrix remodeling by inhibition of MMP-9 activity [30]. An imbalance of MMP/TIMP has been implicated in structural and functional changes in hypertensive heart disease [31]. The pathological role of MMPs in left ventricular remodeling and heart failure has been extensively reported in both pre-clinical and clinical studies [32]. In spontaneously hypertensive rats, MMP inhibition was shown to attenuate pathological cardiac remodeling during hypertension [33]. In a recent study, MMP-2 activity was found to be responsible for the development of left ventricular hypertrophy in a two kidney, one clip hypertensive rat model [34]. In the same model, temporal changes in MMP-2 activity was associated with simultaneous cardiac remodeling along with increased expression of TGF-β and reactive oxygen species (ROS) [35]. Treatment with antioxidant, Tempol alleviated the cardiac remodeling by decreasing the TGF-β and MMPs expression [35]. Although significant role of MMP-2 in remodeling has been extensively reported, recent report suggests that the infusion of recombinant human MMP-2 in lambs did not alter any hemodynamic parameters but for the impairment of beta adrenoceptor activation response [36].
The involvement of MMPs in left ventricular pathology has been extensively reported [37] however, a recent study in acute pulmonary thromboembolism (APT) showed a similar role in right ventricular remodeling [38]. In APT, ROS production was associated with MMP activation and treatment with non-specific MMP inhibitors or antioxidants were found to mitigate MMP induced remodeling [39, 40].
MMP inhibitors
MMP inhibitors are classified as specific and non-specific inhibitors. Non-specific inhibitors act through chelation of Zn2+ ion [41]. Nonspecific inhibitors such as batimastat, marimastat, GM-6001 (ilomastat or gelardin), PD-166793 and ONO-4817 [42] have been extensively used in various experimental models of disease. Lately, though peptides containing the HWGF motif, CRRHWGFEFC and CTTHWGFTLC, were found to be selective inhibitors of MMP-9 and MMP-2 [41], their clinical usage is highly questionable because of their susceptibility to proteolysis inside the body. Tetracyclines are a group of antibiotics that are found to have MMP inhibition property. Chemically modified tetracyclines (CMTs) are devoid of antimicrobial property but retain the MMP inhibition function [43]. CMT 3 has been shown to inhibit MMP-2 and -9 activities along with collagenase activity and ameliorate pathological cardiovascular remodeling. This is the only CMT administered to humans in clinical trials [44].
Cross-talk between MMPs and miRNAs in cardiac matrix
Cardiac matrix is implicated in conferring biochemical stability of growth factors, facilitating signal transduction and inducing apoptosis [23]. Since MMPs degrade cardiac matrix, they also disrupt the endothelial–myocyte coupling, signaling cascade and regulatory machinery necessary for maintaining normal function [9]. MicroRNAs (miRNAs) are small (20-23 nucleotide long), non-coding RNAs that regulate gene expression either by mRNA degradation or translational repression [45, 46]. Nearly 2000 miRNAs have been reported in humans. MicroRNAs play a pivotal role in the regulation of cardiovascular diseases [6, 47]. MMPs also alter the expression of miRNAs. The first report of regulation of miRNAs by MMPs was documented in MMP-9KO hearts [7]. This study revealed several key miRNAs that are down regulated in the failing heart, are up regulated in the MMP-9KO hearts [7]. However, the mechanism of miRNA regulation by MMPs is still not clear. Nevertheless, this finding opens a new avenue to explore the complex regulatory network between MMPs and miRNAs and dissect the mechanism of cardiac matrix remodeling in pathological conditions.
Cardiac ECM, MMPs, miRNAs and epigenetic modifications
ECM is the key component of the myocardium which not only maintains the structural integrity and plasticity of the heart but also provides the micro-environment for signaling cascade required for cardiac homeostasis [48]. Collagen provides stiffness to ECM and is involved in structural remodeling leading to heart failure [8, 9, 48-50]. Collagen is a substrate for both MMP-2 and MMP-9 [10]. MMP-9 is found to be robust and degraded ECM in a failing heart [51-53]. Targeted deletion of MMP-9 mitigated fibrosis in diabetic hearts [8]. During the remodeling process, though both collagen and elastin are resynthesized as a compensatory mechanism, collagen turnover is faster than elastin and contributes to cardiac fibrosis. The cardiac fibrosis impairs endothelium-cardiomyocyte (E-C) coupling leading to ventricular dysfunction [9]. The role of MMP-9 on contractility of the heart was elucidated by an ex-vivo experiment, in which treatment with MMP-9 decreases rate of contraction and relaxation (±dL/dt) of cardiomyocytes and inhibition of MMP-9 by tissue inhibitor of metalloproteinase-4 (TIMP4) ameliorated impaired contractility [7]. The targeted deletion of MMP-9 also improved contractility of cardiomyocytes at in vivo studies [7].
Micro RNA-133 is attenuated in heart failure. Inhibition of miR-133 induced cardiac hypertrophy [54] while transgenic expression of miR-133 attenuated cardiac fibrosis in trans-aortic constriction model of heart failure [55]. Interestingly, targeted deletion of MMP-9 up-regulated the expression of miR-133 in a failing heart [7]. Recently, it was reported that abrogation of miR-133 induces DNA methyl transferases (DNMTs) and over expression of miR-133 inhibits DNMTs in cardiomyocytes [56]. Surprisingly, up-regulation of miR-133 down regulates hyperglycemia mediated induction of DNMTs (DNMT-1) suggesting a crucial role of miR-133 in epigenetic modification in diabetic hearts [56]. Although ablation of MMP-9 induces miR-133, the underlying mechanism is unclear. The plausible mechanism of MMP-9 mediated amelioration of heart failure could be that the deletion of MMP-9 gene provides a favorable cardiac matrix micro-environment required for increased transcription of specific miRNAs that mitigates cardiac dysfunction. Further understanding of cross-talk between ECM, MMPs and miRNAs will open a new avenue to elucidate the complex regulatory network in the heart that can be exploited for therapeutic endeavor.
Elasticity of cardiac matrix and stem cell survival and differentiation
Stem cells are recognized by three salient properties, (i) self-renewal, (ii) pluripotent, and (iii) clonogenic [57]. The human heart is a self-renewing organ [3] because of its endogenous cardiac stem cells and self-renewal is dependent on the elasticity of the surrounding matrix [1, 2]. The ECM elasticity determines stem cell lineage specification, expansion and differentiation [1, 8]. In the heart, ECM exhibits all the mechanical properties and provides a frame work required for differentiation of cardiac stem cells [58]. The contractility of cardiomyocytes also depends on the elasticity of matrix [2]. It is documented that embryonic cardiomyocytes beat best on a matrix with heart like elasticity [2]. As MMP-9 deletion contributes to the elasticity of ECM and up regulates several miRNAs involved in stem cell differentiation, we determined the role of MMP-9 in the survival and differentiation of cardiac stem cells [8]. The comparative analysis of c-kit (a stem cell marker) and troponin I (cardiomyocytes marker) in the WT, diabetic Ins2+/- Akita and Ins2+/- /MMP-9-/- (diabetic mice without MMP-9 gene) revealed that MMP-9 deletion enhances stem cell survival and differentiation to cardiomyocytes in the heart [8]. It points to a complex interaction amongst MMPs (at least MMP-9), ECM elasticity and miRNAs that determines the cardiac stem cell survival and differentiation. ECM might be crucial for regulating stem cell differentiation signaling in autocrine and paracrine fashion for regenerating the myocardium that is pivotal in stem cell therapy (Figure 2).
Figure 2.

Activation of MMP-9 increases ECM turn over, attenuates miRNAs, induces cardiac stem cell apoptosis and inhibits their differentiation leading to pathological cardiac remodeling.
MMPs, angiogenesis, cardiovascular remodeling and repair
Aberrant remodeling in the myocardial ECM results in heart failure. MMPs play a pivotal role in regulation of angiogenesis by altering the balance between angiogenic and anti-angiogenic factors [16, 59, 60]. Brooks and colleagues reported that an angiogenic stimulus induces vascular remodeling and defines the role of MMP-2 and integrin (αvβ3) [61, 62]. During hypertrophic remodeling and angiogenesis, MMP-2 is constitutively expressed [16, 49]. Alternatively, MMP-9 is expressed in the failing heart and induces the expression of anti-angiogenic factors - endostatin and angiostatin [16, 50, 63]. Previous reports demonstrated that the therapeutic angiogenesis using growth promoting factors can increase blood supply to the ischemic myocardium [64-66].
Studies in cardiac specific inducible protein kinase B (AKT1) transgenic mice show decreased angiogenesis during pathological remodeling and suggested that both heart size and cardiac function are angiogenesis dependent. Additionally, the disruption of coordinated cardiac hypertrophy and angiogenesis plays a crucial role in the pathogenesis of heart failure [67]. Although role of MMPs in acute coronary syndrome (ACS) is documented [23, 24], the inhibition of vascular versus cardiac MMP is counter intuitive. The comparative analyses of angiostatin, endostatin and coronary collateral formation in myocardial tissue harvested from diabetic and non-diabetic coronary artery disease (CAD) patients revealed that angiostatin and endostatin are induced in diabetic CAD patients compared with non-diabetic patients and negatively correlates with coronary collateralization [63]. Although MMPs are integral regulators of angiogenesis and anti-angiogenesis, whether all MMPs have similar or differential function is nebulous. MMP-2 and MMP-9 are mostly studied MMPs in angiogenesis; however contrasting roles have been reported. We and several others believe that MMP-2 is pro-angiogenic but MMP-9 is anti-angiogenic in the heart [16, 63, 68]. The role of MMP-9 as anti-angiogenic factor is also supported by the finding that abrogation of MMP-9 attenuates cardiac hypertrophy and collagen accumulation in the heart after myocardial infarction [69]. It is documented that endothelial mesenchymal transition (EndMT) similar to that of epithelial mesenchymal transition (EMT) plays an important role in synthesizing new fibroblasts resulting in increased deposition of fibrosis in endocardium and microvascular endothelium [70-72]. However, ablation of MMP-9 gene inhibits EMT [71, 73]. These findings points to the role of MMP-9 as inducer of EndMT in cardiac and vascular endothelium that promotes cardiac fibrosis along with increased expression of anti-angiogenic factors such as angiostatin and endostatin in an experimental ascending aortic banding model (Figure 3).
Figure 3.
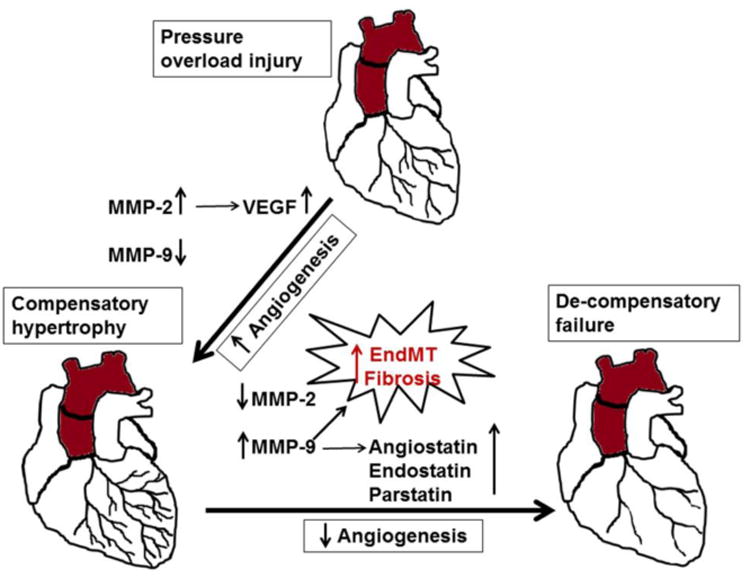
Mechanism of pressure overload (ascending aortic banding) mediated compensatory cardiac hypertrophy to de-compensatory heart failure. During early stages of aortic banding, angiogenesis is increased due to up-regulation of MMP-2, vascular endothelial growth factor (VEGF) and inhibition of MMP-9. However, sustained overload results in de-compensatory heart failure due to anti-angiogenesis, where expression of MMP-9 supersedes that of MMP-2 and promotes release of anti-angiogenic factors such as angiostatin, endostatin and parstatin. MMP-9 also stimulate endothelial mesenchymal transition (EndMT) leading to fibrosis and end stage heart failure.
Imbalance of angiogenesis and anti-angiogenesis could be one of the major pathogenic mechanisms during cardiac injury such as pressure overload, ischemia and infarction. Restoration of angiogenesis may potentiate myocardial recovery from an insult or injury. We and others have reported that therapeutic doses of hydrogen sulfide (H2S) promote angiogenesis and have cardio protective role in pressure overload models [68, 74, 75]. Since homocysteine is a precursor for H2S, the treatment with H2S can mitigate hyperhomocysteinemia mediated cardiac toxicity. Although angiogenic therapy for the cardiac repair is promising, successful clinical data is still missing and targeted induction of angiogenesis in the localized area of heart could be a viable approach for cardiac repair. Empirical studies revealed that administration of self-assembling peptide nanofiber combined with VEGF in the post myocardial infarction heart stabilized VEGF locally for more than 14 days and improved angiogenesis, arteriogenesis and cardiac performance [76]. This study upholds the cardiac angiogenesis therapy for future clinical trials. However, understanding the types of VEGF and their functions in relation to cardiac repair is crucial as different VEGFs are involved in vasculogenesis, angiogenesis and lymphangiogenesis (Figure 4).
Figure 4.
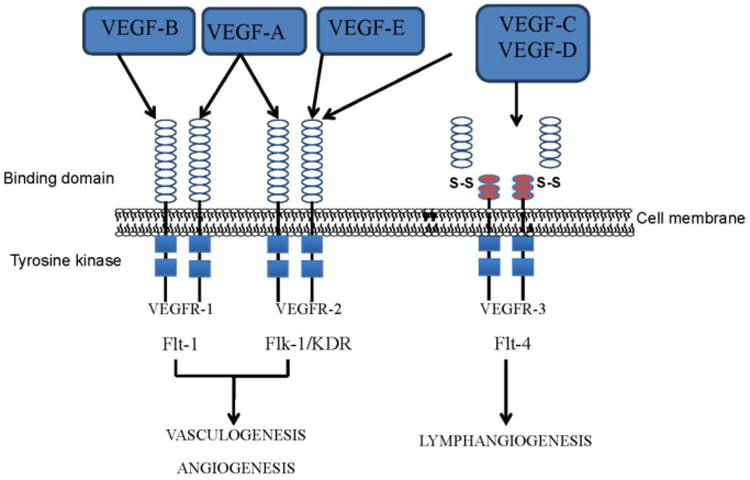
The effect of different vascular endothelial growth factors (VEGF) following their binding to corresponding receptors (VEGFR). VEGF1 and 2 up regulates vasculogenesis and angiogenesis through Flt -1and Flk-1/KDR pathway, whereas VEGF3 enhances lymphangiogenesis through Flt-4 pathway.
Exploiting cardiac matrix for future therapy
Cardiac matrix contains MMPs, TIMPs, miRNAs, DNMTs, cardiac stem cells, angiogenic and anti-angiogenic factors, signaling molecules involved in autophagy, apoptosis, epigenetic modifications, fibrosis and hypertrophy. The unique interactions of these factors in the favorable cardiac matrix milieu maintain synchronized beating of cardiomyocytes and contractility of the heart. However, differential expression of even a single molecule disrupts the sophisticated regulatory network by altering the matrix milieu that leads to pathological remodeling. For example, inhibition of only miR-133 causes cardiac hypertrophy [54]. Similarly, ablation of MMP-9 reduces cardiac fibrosis [8]. In the diabetic heart, miR-133 is attenuated that induces DNA methylation [56] but MMP-9 is up regulated that contributes to cardiac fibrosis [9]. Attenuation of miR-133 is also responsible for cardiac fibrosis [55]. Therefore, the interactions of miR-133, MMP-9, epigenetic modifications and cardiac fibrosis are somehow closely related. Similarly, stem cell function is regulated by miRNAs [5] and MMP-9 regulates stem cell survival [8] and miRNA levels [7]. Therefore, CSC survival, differentiation and function depend on interactions of miRNAs and MMP-9. As elucidated above, there are empirical evidences suggesting the role of MMP-9 and MMP-2 in angiogenesis, which is a promising area for therapy of cardiovascular diseases. The interactions of these factors constitute the dynamic cardiac matrix. There is very little known about how the dynamic cardiac matrix behaves during myocardial regeneration, pathological cardiac remodeling and compensatory and de-compensatory stages of heart failure. The detailed insights of signaling cascade of apoptosis, autophagy, epigenetic modifications and angiogenesis by cardiac matrix in the failing heart and their regulation by miRNAs, MMPs and TIMPs will provide a clue for future therapy for heart failure (Figure 5).
Figure 5.
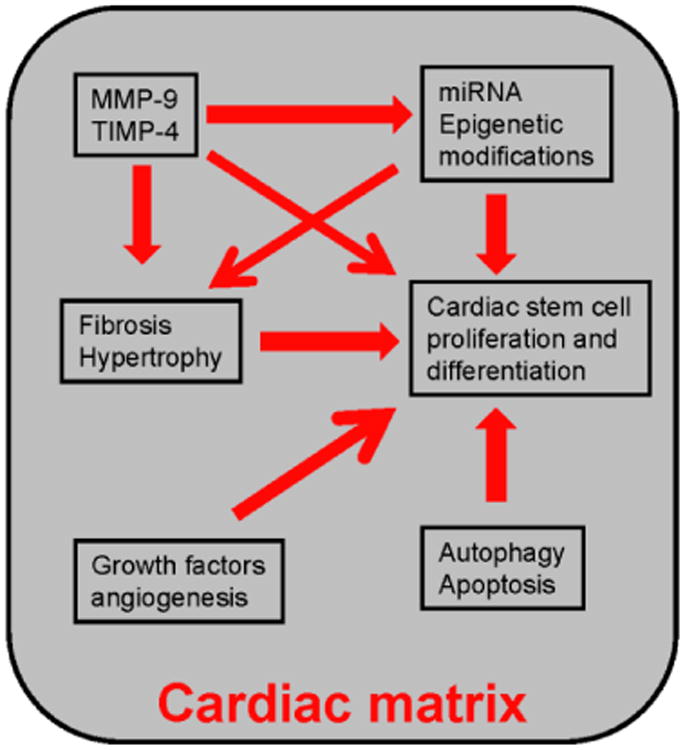
The unique cross –talk amongst MMP-9/TIMP4, miRNA, epigenetic modification, fibrosis, hypertrophy, cardiac stem cell proliferation and differentiation, growth factors, autophagy and apoptosis is maintained in the cardiac matrix. The interactions of these molecules alter in pathological remodeling due to change in matrix milieu. Dissecting the causative factor (s) and their interactions will provide a clue to for future therapy.
Highlights.
MMPs and TIMPs in Cardiac matrix.
MMP-9 regulates differential expression of Micro RNAs.
Cardiac matrix regulating cardiac disease pathology.
Acknowledgments
The financial supports from American Heart Association grant (11BGIA 7690055) and National Institute of Health, HL-113281 to P.K.M. and HL-108621 and HL-74185 to S.C.T. is gratefully acknowledged.
Abbreviations
- CVD
cardiovascular disease
- CSC
cardiac stem cell
- CPC
cardiac progenitor cell
- miRNA
MicroRNA
- MMP
matrix metalloproteinase
- TIMP
tissue inhibitor of MMP
- SERCA
sarco endoplasmic reticulum ca2+ATPase
- ECM
extracellular matrix
- ROS
reactive oxygen species
- APT
acute pulmonary thromboembolism
- DNMT
DNA methyl transferase
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P, Nguyen NK, Thrun S, Lutolf MP, Blau HM. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kajstura J, Hosoda T, Bearzi C, Rota M, Maestroni S, Urbanek K, Leri A, Anversa P. The human heart: a self-renewing organ. Clin Transl Sci. 2008;1:80–86. doi: 10.1111/j.1752-8062.2008.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy SCIPIO: initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Hu S, Huang M, Nguyen PK, Gong Y, Li Z, Jia F, Lan F, Liu J, Nag D, Robbins RC, Wu JC. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124:S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tyagi AC, Sen U, Mishra PK. Synergy of microRNA and stem cell: a novel therapeutic approach for diabetes mellitus and cardiovascular diseases. Curr Diabetes Rev. 2011;7:367–376. doi: 10.2174/157339911797579179. [DOI] [PubMed] [Google Scholar]
- 7.Mishra PK, Metreveli N, Tyagi SC. MMP-9 gene ablation and TIMP-4 mitigate PAR-1-mediated cardiomyocyte dysfunction: a plausible role of dicer and miRNA. Cell Biochem Biophys. 2010;57:67–76. doi: 10.1007/s12013-010-9084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishra PK, Chavali V, Metreveli N, Tyagi SC. Ablation of MMP9 induces survival and differentiation of cardiac stem cells into cardiomyocytes in the heart of diabetics: a role of extracellular matrix. Can J Physiol Pharmacol. 2012;90:353–360. doi: 10.1139/y11-131. [DOI] [PubMed] [Google Scholar]
- 9.Mishra PK, Tyagi N, Sen U, Joshua IG, Tyagi SC. Synergism in hyperhomocysteinemia and diabetes: role of PPAR gamma and tempol. Cardiovasc Diabetol. 2010;9:49. doi: 10.1186/1475-2840-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyagi SC, Ratajska A, Weber KT. Myocardial matrix metalloproteinase(s): localization and activation. Mol Cell Biochem. 1993;126:49–59. doi: 10.1007/BF01772207. [DOI] [PubMed] [Google Scholar]
- 11.Dhanaraj V, Ye QZ, Johnson LL, Hupe DJ, Ortwine DF, Dunbar JB, Jr, Rubin JR, Pavlovsky A, Humblet C, Blundell TL. X-ray structure of a hydroxamate inhibitor complex of stromelysin catalytic domain and its comparison with members of the zinc metalloproteinase superfamily. Structure. 1996;4:375–386. doi: 10.1016/s0969-2126(96)00043-3. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein M, Sloutskis D, Kumanyika S, Sparti A, Schutz Y, Morabia A. Data-based approach for developing a physical activity frequency questionnaire. Am J Epidemiol. 1998;147:147–154. doi: 10.1093/oxfordjournals.aje.a009427. [DOI] [PubMed] [Google Scholar]
- 13.Moshal KS, Tyagi N, Moss V, Henderson B, Steed M, Ovechkin A, Aru GM, Tyagi SC. Early induction of matrix metalloproteinase-9 transduces signaling in human heart end stage failure. J Cell Mol Med. 2005;9:704–713. doi: 10.1111/j.1582-4934.2005.tb00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moshal KS, Tipparaju SM, Vacek TP, Kumar M, Singh M, Frank IE, Patibandla PK, Tyagi N, Rai J, Metreveli N, Rodriguez WE, Tseng MT, Tyagi SC. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am J Physiol Heart Circ Physiol. 2008;295:H890–H897. doi: 10.1152/ajpheart.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moshal KS, Metreveli N, Frank I, Tyagi SC. Mitochondrial MMP activation, dysfunction and arrhythmogenesis in hyperhomocysteinemia. Curr Vasc Pharmacol. 2008;6:84–92. doi: 10.2174/157016108783955301. [DOI] [PubMed] [Google Scholar]
- 16.Givvimani S, Tyagi N, Sen U, Mishra PK, Qipshidze N, Munjal C, Vacek JC, Abe OA, Tyagi SC. MMP-2/TIMP-2/TIMP-4 versus MMP-9/TIMP-3 in transition from compensatory hypertrophy and angiogenesis to decompensatory heart failure. Arch Physiol Biochem. 2010;116:63–72. doi: 10.3109/13813451003652997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Z, Chang MJ, Shah R, Adamski J, Zhao X, Benveniste EN. Brg-1 is required for maximal transcription of the human matrix metalloproteinase-2 gene. J Biol Chem. 2004;279:46326–46334. doi: 10.1074/jbc.M405438200. [DOI] [PubMed] [Google Scholar]
- 18.Qin H, Sun Y, Benveniste EN. The transcription factors Sp1, Sp3, and AP-2 are required for constitutive matrix metalloproteinase-2 gene expression in astroglioma cells. J Biol Chem. 1999;274:29130–29137. doi: 10.1074/jbc.274.41.29130. [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez FR, Lalu MM, Mariano FS, Milanezi CM, Cena J, Gerlach RF, Santos JE, Torres-Duenas D, Cunha FQ, Schulz R, Silva JS. Increased activities of cardiac matrix metalloproteinases matrix metalloproteinase MMP-2 and MMP-9 are associated with mortality during the acute phase of experimental Trypanosoma cruzi infection. J Infect Dis. 2008;197:1468–1476. doi: 10.1086/587487. [DOI] [PubMed] [Google Scholar]
- 20.Wang GY, Bergman MR, Nguyen AP, Turcato S, Swigart PM, Rodrigo MC, Simpson PC, Karliner JS, Lovett DH, Baker AJ. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res. 2006;69:688–696. doi: 10.1016/j.cardiores.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Li L, Zielke HR, Cheng L, Xiao R, Crow MT, Stetler-Stevenson WG, Froehlich J, Lakatta EG. Increased expression of 72-kd type IV collagenase MMP-2 in human aortic atherosclerotic lesions. Am J Pathol. 1996;148:121–128. [PMC free article] [PubMed] [Google Scholar]
- 22.Lovett DH, Mahimkar R, Raffai RL, Cape L, Maklashina E, Cecchini G, Karliner JS. A novel intracellular isoform of matrix metalloproteinase-2 induced by oxidative stress activates innate immunity. PloS one. 2012;7:e34177. doi: 10.1371/journal.pone.0034177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Libby P, Lee RT. Matrix matters. Circulation. 2000;102:1874–1876. doi: 10.1161/01.cir.102.16.1874. [DOI] [PubMed] [Google Scholar]
- 24.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 25.Spoto B, Testa A, Parlongo RM, Tripepi G, D'Arrigo G, Mallamaci F, Zoccali C. Tissue inhibitor of metalloproteinases TIMP-1, genetic markers of insulin resistance and cardiomyopathy in patients with kidney failure. Nephrol Dial Transplant. 2012;27:2440–2445. doi: 10.1093/ndt/gfr710. [DOI] [PubMed] [Google Scholar]
- 26.Seo DW, Saxinger WC, Guedez L, Cantelmo AR, Albini A, Stetler-Stevenson WG. An integrin-binding N-terminal peptide region of TIMP-2 retains potent angio-inhibitory and anti-tumorigenic activity in vivo. Peptides. 2011;32:1840–1848. doi: 10.1016/j.peptides.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 28.Kallio JP, Hopkins-Donaldson S, Baker AH, Kahari VM. TIMP-3 promotes apoptosis in nonadherent small cell lung carcinoma cells lacking functional death receptor pathway. Int J Cancer. 2011;128:991–996. doi: 10.1002/ijc.25404. [DOI] [PubMed] [Google Scholar]
- 29.Finan KM, Hodge G, Reynolds AM, Hodge S, Holmes MD, Baker AH, Reynolds PN. In vitro susceptibility to the pro-apoptotic effects of TIMP-3 gene delivery translates to greater in vivo efficacy versus gene delivery for TIMPs-1 or -2. Lung Cancer. 2006;53:273–284. doi: 10.1016/j.lungcan.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Koskivirta I, Kassiri Z, Rahkonen O, Kiviranta R, Oudit GY, McKee TD, Kyto V, Saraste A, Jokinen E, Liu PP, Vuorio E, Khokha R. Mice with tissue inhibitor of metalloproteinases 4 (Timp4) deletion succumb to induced myocardial infarction but not to cardiac pressure overload. J Biol Chem. 2010;285:24487–24493. doi: 10.1074/jbc.M110.136820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–2096. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 32.Spinale FG, Janicki JS, Zile MR. Membrane-associated matrix proteolysis and heart failure. Circulation research. 2013;112:195–208. doi: 10.1161/CIRCRESAHA.112.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson JT, Hallak H, Johnson L, Li H, O'Brien PM, Sliskovic DR, Bocan TM, Coker ML, Etoh T, Spinale FG. Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation. 2001;103:2303–2309. doi: 10.1161/01.cir.103.18.2303. [DOI] [PubMed] [Google Scholar]
- 34.Rizzi E, Castro MM, Prado CM, Silva CA, Fazan R, Jr, Rossi MA, Tanus-Santos JE, Gerlach RF. Matrix metalloproteinase inhibition improves cardiac dysfunction and remodeling in 2-kidney, 1-clip hypertension. Journal of cardiac failure. 2010;16:599–608. doi: 10.1016/j.cardfail.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Rizzi E, Ceron CS, Guimaraes DA, Prado CM, Rossi MA, Gerlach RF, Tanus-Santos JE. Temporal changes in cardiac matrix metalloproteinase activity, oxidative stress, and TGF-beta in renovascular hypertension-induced cardiac hypertrophy. Experimental and molecular pathology. 2013;94:1–9. doi: 10.1016/j.yexmp.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 36.Ferraz KC, Sousa-Santos O, Neto-Neves EM, Rizzi E, Muniz JJ, Gerlach RF, Tanus-Santos JE. Recombinant human matrix metalloproteinase-2 impairs cardiovascular beta-adrenergic responses. Basic & clinical pharmacology & toxicology. 2013;112:103–109. doi: 10.1111/bcpt.12001. [DOI] [PubMed] [Google Scholar]
- 37.Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. The Canadian journal of cardiology. 2007;23:727–733. doi: 10.1016/s0828-282x(07)70818-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neto-Neves EM, Kiss T, Muhl D, Tanus-Santos JE. Matrix metalloproteinases as drug targets in acute pulmonary embolism. Current drug targets. 2013;14:344–352. doi: 10.2174/1389450111314030006. [DOI] [PubMed] [Google Scholar]
- 39.Neto-Neves EM, Dias-Junior CA, Rizzi E, Castro MM, Sonego F, Gerlach RF, Tanus-Santos JE. Metalloproteinase inhibition protects against cardiomyocyte injury during experimental acute pulmonary thromboembolism. Critical care medicine. 2011;39:349–356. doi: 10.1097/CCM.0b013e3181fa3dfe. [DOI] [PubMed] [Google Scholar]
- 40.Sousa-Santos O, Neto-Neves EM, Ferraz KC, Ceron CS, Rizzi E, Gerlach RF, Tanus-Santos JE. Antioxidant treatment protects against matrix metalloproteinase activation and cardiomyocyte injury during acute pulmonary thromboembolism. Naunyn-Schmiedeberg's archives of pharmacology. 2012;385:685–696. doi: 10.1007/s00210-012-0748-9. [DOI] [PubMed] [Google Scholar]
- 41.Castro MM, Kandasamy AD, Youssef N, Schulz R. Matrix metalloproteinase inhibitor properties of tetracyclines: therapeutic potential in cardiovascular diseases. Pharmacological research : the official journal of the Italian Pharmacological Society. 2011;64:551–560. doi: 10.1016/j.phrs.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Yamada A, Uegaki A, Nakamura T, Ogawa K, et al. ONO-4817, an orally active matrix metalloproteinase inhibitor, prevents lipopolysaccharide-induced proteoglycan release from the joint cartilage in guinea pigs. Inflammation research : official journal of the European Histamine Research Society … [et al.] 2000;49:144–146. doi: 10.1007/s000110050573. [DOI] [PubMed] [Google Scholar]
- 43.Golub LM, McNamara TF, D'Angelo G, Greenwald RA, Ramamurthy NS. A non-antibacterial chemically-modified tetracycline inhibits mammalian collagenase activity. Journal of dental research. 1987;66:1310–1314. doi: 10.1177/00220345870660080401. [DOI] [PubMed] [Google Scholar]
- 44.Richards C, Pantanowitz L, Dezube BJ. Antimicrobial and non-antimicrobial tetracyclines in human cancer trials. Pharmacological research : the official journal of the Italian Pharmacological Society. 2011;63:151–156. doi: 10.1016/j.phrs.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 46.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778–789. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pelouch V, Dixon IM, Golfman L, Beamish RE, Dhalla NS. Role of extracellular matrix proteins in heart function. Mol Cell Biochem. 1993;129:101–120. doi: 10.1007/BF00926359. [DOI] [PubMed] [Google Scholar]
- 49.Tyagi SC. Proteinases and myocardial extracellular matrix turnover. Mol Cell Biochem. 1997;168:1–12. doi: 10.1023/a:1006850903242. [DOI] [PubMed] [Google Scholar]
- 50.Tyagi SC, Kumar SG, Haas SJ, Reddy HK, Voelker DJ, Hayden MR, Demmy TL, Schmaltz RA, Curtis JJ. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol. 1996;28:1415–1428. doi: 10.1006/jmcc.1996.0132. [DOI] [PubMed] [Google Scholar]
- 51.Felkin LE, Birks EJ, George R, Wong S, Khaghani A, Yacoub MH, Barton PJ. A quantitative gene expression profile of matrix metalloproteinases (MMPS) and their inhibitors (TIMPS) in the myocardium of patients with deteriorating heart failure requiring left ventricular assist device support. J Heart Lung Transplant. 2006;25:1413–1419. doi: 10.1016/j.healun.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Ovechkin AV, Tyagi N, Rodriguez WE, Hayden MR, Moshal KS, Tyagi SC. Role of matrix metalloproteinase-9 in endothelial apoptosis in chronic heart failure in mice. J Appl Physiol. 2005;99:2398–2405. doi: 10.1152/japplphysiol.00442.2005. [DOI] [PubMed] [Google Scholar]
- 53.Pavlovsky AG, Williams MG, Ye QZ, Ortwine DF, Purchase CF, White AD, Dhanaraj V, Roth BD, Johnson LL, Hupe D, Humblet C, Blundell TL. X-ray structure of human stromelysin catalytic domain complexed with nonpeptide inhibitors: implications for inhibitor selectivity. Protein Sci. 1999;8:1455–1462. doi: 10.1110/ps.8.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nature medicine. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 55.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW., 2nd MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circulation research. 2010;106:166–175. doi: 10.1161/CIRCRESAHA.109.202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chavali V, Tyagi SC, Mishra PK. MicroRNA-133a regulates DNA methylation in diabetic cardiomyocytes. Biochem Biophys Res Commun. 2012 doi: 10.1016/j.bbrc.2012.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mishra PK, Singh SR, Joshua IG, Tyagi SC. Stem cells as a therapeutic target for diabetes. Front Biosci. 2010;15:461–477. doi: 10.2741/3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eitan Y, Sarig U, Dahan N, Machluf M. Acellular cardiac extracellular matrix as a scaffold for tissue engineering: in vitro cell support, remodeling, and biocompatibility. Tissue Eng Part C Methods. 2010;16:671–683. doi: 10.1089/ten.TEC.2009.0111. [DOI] [PubMed] [Google Scholar]
- 59.Friehs I, Margossian RE, Moran AM, Cao-Danh H, Moses MA, del Nido PJ. Vascular endothelial growth factor delays onset of failure in pressure-overload hypertrophy through matrix metalloproteinase activation and angiogenesis. Basic Res Cardiol. 2006;101:204–213. doi: 10.1007/s00395-005-0581-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sang QX. Complex role of matrix metalloproteinases in angiogenesis. Cell Res. 1998;8:171–177. doi: 10.1038/cr.1998.17. [DOI] [PubMed] [Google Scholar]
- 61.Brooks PC, Silletti S, von Schalscha TL, Friedlander M, Cheresh DA. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998;92:391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- 62.Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 63.Sodha NR, Clements RT, Boodhwani M, Xu SH, Laham RJ, Bianchi C, Sellke FW. Endostatin and angiostatin are increased in diabetic patients with coronary artery disease and associated with impaired coronary collateral formation. Am J Physiol Heart Circ Physiol. 2009;296:H428–H434. doi: 10.1152/ajpheart.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laham RJ, Sellke FW, Edelman ER, Pearlman JD, Ware JA, Brown DL, Gold JP, Simons M. Local perivascular delivery of basic fibroblast growth factor in patients undergoing coronary bypass surgery: results of a phase I randomized, double-blind, placebo-controlled trial. Circulation. 1999;100:1865–1871. doi: 10.1161/01.cir.100.18.1865. [DOI] [PubMed] [Google Scholar]
- 65.Rosengart TK, Lee LY, Patel SR, Kligfield PD, Okin PM, Hackett NR, Isom OW, Crystal RG. Six-month assessment of a phase I trial of angiogenic gene therapy for the treatment of coronary artery disease using direct intramyocardial administration of an adenovirus vector expressing the VEGF121 cDNA. Ann Surg. 1999;230:466–470. doi: 10.1097/00000658-199910000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher B, Pecher P, von Specht BU, Stegmann T. Induction of neoangiogenesis in ischemic myocardium by human growth factors: first clinical results of a new treatment of coronary heart disease. Circulation. 1998;97:645–650. doi: 10.1161/01.cir.97.7.645. [DOI] [PubMed] [Google Scholar]
- 67.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Givvimani S, Munjal C, Gargoum R, Sen U, Tyagi N, Vacek JC, Tyagi SC. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J Appl Physiol. 2011;110:1093–1100. doi: 10.1152/japplphysiol.01064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hua JY, Zhang ZC, Jiang XH, He YZ, Chen P. Relationship between endothelial-to-mesenchymal transition and cardiac fibrosis in acute viral myocarditis. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2012;41:298–304. [PubMed] [Google Scholar]
- 71.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 73.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y. Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest. 2002;110:1525–1538. doi: 10.1172/JCI16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lefer DJ. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci USA. 2007;104:17907–17908. doi: 10.1073/pnas.0709010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qipshidze N, Metreveli N, Mishra PK, Lominadze D, Tyagi SC. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int J Biol Sci. 2012;8:430–441. doi: 10.7150/ijbs.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lin YD, Luo CY, Hu YN, Yeh ML, Hsueh YC, Chang MY, Tsai DC, Wang JN, Tang MJ, Wei EI, Springer ML, Hsieh PC. Instructive nanofiber scaffolds with VEGF create a microenvironment for arteriogenesis and cardiac repair. Sci Transl Med. 2012;4:146ra109. doi: 10.1126/scitranslmed.3003841. [DOI] [PubMed] [Google Scholar]