

Abstract
Background
Despite medical advances, children with dilated cardiomyopathy (DCM) remain at high risk of death or need for cardiac transplantation. We sought to identify predictors of disease progression in pediatric DCM.
Methods and Results
The Pediatric Heart Network evaluated chronic DCM patients with prospective echocardiographic and clinical data collection during an 18-month follow-up. Inclusion criteria were age <22 years and DCM disease duration >2 months. Patients requiring intravenous inotropic/mechanical support or listed status 1A/1B for transplant were excluded. Disease progression was defined as an increase in transplant listing status, hospitalization for heart failure, intravenous inotropes, mechanical support, or death. Predictors of disease progression were identified using Cox proportional hazards modeling and classification and regression tree analysis. Of the 127 patients, 28 (22%) had disease progression during the 18-month follow-up. Multivariable analysis identified older age at diagnosis (hazard ratio=1.14 per year; P<0.001), larger left ventricular (LV) end-diastolic M-mode dimension z-score (hazard ratio=1.49; P<0.001), and lower septal peak systolic tissue Doppler velocity z-score (hazard ratio=0.81; P=0.01) as independent predictors of disease progression. Classification and regression tree analysis stratified patients at risk of disease progression with 89% sensitivity and 94% specificity based on LV end-diastolic M-mode dimension z-score ≥7.7, LV ejection fraction <39%, LV inflow propagation velocity (color M-mode) z-score <-0.28, and age at diagnosis ≥8.5 months.
Conclusions
In children with chronic stable DCM, a combination of diagnosis after late infancy and echocardiographic parameters of larger LV size and systolic and diastolic function predicted disease progression.
Keywords: cardiomyopathies, heart transplantation, pediatrics
Dilated cardiomyopathy (DCM) is the most common pediatric cardiomyopathy, an important cause of heart failure (HF) and a leading cause of heart transplantation in children.1–4 Despite recent medical advances, event-free survival remains poor, with 5-year rates of death or transplantation reported as high as 46%.1 A limited number of risk factors have been consistently identified to help predict outcomes; older age at presentation and decreased indices of systolic left ventricular (LV) function have been associated with a worse prognosis in multiple studies.5–13 However, the sensitivity of these parameters in early risk stratification and prediction of adverse events remains limited. Newer echocardiographic modalities have shown promise in identifying abnormalities that may be associated with adverse events, such as comprehensive assessment of ventricular dysfunction with tissue Doppler imaging (TDI); however, these modalities have not yet been studied in a comprehensive manner in a large prospective cohort of pediatric patients with DCM.14–17 Early risk stratification would help guide frequency of monitoring and optimize timing and type of interventions, including medications or device therapies and, ultimately, cardiac transplantation. The purpose of this study was, therefore, to identify clinical and echocardiographic factors associated with disease progression in children with DCM.
Methods
The study was part of the Ventricular Volume Variability (VVV) Study conducted through the Pediatric Heart Network, a multicenter clinical research consortium. The VVV study is a multicenter, observational study of a prospectively enrolled cohort of children with DCM. Subjects were enrolled at 8 study centers between May 2005 and July 2007. The study was approved by an institutional review board at all sites with informed consent obtained from all subjects. The core laboratory measurements from echocardiograms performed at study enrollment were used in the analysis. Demographic information and clinical data were obtained during regularly scheduled visits throughout the 18-month follow-up period. The primary aim of this report was to identify echocardiographic and clinical variables present at the time of enrollment that correlated with subsequent disease progression.
Significant disease progression was defined as any of the following: hospitalization for HF, initiation of intravenous inotropic support, transplant listing or increase in listing status, decompensated HF requiring mechanical circulatory support, or death. Patient enrollment criteria included age <22 years, diagnosis of DCM based on the first study echocardiogram with an LV end-diastolic dimension (EDD) >5.5 cm (or z-score for age >2) and LV ejection fraction (EF) <50% or shortening fraction <28% (or z-score for age <-2), disease duration >2 months, anticipated ongoing evaluation at the same institution, and informed consent. Exclusion criteria included other forms of cardiomyopathy including noncompaction, congenital heart disease, frequent ectopy, need for intravenous or mechanical hemodynamic support, and transplant listing status of 1A or 1B at the time of screening. Only 2 patients without an event, of the 127 total, had <18 months of follow-up because of early withdrawal from the study. Patient data collected included age, sex, height, weight, blood pressure, race, cause of DCM, and medication type and dose. Body surface area was calculated using the Haycock formula.18
All centers followed a standardized protocol for echocardiographic image acquisition. Baseline transthoracic echocardiograms performed at study enrollment were submitted to the core laboratory for measurement of echocardiographic variables (M-mode, 2-dimensional, Doppler, and TDI) by 2 experienced readers. For each variable, measurements were performed on 3 sequential cardiac cycles. Thirty-five parameters of LV dimension, mass, and systolic and diastolic function obtained from the enrollment echocardiograms and 28 clinical factors were included for analysis as predictors of disease progression (please refer to the Table in the online-only Data Supplement for detailed list). A previous VVV analysis on the effect of beat averaging on reproducibility of echocardiographic variables showed that use of 3-beat averaging reduced inter- and intrareader variability, and thus the 3-beat average measurements using the primary core reader's interpretation were used in this analysis.19 All echocardiographic measurements were made using custom DICOM software (EchoTrace, Marcus Laboratories, Boston, MA) according to previously published techniques.19 Z-scores on echocardiographic measurements (for body surface area or age) were used where applicable. LV flow propagation z-scores were calculated using the following formula: z-score=(LV flow propagation– 0.191×age+66.7)/(24.5–0.132×age). Additional data points were assessed as both percentages and z-scores, including shortening fraction and EF. Further analysis of mitral regurgitation was undertaken; vena contracta width (VCW) was calculated as the average of the dimensions from orthogonal planes (parasternal long-axis and apical 4-chamber views). Mitral regurgitation grade was evaluated subjectively based on visual appearance and objectively based on VCW adjusted to body surface area as adjusted VCW (mm/m)=VCW/body surface area0.5.20 Severity categories were defined as mild, moderate, and severe for adjusted VCW <3, 3 to 4.5, and >4.5, respectively. Patients with adjusted VCW within 0.3 mm/m of a boundary value were adjudicated based on visual estimate of severity.
Univariate predictors of time to disease progression were identified using Cox proportional hazards regression. Multivariable modeling used a stepwise Cox proportional hazards procedure for all variables with a univariate P<0.2. A P<0.1 was then required to remain in the model and determine independent predictors. Certain candidate predictors were not included in the multivariate analysis as a result of excessive missing data (see starred items in the online-only Data Supplement), including Doppler variables that were only measurable in the absence of a mitral valve inflow summation wave. Logistic regression analysis was also performed, with identical significant findings demonstrating that use of a dichotomous outcome measure for this analysis is robust to small differences in length of follow-up. As such, a classification and regression tree (CART) analysis was performed to construct a risk stratification algorithm to predict disease progression.21 CART is a nonparametric technique that produces a decision tree by a series of binary splits (recursive partitioning) of the data. The CART model selects variables in order of magnitude of improvement in prediction of the outcome, and the variables that contribute the most to the outcome are listed first at the top of the tree. Analyses were performed using SAS version 9.2 (SAS Institute, Inc, Cary, NC) and an open-source adaptation of the CART algorithm (RPART library in R version 2.14.1).
Results
Of the 127 patients in the analytic cohort, median age at enrollment was 9.2 years (interquartile range, 4.0–15.0 years). Table 1 summarizes the demographic information of patients who subsequently developed disease progression compared with those who did not. At the time of enrollment, most patients were classified as having New York Heart Association/Ross class I or II HF symptoms. Medical therapy included angiotensin-converting enzyme inhibition (83%), digoxin (59%), diuretics (54%), and β-blockers (53%). Of the 28 patients (22%) who met criteria for disease progression, 3 died, 16 were transplanted, 3 required initiation of intravenous inotropes, 1 received an LV assist device, 7 had an increase in listing status, and 14 were hospitalized for worsened HF (not mutually exclusive). The majority of patients with disease progression had a diagnosis of idiopathic DCM (n=20), followed by Adriamycin-associated cardiotoxicity (n=3); no patients with myocarditis subsequently developed disease progression in this follow-up period. Figure 1 depicts the freedom from disease progression for the entire cohort; 6- and 12-month rates of disease progression were 15% and 22%, respectively. Table 2 outlines the univariate associations between baseline clinical and echocardiographic variables in patients with disease progression with a P<0.2 (for a comprehensive listing of all predictors assessed, refer to the Table in the online-only Data Supplement). Clinical factors associated with disease progression on univariate analysis included older age at diagnosis, more recent diagnosis of DCM, male sex, and symptomatic HF—specifically increased symptoms of exercise intolerance, feeding difficulties, dyspnea, need for hospitalization, and intravenous inotropes in the 6 months before enrollment. Echocardiographic indices associated with disease progression included measures of LV size, degree of mitral regurgitation, as well as dimensional and volumetric assessments of systolic function. Diastolic assessments using mitral inflow measurements and TDI were also significantly associated with disease progression; however, many of these parameters were not included in subsequent multivariate analysis as a result of incomplete separation of early and late diastolic flows or annular motion (Doppler summation waves) resulting in missing data (see asterisked variables in the online-only Data Supplement).
Table 1. Demographics by Disease Progression.
| Stable Disease | Progressive Disease | P Value* | |
|---|---|---|---|
| n | 99 | 28 | |
| Male | 39 (39%) | 17 (61%) | 0.054 |
| Age at diagnosis, y | 1.1 (0.3–10.2) | 7.5 (0.9–13.5) | 0.04 |
| Age at enrollment, y | 8.8 (4.0–15.0) | 9.4 (3.6 –14.8) | 0.869 |
| Time from diagnosis of cardiomyopathy to enrollment, y | 3.5 (0.6–6.8) | 0.8 (0.3–3.0) | 0.006 |
| Length of follow-up, y | 1.5 (1.1,1.5) | 0.5 (0.2,1.3) | <0.001 |
| Ross/NYHA Heart Failure Classification | 0.002 <0.001† | ||
| Class I | 73 (74%) | 11 (39%) | |
| Class II | 23 (23%) | 14 (50%) | |
| Class III | 3 (3%) | 3 (11%) | |
| Class IV | 0 (0%) | 0 (0%) | |
| Primary cause of dilated cardiomyopathy | 0.329 | ||
| Metabolic disorder | 2 (2%) | 0 (0%) | |
| Mitochondrial disorder | 1 (1%) | 1 (4%) | |
| Neuromuscular disease associated with cardiomyopathy | 2 (2%) | 2 (7%) | |
| Single gene defect | 3 (3%) | 1 (4%) | |
| Adriamycin-associated cardiotoxicity | 11 (11%) | 3 (11%) | |
| Idiopathic | 65 (66%) | 20 (71%) | |
| Myocarditis | 6 (6%) | 0 (0%) | |
| Other | 9 (9%) | 1 (4%) |
Median (IQR) or frequency (%) is shown. IQR indicates interquartile range; and NYHA, New York Heart Association.
Wilcoxon rank-sum test for medians or a Fisher exact test for frequencies.
Mantel–Haenszel test for trend.
Figure 1.
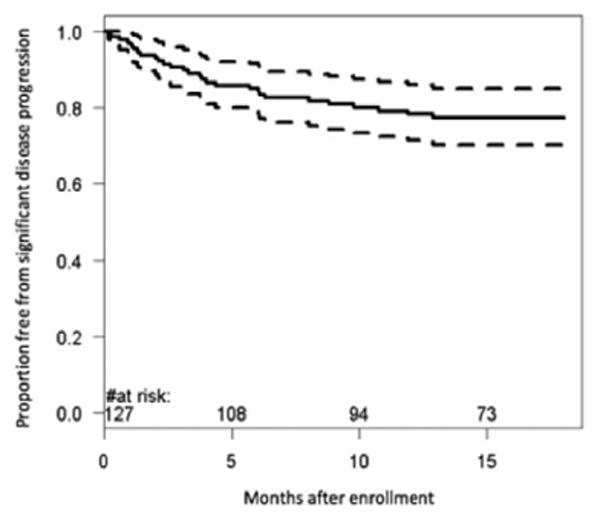
Kaplan–Meier curve demonstrating percentages of patient cohort meeting criteria for clinically significant disease progression during the 18-month time interval. Within 9.8 months after enrollment, 20% of the population developed significant disease progression.
Table 2. Univariate Predictors of Significant Disease Progression With P<0.2 Using Cox Proportional Hazards Process.
| Variable* | n | Stable Disease (n=99) |
n | Progressive Disease (n=28) |
P Value† |
|---|---|---|---|---|---|
| Median age at diagnosis of cardiomyopathy, y (IQR) | 99 | 1.1 (0.3–10.2) | 28 | 7.5 (0.9–13.5) | 0.03 |
| Median time from diagnosis to baseline echo, y (IQR) | 99 | 3.47 (0.56–6.84) | 28 | 0.79 (0.27–2.97) | 0.01 |
| Male | 99 | 39 (39%) | 28 | 17 (61%) | 0.04 |
| Race | 99 | 28 | 0.06 | ||
| White | 69 (70%) | 14 (50%) | |||
| Other or unknown race | 30 (30%) | 14 (50%) | |||
| Hospitalized in previous 6 mo | 99 | 26 (26%) | 28 | 15 (54%) | 0.007 |
| No. of hospitalizations related to heart failure | 99 | 28 | <0.001 | ||
| 0 | 86 (87%) | 17 (61%) | |||
| 1 | 11 (11%) | 6 (21%) | |||
| 2 | 2 (2%) | 5 (18%) | |||
| Failure to thrive in 6 mo before enrollment | 98 | 9 (9%) | 28 | 6 (21%) | 0.08 |
| Dyspnea in 6 mo before enrollment | 99 | 11 (11%) | 27 | 12 (44%) | <0.001 |
| Feeding difficulties or exercise intolerance in 6 mo before enrollment | 99 | 32 (32%) | 28 | 18 (64%) | 0.02 |
| Palpitations in 6 mo before enrollment | 91 | 9 (10%) | 24 | 5 (21%) | 0.15 |
| Intravenous inotropic medications in 6 mo before enrollment | 90 | 8 (9%) | 27 | 7 (26%) | 0.01 |
| Weight-for-age z-score | 97 | 28 | 0.18 | ||
| Z<-1 | 25 (26%) | 11 (39%) | |||
| −1 ≤Z≤1 | 53 (55%) | 10 (36%) | |||
| Z>1 | 19 (20%) | 7 (25%) | |||
| End-diastolic short-axis dimension, M-mode z-score | 99 | 3.4±1.8 | 26 | 7.4±3.7 | <0.001 |
| End-diastolic short-axis dimension, 2D z-score | 99 | 3.6±2.4 | 27 | 7.8±4.3 | <0.001 |
| Posterior wall thickness to dimension ratio, M-mode z-score | 98 | −1.8±1.0 | 27 | −2.6±1.0 | <0.001 |
| Posterior wall thickness to dimension ratio, 2D z-score | 99 | −1.5±1.2 | 28 | −2.2±1.3 | 0.004 |
| LV mass z-score, M-mode | 99 | 1.3±1.5 | 26 | 3.2±2.2 | <0.001 |
| LV mass z-score, 2D | 98 | 1.9±1.7 | 27 | 4.6±2.3 | <0.001 |
| Mass to volume ratio z-score | 99 | −1.0±1.1 | 28 | −1.7±1.2 | 0.003 |
| Sphericity index z-score | 99 | 1.0±1.3 | 28 | 2.0±1.4 | <0.001 |
| Median eccentricity index (IQR) | 99 | 0.93 (0.92–0.94) | 28 | 0.92 (0.90–0.93) | 0.001 |
| Shortening fraction, M-mode, % | 99 | 21.7±6.4 | 27 | 12.8±4.8 | <0.001 |
| Median IQR | 99 | 21.9 (18.0–27.0) | 27 | 12.0 (8.7–16.7) | |
| Shortening fraction, M-mode z-score | 99 | −6.1±3.9 | 27 | −12.1±4.8 | <0.001 |
| Shortening fraction, 2D % | 99 | 18.7±6.2 | 28 | 11.6±5.3 | <0.001 |
| Median IQR | 99 | 18.6 (14.3–23.7) | 28 | 11.7 (8.3–13.2) | |
| Shortening fraction, 2D z-score | 99 | −5.1±1.9 | 28 | −7.2±1.8 | <0.001 |
| Aortic ejection time, M-mode, ms | 99 | 263.6±36.3 | 28 | 241.4±33.1 | 0.003 |
| Ejection fraction, % | 99 | 43.1±9.7 | 28 | 27.9±11.4 | <0.001 |
| Median (IQR) | 99 | 44.0 (37.6–51.0) | 28 | 27.1 (19.1–35.0) | |
| Ejection fraction z-score | 99 | −4.3±2.1 | 28 | −7.5±2.4 | <0.001 |
| Median mitral regurgitation jet width/BSA0.5 (IQR) | 98 | 0.2 (0.0–0.3) | 28 | 0.3 (0.2–0.5) | <0.001 |
| Severity of mitral valve regurgitation | 98 | 28 | 0.003 | ||
| None | 33 (34%) | 3 (11%) | |||
| Mild | 44 (45%) | 9 (32%) | |||
| Moderate | 13 (13%) | 8 (29%) | |||
| Severe | 8 (8%) | 8 (29%) | |||
| Severity of mitral valve regurgitation | 98 | 28 | <0.001 | ||
| None/mild | 77 (79%) | 12 (43%) | |||
| Moderate/severe | 21 (21%) | 16 (57%) | |||
| Left lateral AV valve peak systolic velocity z-score | 99 | −1.2±1.4 | 26 | −2.3±1.2 | <0.001 |
| Tei index | 99 | 0.42±0.16 | 28 | 0.54±0.25 | 0.001 |
| Septal AV valve peak systolic velocity z-score | 99 | −1.4±2.1 | 27 | −3.1±2.2 | <0.001 |
| Right lateral AV valve peak systolic velocity z-score | 98 | −0.2±1.4 | 26 | −1.1±1.4 | <0.001 |
| Left ventricular flow propagation velocity z-score | 94 | −0.3±0.9 | 28 | −0.7±0.5 | 0.02 |
All echocardiographic measures are from the left ventricle, where applicable. 2D indicates 2-dimensional; AV, atrioventricular; IQR, interquartile range; and LV, left ventricular.
All measures are from the time of study enrollment unless otherwise noted.
P value is from Cox proportional hazards comparing stable disease with significant disease progression.
Independent predictors of disease progression on multivariate analysis included older age at diagnosis (P<0.001), larger M-mode LV EDD z-score (P<0.001), and lower septal peak systolic TDI velocity z-score at enrollment (P=0.01; Figure 2). Older age at diagnosis was associated with disease progression with a hazard ratio (HR) of 1.14 per 1 year increase in age at enrollment (confidence interval, 1.06–1.23). Echocardiographic evidence of larger LV EDD at enrollment was the strongest predictor of disease progression with an HR of 1.49 for every unit z-score increase (confidence interval, 1.32–1.69). In addition, a higher septal peak systolic TDI velocity z-score was protective against disease progression with an HR of 0.81 for every unit z-score increase (confidence interval, 0.69–0.95).
Figure 2.
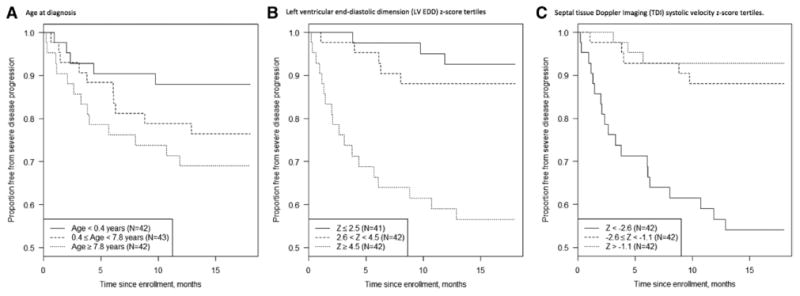
Box plots demonstrating the 3 independent predictors of disease progression: (A) age at diagnosis, (B) left ventricular end-diastolic dimension (LV EDD) z-score, and (C) septal tissue Doppler imaging (TDI) systolic velocity z-score.
Given the relatively large number of associated variables on Cox proportional hazards modeling, a CART analysis was performed to identify the variables with the highest discriminatory power to predict disease progression. This analysis identified a combination of 3 echocardiographic and 1 clinical factor to be useful in risk stratifying these patients (Figure 3). M-mode LV EDD z-score was identified as the top discriminator of disease progression, with 14 of 15 (93%) patients with LV EDD z-score >7.7 showing disease progression compared with 13% of patients with LV EDD z-score <7.7. In patients with LV EDD z-score <7.7, LV EF emerged as a predictor of disease progression, with 12 of 36 (33%) patients with LV EF <38% showing disease progression compared with only 3% of those with a higher EF. LV flow propagation velocity z-score served as a further discriminator; among those with an LV EDD z-score <7.7 and LV EF <38%, 12 of 24 (50%) patients with an LV flow propagation velocity z-score <-0.28 had disease progression compared with none of the 12 patients with a z-score >-0.28. The final discriminator was age at diagnosis. In children with an LV EDD z-score <7.7, an LV EF <38%, and an LV flow propagation z-score <-0.28, 11 of 16 (69%) patients >8.5 months at diagnosis had disease progression compared with 1 of 8 (12%) patients <8.5 months at diagnosis. This risk algorithm tree had a sensitivity of 89%, a specificity of 94%, and a predictive accuracy of 93% for identifying patients who experienced disease progression (Table 3). Figure 4 depicts the Kaplan–Meier curve comparing those individuals identified by CART analysis to be at high risk of disease progression with those at low risk of progression.
Figure 3.
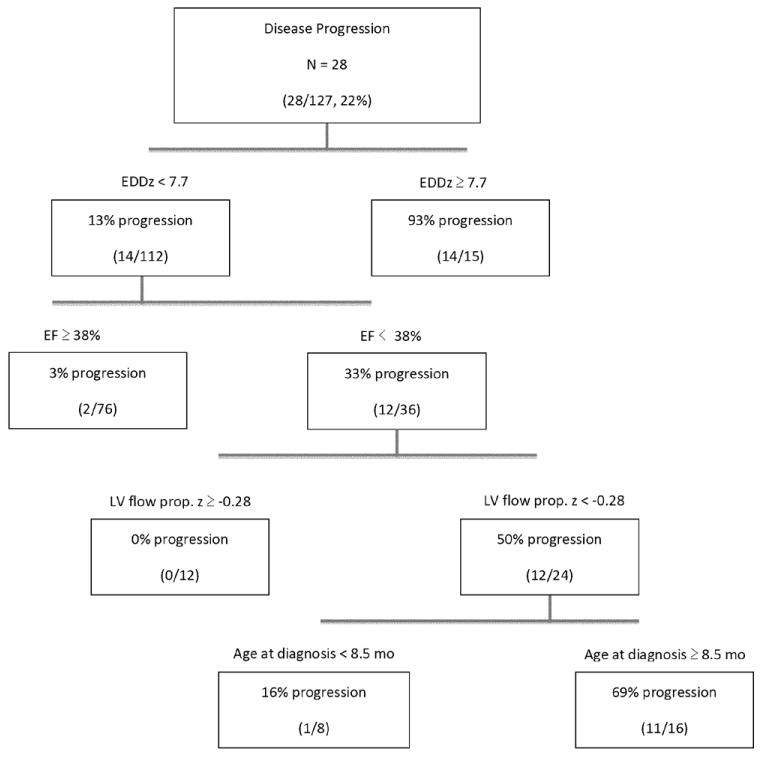
Classification and regression tree analysis risk stratifying patients with significant disease progression by 3 echocardiographic and 1 clinical factor. EDDz indicates left ventricular end-diastolic dimension z-score; EF, ejection fraction; and LV flow prop., left ventricular propagation velocity slope.
Table 3. Operating Characteristics of Each CART Analysis Branch.
| Variables in Tree | Sensitivity | Specificity | PPV | NPV | Accuracy |
|---|---|---|---|---|---|
| EDDz ≥7.7 only | 50.0% | 99.0% | 93.3% | 87.5% | 88.2% |
| EDDz ≥7.7 and EF <38.2 | 92.9% | 74.7% | 51.0% | 97.4% | 78.7% |
| EDDz ≥7.7, EF <38.2, and LV flow prop. z <-0.281 | 92.9% | 86.9% | 66.7% | 97.7% | 88.2% |
| All 4 variables | 89.3% | 93.9% | 80.6% | 96.9% | 92.9% |
CART indicates classification and regression tree; EDDz, left ventricular end-diastolic dimension z-score; EF, ejection fraction; LV, left ventricular; NPV, negative predictive value; PPV, positive predictive value; and prop., propagation velocity slope.
Figure 4.
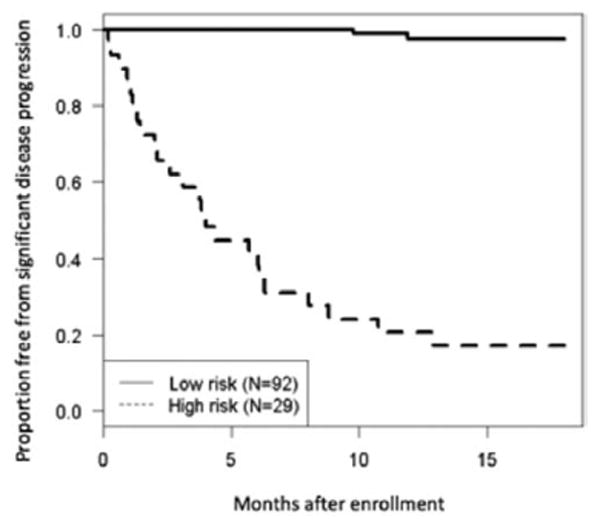
Kaplan–Meier curve demonstrating patients at high risk of disease progression versus low risk. Definition of risk was based on whether patients met criteria on the classification and regression tree analysis for significant disease progression using all 4 tiers
Discussion
Transplant-free survival in children with DCM has not appreciably improved in recent decades.11–13 In adults with HF, models have been devised to accurately assess patient risk of mortality largely based on prognostic factors incorporating patient demographics (age, sex), clinical parameters of HF (New York Heart Association class, EF, blood pressure, maximal oxygen consumption [Vo2]), medical therapies used (drug dosing, devices), and serum biomarkers of disease severity (hemoglobin, lymphocyte counts, sodium levels, total cholesterol, and uric acid).22,23 Robust predictors of disease progression in pediatric DCM are lacking. Our study assessed many of the above parameters, with the exception of laboratory markers and Vo2, in an attempt to identify prognostic factors for pediatric DCM disease progression in chronically stable outpatients. This analysis confirms the importance of age at diagnosis and echocardiographic evidence of LV size and impaired systolic and diastolic function as predictors of disease progression and associated adverse events. Unlike most published studies that have used a retrospective or registry design, our prospective study has unique strengths and allowed for the development of a model predictive of disease progression with high sensitivity, specificity, and predictive accuracy. If validated in additional populations, this model may provide a clinically useful decision making tool.
Patients who showed disease progression were more symptomatic and had more severe echocardiographic abnormalities at enrollment. However, older age at diagnosis in this study was the only clinical parameter that remained independently associated with disease progression on multivariate analysis. Patient age has long been identified as an important predictor of outcome in both children and adults with DCM, with older school-aged children at increased risk in pediatric DCM studies.1–3,5,9,11–13 In addition, there is some evidence that infants may also be at risk. Tsirka et al11 identified 2 age groups at risk for death/transplantation: those aged <1 year at diagnosis (HR, 7.1) and an older subset aged >12 years (HR, 4.5).
A pediatric cohort from Australia also pointed to a possible bimodal distribution of age-related risk, with a trend toward worse survival in children aged <1 year.2 The younger subset of patients in our study cohort was not identified to be at increased risk, but the inclusion criteria requiring a minimum of 2-month duration with the diagnosis of DCM and the relatively small patient sample in this age range may have limited our ability to extrapolate the effect of a diagnosis of DCM in early infancy on long-term prognosis. Interestingly, no other demographic parameters or clinical symptoms were independently predictive of worse outcome in this analysis; this contrasts with the recent study by Alvarez et al13 that used the Pediatric Cardiomyopathy Registry of patients with idiopathic DCM in which the severity of HF symptoms at diagnosis was the single strongest predictor of death or transplantation. This difference could be because of the focus in the VVV study on a cohort of chronic stable DCM outpatients, thus excluding those patients with symptomatic HF requiring inotropic support at the time of enrollment. The emergence of echocardiographic parameters as independent predictors of progression suggests that echocardiography provides a more objective assessment of disease severity compared with clinical symptomatology.
Echocardiography plays a prominent role in both diagnosis and ongoing evaluation of patients with DCM. This analysis highlights the immense value that standard imaging techniques can add in identifying patients at risk for a poor outcome. The prospective nature of this study allowed for a standardized and comprehensive assessment of a relatively larger number of echocardiographic variables than has previously been studied in children. Our study confirmed previously identified associations of larger LV dimensions, decreased indices of LV systolic and diastolic function, as well as degree of mitral regurgitation as important univariate predictors of disease progression.5–14,24,25 On multivariate analysis, the strongest predictor of disease progression was larger LV EDD, with further evidence of its importance found in the CART analysis algorithm. Increasing LV EDD is a part of the cardiac remodeling process in patients with decreased systolic function and DCM. Larger LV EDD has long been found to correlate with adverse clinical outcome1,2,11,13,25,26 and has been associated with increased mortality in patients with DCM listed for cardiac transplantation, particularly the infant subset, as well as affecting survival in the first 6 months after transplant.25 The Pediatric Cardiomyopathy Registry found that a larger LV EDD was associated with increased risk of transplantation but a decreased risk of death.13 This finding may be because of an aspect of referral bias, where children with larger LV EDD may be directed toward transplantation earlier, potentially limiting true assessment of their overall mortality risk. Regardless, the use of LV EDD has repeatedly been identified as a powerful prognosticating tool in pediatric DCM.
Our study also found that lower peak septal systolic velocity z-scores by TDI were associated with worse clinical outcome on multivariate analysis. This TDI parameter highlights both the role of diminished systolic function in these patients and may further signify the importance of biventricular structural interdependence and functional augmentation that can be lost as a consequence of DCM. As one of the newer imaging modalities, TDI has proven useful in evaluating both systolic and diastolic function in several disease processes and now has become a standard part of echocardiographic assessment. McMahon et al14 found that TDI velocities were significantly reduced in children with DCM compared with normal controls and that tricuspid velocity during early diastole was a predictor of patients who subsequently died or needed transplantation. In adult patients with chronic systolic HF, septal TDI measurements were found to be more reliable and clinically relevant compared with lateral TDI measurements, with higher septal E/Ea ratios being correlated with natriuretic peptide levels and adverse cardiac events.27 In adults with HF, septal TDI has also been shown to be of value in identifying patients who may benefit from cardiac resynchronization therapy.28
CART modeling and analysis identified a combination of LV dimension, LV systolic and diastolic function, and the clinical parameter of age at diagnosis as valuable in risk stratifying this cohort of patients with high sensitivity, specificity, and predictive accuracy. Although each of the 4 parameters shows some discriminating value, it is the combination of all 4 components that yields the best prognostic value (Table 3). LV dimension, specifically an LV EDD z-score ≥7.7, remained as the top predictor on CART analysis. The degree of LV systolic dysfunction added an additional important determinant of prognosis, with an LV EF <38% demonstrating incremental value in further subcategorizing patients with a less dilated phenotype (LV EDD <7.7 z-score) of DCM. Measures of decreased systolic function at presentation, both by shortening fraction and by EF, previously have been associated with worse prognosis in patients with DCM.1,2,11,12,25,29 Kantor et al12 determined that patients with a lower EF at presentation exhibited a substantially increased risk of death or transplantation, with an incremental increased risk of 35% for every 10% decrease in LV EF at presentation. Furthermore, patients who show no significant improvement in LV function after initiation of medical therapy are at increased risk, with persistently decreased shortening fraction <20% in long-term follow-up strongly associated with poor outcome in the pediatric DCM population.2
The final echocardiographic component in the CART algorithm used the diastolic assessment of LV flow propagation to characterize patients at risk for disease progression. This measurement of the slope of the color Doppler mitral inflow wave during early LV filling has been used as a non-invasive assessment of LV relaxation. Although this calculation is not regularly performed as a part of the standard pediatric echocardiogram, it has proven useful in evaluating patients with abnormal LV relaxation, including different forms of cardiomyopathy and systemic hypertension. Brun et al30 found that the velocity of flow propagation was lower in patients with DCM and proved a useful tool in studying diastolic function. Although LV flow propagation was not identified as an independent predictor of disease progression in this patient population, it provides additional stratifying power if the z-score is <-0.28 for determining patients at risk for worse outcomes when used in conjunction with LV EDD and EF. Prospective collection of an extensive list of echocardiographic parameters of cardiac size and function with concurrent clinical factors allowed for the CART analysis and formulation of a well-designed risk stratification tool that, once further validated in other patient groups, may help practitioners enhance their ability to prognosticate disease progression in their patients.
Study Limitations
The purpose of this study was to evaluate patients with chronic, stable DCM that resulted in exclusion of those who at the time of enrollment manifested rapidly advancing or end-stage HF. Thus, the results of this study may not be generalizable to patients acutely presenting with DCM or those requiring intravenous inotropic or mechanical support early in their clinical course. Furthermore, the robustness of these predictors is limited by the small sample size. The follow-up period to determine disease progression was relatively short compared with other studies,1,2 and longer evaluation might prove useful to better understand the large number of factors affecting the natural history of pediatric DCM. As a result of limited numbers of patients with additional laboratory markers, such as natriuretic peptides or exercise testing Vo2 measurements, the influence of these markers of HF could not be assessed in our study. Independent validation of our CART risk algorithm in other populations of patients with pediatric DCM is needed to confirm its use as a risk-stratifying tool.
Conclusions
In children with chronic stable DCM, diagnosis after late infancy and echocardiographic parameters of LV size and systolic and diastolic dysfunction were independently associated with disease progression. If confirmed in other populations, the proposed CART algorithm may be useful to reliably risk stratify patients with DCM and identify those who might benefit from more frequent monitoring, intensification of medical treatment, or earlier consideration of mechanical support and transplant evaluation and listing.
Supplementary Material
Table. Online supplement presenting all candidate predictors assessed for disease progression. Mean±SD or Frequency (%) shown unless otherwise noted.
Clinical Perspective.
Predictors of disease progression in children with clinically stable heart failure are lacking. Early risk stratification may help guide frequency of monitoring and help optimize timing of medical and device therapy and of cardiac transplantation. In this prospective study, we identified clinical and echocardiographic factors including age at diagnosis, left ventricular M-mode dimension z-score, and septal systolic tissue Doppler imaging velocity z-score as being associated with disease progression in 127 pediatric patients with chronic stable dilated cardiomyopathy. A model was developed with high sensitivity and specificity in identifying patients at risk of disease progression. This model requires further validation in additional cohorts for application as a clinical algorithm for risk stratification of pediatric patients with chronic stable dilated cardiomyopathy.
Acknowledgments
Sources of Funding: This work was supported by U01 grants from the National Heart, Lung, and Blood Institute (NHLBI; HL068269, HL068270, HL068279, HL068281, HL068285, HL068292, HL068290, HL068288). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI.
Footnotes
The online-only Data Supplement is available at http://circheartfailure.ahajournals.org/lookup/suppl/doi:10.1161/CIRCHEARTFAILURE.113.000125/-/DC1.
Disclosures: None.
Contributor Information
Kimberly M. Molina, University of Utah School of Medicine, Salt Lake City.
Peter Shrader, New England Research Institutes, Watertown, MA.
Steven D. Colan, Boston Children's Hospital and Harvard Medical School, MA.
Seema Mital, The Hospital for Sick Children, Toronto, Canada.
Renee Margossian, Boston Children's Hospital and Harvard Medical School, MA.
Lynn A. Sleeper, New England Research Institutes, Watertown, MA.
Girish Shirali, Children's Mercy Hospitals and Clinics, Kansas City, MO.
Piers Barker, Duke University Medical Center, Durham, NC.
Charles E. Canter, Washington University in St. Louis, MO.
Karen Altmann, Children's Hospital of New York.
Elizabeth Radojewski, The Hospital for Sick Children, Toronto, Canada.
Elif Seda Selamet Tierney, Lucille Packard Children's Hospital, Palo Alto, CA.
Jack Rychik, Children's Hospital of Philadelphia, PA.
Lloyd Y. Tani, University of Utah School of Medicine, Salt Lake City.
References
- 1.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 2.Daubeney PE, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114:2671–2678. doi: 10.1161/CIRCULATIONAHA.106.635128. [DOI] [PubMed] [Google Scholar]
- 3.Hsu DT, Canter CE. Dilated cardiomyopathy and heart failure in children. Heart Fail Clin. 2010;6:415–432. vii. doi: 10.1016/j.hfc.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Kirk R, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dobbels F, Rahmel AO, Stehlik J, Hertz MI. The registry of the international society for heart and lung transplantation: fourteenth pediatric heart transplantation report-2011. J Heart Lung Transplant. 2011;30:1095–1103. doi: 10.1016/j.healun.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez JA, Wilkinson JD, Lipshultz SE. Outcome predictors for pediatric dilated cardiomyopathy: a systematic review. Prog Pediatr Cardiol. 2007;23:25–32. doi: 10.1016/j.ppedcard.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taliercio CP, Seward JB, Driscoll DJ, Fisher LD, Gersh BJ, Tajik AJ. Idiopathic dilated cardiomyopathy in the young: clinical profile and natural history. J Am Coll Cardiol. 1985;6:1126–1131. doi: 10.1016/s0735-1097(85)80319-3. [DOI] [PubMed] [Google Scholar]
- 7.Lewis AB, Chabot M. Outcome of infants and children with dilated cardiomyopathy. Am J Cardiol. 1991;68:365–369. doi: 10.1016/0002-9149(91)90833-7. [DOI] [PubMed] [Google Scholar]
- 8.Akagi T, Benson LN, Lightfoot NE, Chin K, Wilson G, Freedom RM. Natural history of dilated cardiomyopathy in children. Am Heart J. 1991;121:1502–1506. doi: 10.1016/0002-8703(91)90158-e. [DOI] [PubMed] [Google Scholar]
- 9.Burch M, Siddiqi SA, Celermajer DS, Scott C, Bull C, Deanfield JE. Dilated cardiomyopathy in children: determinants of outcome. Br Heart J. 1994;72:246–250. doi: 10.1136/hrt.72.3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol. 1997;146:385–393. doi: 10.1093/oxfordjournals.aje.a009291. [DOI] [PubMed] [Google Scholar]
- 11.Tsirka AE, Trinkaus K, Chen SC, Lipshultz SE, Towbin JA, Colan SD, Exil V, Strauss AW, Canter CE. Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J Am Coll Cardiol. 2004;44:391–397. doi: 10.1016/j.jacc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 12.Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN. The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:1377–1384. doi: 10.1016/j.jacc.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 13.Alvarez JA, Orav EJ, Wilkinson JD, Fleming LE, Lee DJ, Sleeper LA, Rusconi PG, Colan SD, Hsu DT, Canter CE, Webber SA, Cox GF, Jefferies JL, Towbin JA, Lipshultz SE. Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy-results from the pediatric cardiomyopathy registry. Circulation. 2011;124:814–823. doi: 10.1161/CIRCULATIONAHA.110.973826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahon CJ, Nagueh SF, Eapen RS, Dreyer WJ, Finkelshtyn I, Cao X, Eidem BW, Bezold LI, Denfield SW, Towbin JA, Pignatelli RH. Echocardiographic predictors of adverse clinical events in children with dilated cardiomyopathy: a prospective clinical study. Heart. 2004;90:908–915. doi: 10.1136/hrt.2003.020966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alehan F, Ozkutlu S, Alehan D, Saraçlar M. Echocardiographic assessment of left and right ventricular diastolic functions in children with dilated cardiomyopathy. Turk J Pediatr. 1998;40:337–346. [PubMed] [Google Scholar]
- 16.Friedberg MK, Roche SL, Mohammed AF, Balasingam M, Atenafu EG, Kantor PF. Left ventricular diastolic mechanical dyssynchrony and associated clinical outcomes in children with dilated cardiomyopathy. Circ Cardiovasc Imaging. 2008;1:50–57. doi: 10.1161/CIRCIMAGING.108.782086. [DOI] [PubMed] [Google Scholar]
- 17.Mohammed A, Mertens L, Friedberg MK. Relations between systolic and diastolic function in children with dilated and hypertrophic cardiomyopathy as assessed by tissue Doppler imaging. J Am Soc Echocardiogr. 2009;22:145–151. doi: 10.1016/j.echo.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Haycock GB, Schwartz GJ, Wisotsky DH. Geometric method for measuring body surface area: a heightweight formula validated in infants, children and adults. J Pediatr. 1978;93:62–66. doi: 10.1016/s0022-3476(78)80601-5. [DOI] [PubMed] [Google Scholar]
- 19.Colan SD, Shirali G, Margossian R, Gallagher D, Altmann K, Canter C, Chen S, Golding F, Radojewski E, Camitta M, Carboni M, Rychik J, Stylianou M, Tani LY, Selamet Tierney ES, Wang Y, Sleeper LA. The Ventricular Volume Variability study of the Pediatric Heart Network: study design and impact of beat averaging and variable type on the reproducibility of echocardiographic measurements in children with chronic dilated cardiomyopathy. J Am Soc Echocardiogr. 2012;25:842–854. doi: 10.1016/j.echo.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zoghbi WA, Enriquez-Sarano M, Foster E, Grayburn PA, Kraft CD, Levine RA, Nihoyannopoulos P, Otto CM, Quinones MA, Rakowski H, Stewart WJ, Waggoner A, Weissman NJ. Recommendations for evaluation of the severity of native valvular regurgitation with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr. 2003;16:777–802. doi: 10.1016/S0894-7317(03)00335-3. [DOI] [PubMed] [Google Scholar]
- 21.Breiman L. Classification and Regression Trees. Belmont, CA: Wadsworth International Group; 1984. [Google Scholar]
- 22.Levy WC, Mozaffarian D, Linker DT, Sutradhar SC, Anker SD, Cropp AB, Anand I, Maggioni A, Burton P, Sullivan MD, Pitt B, Poole-Wilson PA, Mann DL, Packer M. The Seattle Heart Failure Model: prediction of survival in heart failure. Circulation. 2006;113:1424–1433. doi: 10.1161/CIRCULATIONAHA.105.584102. [DOI] [PubMed] [Google Scholar]
- 23.Aaronson KD, Schwartz JS, Chen TM, Wong KL, Goin JE, Mancini DM. Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation. Circulation. 1997;95:2660–2667. doi: 10.1161/01.cir.95.12.2660. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes FP, Manlhiot C, McCrindle BW, Mertens L, Kantor PF, Friedberg MK. Usefulness of mitral regurgitation as a marker of increased risk for death or cardiac transplantation in idiopathic dilated cardiomyopathy in children. Am J Cardiol. 2011;107:1517–1521. doi: 10.1016/j.amjcard.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 25.Azevedo VM, Albanesi Filho FM, Santos MA, Castier MB, Tura BR. How can the echocardiogram be useful for predicting death in children with idiopathic dilated cardiomyopathy? Arq Bras Cardiol. 2004;82:505–514. doi: 10.1590/s0066-782x2004000600003. [DOI] [PubMed] [Google Scholar]
- 26.Singh TP, Sleeper LA, Lipshultz S, Cinar A, Canter C, Webber SA, Bernstein D, Pahl E, Alvarez JA, Wilkinson JD, Towbin JA, Colan SD. Association of left ventricular dilation at listing for heart transplant with postlisting and early posttransplant mortality in children with dilated cardiomyopathy. Circ Heart Fail. 2009;2:591–598. doi: 10.1161/CIRCHEARTFAILURE.108.839001. [DOI] [PubMed] [Google Scholar]
- 27.Tang WH, Shrestha K, Mullens W, Borowski AG, Martin MG, Troughton RW, Klein AL. Impact of left ventricular remodeling on diagnostic and prognostic value of tissue Doppler indices in chronic systolic heart failure. J Card Fail. 2011;17:128–134. doi: 10.1016/j.cardfail.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Tada H, Toide H, Naito S, Kurosaki K, Ito S, Miyaji K, Yamada M, Okaniwa H, Kobayashi Y, Maruyama H, Higuchi R, Nogami A, Oshima S, Taniguchi K. Tissue Doppler imaging and strain Doppler imaging as modalities for predicting clinical improvement in patients receiving biventricular pacing. Circ J. 2005;69:194–200. doi: 10.1253/circj.69.194. [DOI] [PubMed] [Google Scholar]
- 29.Hollander SA, Bernstein D, Yeh J, Dao D, Sun HY, Rosenthal D. Outcomes of children following a first hospitalization for dilated cardiomyopathy. Circ Heart Fail. 2012;5:437–443. doi: 10.1161/CIRCHEARTFAILURE.111.964510. [DOI] [PubMed] [Google Scholar]
- 30.Brun P, Tribouilloy C, Duval AM, Iserin L, Meguira A, Pelle G, Dubois-Rande JL. Left ventricular flow propagation during early filling is related to wall relaxation: a color M-mode Doppler analysis. J Am Coll Cardiol. 1992;20:420–432. doi: 10.1016/0735-1097(92)90112-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table. Online supplement presenting all candidate predictors assessed for disease progression. Mean±SD or Frequency (%) shown unless otherwise noted.