

Abstract
Background.
A substantial proportion of the general population has low lung function, and lung function is known to decrease as we age. Low lung function is a feature of several pulmonary disorders, such as uncontrolled asthma and chronic obstructive pulmonary disease. The objective of this study is to investigate the association of polymorphisms in asthma and chronic obstructive pulmonary disease candidate genes with rates of lung function decline in a general population sample of aging men.
Methods.
We analyzed data from a cohort of 1,047 Caucasian men without known lung disease, who had a mean of 25 years of lung function data, and on whom DNA was available. The cohort was randomly divided into two groups, and we tested a total of 940 single-nucleotide polymorphisms in 44 asthma and chronic obstructive pulmonary disease candidate genes in the first group (testing cohort, n = 545) for association with change in forced expiratory volume in 1 second over time.
Results.
One hundred nineteen single-nucleotide polymorphisms that showed nominal associations in the testing cohort were then genotyped and tested in the second group (replication cohort, n = 502). Evidence for association from the testing and replication cohorts were combined, and after adjustment for multiple testing, seven variants of three genes (DPP10, NPSR1, and ADAM33) remained significantly associated with change in forced expiratory volume in 1 second over time.
Conclusions.
Our findings that genetic variants of genes involved in asthma and chronic obstructive pulmonary disease are associated with lung function decline in normal aging participants suggest that similar genetic mechanisms may underlie lung function decline in both disease and normal aging processes.
Key Words. Genetics, Pulmonary, Normative aging, Successful aging.
For healthy nonsmokers, the forced expiratory volume in 1 second (FEV1), increases from birth and reaches its peak at around the ages of 20 and 25 years (the growth phase), it remains stable until the ages of 30 to 35 years (the plateau phase), and begins to decline with aging (the decline phase) (1–3). In the elderly participants, low lung function is associated with impaired cognitive function, reduced physical activity, and all-cause mortality (4–6). Because a substantial proportion of the general population has unrecognized low lung function, its impact on health and quality of life can easily be underestimated (7).
Segregation studies have suggested a genetic contribution to lung function variability in the general population (8,9). Linkage (10–12) and association (13–15) studies further attempted to localize the genetic loci influencing lung function. Because low lung function is a feature of uncontrolled asthma and chronic obstructive pulmonary disease (COPD), we hypothesized that genetic variants that predispose to asthma and COPD might underlie the rapidity of lung function decline. We conducted a longitudinal lung function study in a cohort of white men having data on DNA and 25 years of lung function analyses, to determine if polymorphisms of asthma and COPD candidate genes are determinants of lung function decline in a healthy aging population.
Methods
The study cohort was a subset of the original Normative Aging Study cohort (16), a longitudinal study of aging established by the Veterans Associations, with recruitment between 1961 and 1970. A total of 1,245 men had lung function data and adequate DNA samples at the time of the study. We removed 198 participants who developed asthma, emphysema, or chronic bronchitis after entry into the cohort, for a total of 1,047 men included in this analysis. Details are provided in the Supplementary Material. The study protocol was approved by the Human Studies Subcommittee of the Department of Veterans Affairs Medical Center and the Institutional Review Board of the Brigham and Women’s Hospital.
Lung function was measured in a standardized manner beginning in 1963 (17). Beginning in 1984, a new spirometer was used along with new standardized protocols that adhered to American Thoracic Society standards for pulmonary function measurement (18), and these protocols were updated subsequently (19,20). Lung function was also expressed as a percent of predicted using spirometric reference values from the third National Health and Nutrition Examination Survey (21). In addition, the proportions of spirometric values that were above and below the lower limit of normal (LLN) at the baseline exam were also determined using LLN equations from National Health and Nutrition Examination Survey (21). Further details are provided in the Supplementary Material. The phenotype of interest was lung function decline defined as the change in FEV1 between two consecutive visits over the number of years between the two visits:
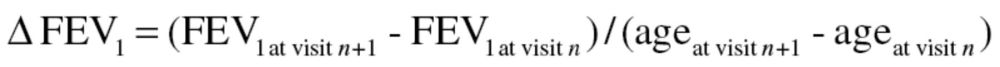 |
Where, n is visit number (1 ≤ n ≤ 13).
Candidate genes were selected based on their known or suspected roles in the pathogeneses of asthma and COPD, from review of the existing literature performed by two of the authors (A.H.P. and A.A.L.). In particular, they are genes that have been identified to be asthma or asthma-related phenotype genes through positional cloning or candidate gene association testing. Single-nucleotide polymorphisms (SNPs) in 44 candidate genes were selected for investigation if they were either (1) tagging SNPs with r 2 < .80 and minor allele frequency >5%, covering 5 kb upstream and downstream of the first and last exons of each gene (2), nonsynonymous amino acid change with minor allele frequency >1%, or (3) known associated variants with asthma, COPD, and related phenotypes. Genotyping of SNPs for the testing cohort (TC) was carried out using the Illumina BeadStation 500G (San Diego, CA). Genotyping for the replication cohort (RC) was carried out using one of two platforms, the Sequenom MassArray MALDI-TOF mass spectrometer (Sequenom, San Diego, CA) and the TaqMan 5′ exonuclease assays (Applied Biosystems, Foster City, CA) (22). Details of genotyping are provided in the Supplementary Material. Briefly, 1,085 SNPs were selected for genotyping; 78 SNPs were removed during the quality control process (see Supplementary Material for details), and 64 SNPs were removed due to deviation from Hardy–Weinberg equilibrium (23), leaving 943 SNPs. An additional three SNPs were removed due to genotyping rates less than 90% (ie, <90% of the samples were successfully genotyped). Thus, a total of 940 SNPs were analyzed in the TC (see below).
A two-stage testing–replication strategy was adopted where the study population was divided into two subsets: a TC and a RC. The entire SNP set of 940 was tested for associations between individual SNP and lung function decline in TC in the presence of potential confounders (height, age, smoking status, and intensity of cigarette smoking in pack–years, as time-varying covariates). Mixed models were implemented in the Mixed Procedure in SAS (SAS Institute, Inc., Cary, NC), using the “Repeated” statement to account for correlations between repeated observations on each participant. Further details of statistical methods can be found in the Supplementary Material. SNPs associated in the TC at p ≤ .1 under either additive or recessive models were genotyped in RC. Associated SNPs were tested by the same statistical method and under the same genetic model as those observed in TC. Analyses in RC were constrained to having the same direction of effect as in TC, thus one-sided testing was employed in the RC. Due to the consistency in direction between associations in TC and RC, two-sided p values from TC and one-sided p values from RC were combined using Fisher’s method (24). We tested 940 SNPs in TC under the additive and recessive models. Because these genotype models are not independent of each other, Bonferroni correction was applied for only 940 SNP tests. Population stratification was assessed using PLINK (see Supplementary Material) (25), and no significant stratification was observed.
Results
Population Characteristics
The study sample consisted of 1,047 participants, who had DNA samples, smoking history, and lung function data, and were randomly divided into 545 and 502 participants for TC and RC, respectively. The two cohorts were comparable in demographic, lung function, and smoking characteristics (Table 1). At baseline, the mean age was 41.3 years (SD = 8.2) in the TC, and was 41.0 years (SD = 7.8) in the RC. Mean FEV1, forced vital capacity (FVC), and FEV1/FVC as percent of predicted were 97.93%, 97.35%, and 100.54% for participants in TC, respectively; and 96.96%, 96.45%, and 100.49% for participants in RC, respectively. Although none of the men had a diagnosis of any lung disease at baseline, in TC, 45 men (8.3%) had FEV1 values that were below the LLN, 43 men (8.9%) had FVC values below the LLN, and 18 (3.3%) men had FEV1/FVC values below the LLN. For RC, 33 (6.6%) had FEV1 values that were below the LLN, 40 (8.0%) had FVC values that were below the LLN, and 19 (3.8%) men had FEV1/FVC values below the LLN. At baseline, the proportions of never, current, and former smokers were also comparable in both cohorts. The mean number of follow-up visits were 8.5 (SD = 2.3) in the TC and 8.6 (SD = 2.2) visits in the RC, with a mean of 24.42 years (± 6.70) of follow-up for the TC and 25.42 years (± 6.20) of follow-up in the RC.
Table 1.
Baseline Population Characteristics
| Testing Cohort | Replication Cohort | |
|---|---|---|
| Number of participants | 545 | 502 |
| Age (y), mean (SD) | 41.3 (8.2) | 41.1 (7.8) |
| Age range (y) | 23.1–70.1 | 23.4–65.4 |
| FEV1% predicted, mean (SD) | 97.93 (12.30) | 96.72 (11.00) |
| FEV1 (L), mean (SD) | 3.96 (0.59) | 3.95 (0.56) |
| FVC %predicted, mean (SD) | 97.35 (10.98) | 96.45 (10.39) |
| FVC (L), mean (SD) | 4.97 (0.71) | 4.96 (0.70) |
| FEV1/FVC, mean (SD) | 0.80 (0.06) | 0.80 (0.58) |
| FEV1/FVC %predicted, mean (SD) | 100.54 (7.14) | 100.49 (7.37) |
| Smoking status (%) | ||
| Nonsmoker | 37.2 | 31.0 |
| Current smoker | 30.4 | 35.5 |
| Former smoker | 32.3 | 33.5 |
| Number of follow-up visits (N), mean (SD) | 7.4 (2.3) | 7.7 (2.2) |
| Number of years of follow-up (y), mean (SD) | 24.64 (6.70) | 25.42 (6.20) |
Notes: FEV1 = forced expiratory volume in 1 s; FVC = forced vital capacity.
SNP Associations
A total of 940 SNPs from 44 candidate genes were genotyped and analyzed in the TC under both additive and recessive models (see Supplementary Table S1). A total of 194 SNPs were associated with change in FEV1 at p value ≤ .1. Twenty-four of the 194 SNPs were associated under both additive and recessive models, and the association under the recessive model for all the 24 SNPs had smaller p values, hence for these 24 SNPs, only the recessive model was tested in the RC. After removal of 11 SNPs that were out of Hardy–Weinberg equilibrium, a total of 119 SNPs were successfully genotyped and analyzed in the RC. Fifty-seven SNPs were found to have the same direction of effect in both the TC and RC, and evidence for association was combined (Supplementary Table S4). A total of seven SNPs remained statistically significant for their association with decline in FEV1, after Bonferroni adjustment (Table 2 and Supplementary Table S5).
Table 2.
Effect Size of Associated SNPs and FEV1 decline*
| Gene | SNP | Genetic Model† | Genotype | Effect Estimate in TC (SE)‡ | p Value | Effect Estimate in RC (SE)‡ | p Value | Combined p Value |
|---|---|---|---|---|---|---|---|---|
| DPP10 | rs17783638 | Recessive | CC | 11.96 (1.63) | 2.93×10−13 | 2.80 (5.80) | .32 | 2.86×10−12 |
| rs17636812 | Recessive | GG | 21.36 (3.89) | 4.35×10−8 | 0.83 (2.92) | .38 | 3.17×10−7 | |
| rs4849383 | Recessive | GG | 4.93 (2.09) | 0.02 | 11.94 (1.81) | 2.31×10−11 | 1.25×10−11 | |
| rs4849384 | Recessive | CC | 17.93 (5.56) | 0.001 | 16.62 (2.56) | 5.40×10−11 | 2.19×10−12 | |
| NPSR1 | rs323917 | Recessive | GG | 16.58 (1.79) | 3.09×10−20 | 10.48 (1.86) | 9.33×10−9 | 1.860×10−26 |
| rs17170012 | Recessive | GG | 22.85 (2.84) | 1.37×10−15 | 4.71 (2.68) | .04 | 2.07×10−15 | |
| ADAM33 | rs3918395 | Recessive | AA | −12.73 (3.10) | 4.24×10−5 | −6.76 (4.69) | .07 | 4.34×10−5 |
Notes: FEV1 = forced expiratory volume in 1 s; RC = replication cohort; SNP = single-nucleotide polymorphism; TC = testing cohort.
*These associations remained significant after Bonferroni correction of 940 tests.
†Genetic model refers to the coding of alleles in the mixed models, as explained in the Methods section.
‡Effect estimates obtained from mixed effects models. Effect estimates are displayed as mL/y change in FEV1; reference group = homozygotes plus heterozygotes of the common allele (recessive model). Positive estimates denote slower decline associated with the genotype, while negative estimates denote faster decline.
Table 2 presents the results of the mixed effects modeling of lung function decline. Under a recessive model, four variants of DPP10 (rs17783638, rs17638612, rs4849383, and rs4849384), two variants of neuropeptide S receptor 1 (NPSR1; rs323917 and rs17170012), and a variant of ADAM33 (rs3918395) were associated with rates of lung function decline. Homozygosity for the minor alleles of DPP10 and NPSR1 variants conferred a slower rate of FEV1 decline compared with carriers of the major allele (also see Supplemetary Material). In contrast, homozygosity of the ADAM33 variant was associated with a faster rate of lung function decline.
In addition, the same variants were also tested for association with rates of FVC decline (Table 3), defined in a similar way as for FEV1 decline. Two of the seven FEV1 decline-associated SNPs (rs177838 and rs484938 of DPP10) were associated with rate of FVC decline (p < .05), and both of these had effects in the same direction as that for the FEV1 analysis.
Table 3.
Effect Size of Associated SNPs and FVC Decline
| Gene | SNP | Genetic Model* | Genotype | Effect Estimate in TC (SE)† | p Value | Effect Estimate in RC (SE)† | p Value | Combined p Value |
|---|---|---|---|---|---|---|---|---|
| DPP10 | rs17783638 | Recessive | CC | 16.37 (3.59) | 5.29×10−6 | 5.63 (4.13) | .08 | 7.17×10−6‡ |
| DPP10 | rs4849383 | Recessive | GG | 10.52 (1.84) | 1.29×10−8 | 27.88 (2.29) | 9.53×10−39 | 1.17×10−39‡ |
Notes: FEV1 = forced expiratory volume in 1 s; FVC = forced vital capacity; RC = replication cohort; SNP = single-nucleotide polymorphism; TC = testing cohort.
*Genetic model refers to the coding of alleles in the mixed models, as explained in the Methods section.
†Effect estimates obtained from mixed effects models. Effect estimates are displayed as mL/y change in FEV1; reference group = homozygotes plus heterozygotes of the common allele (recessive model). Positive estimates denote slower decline associated with the genotype, while negative estimates denote faster decline.
‡These associations remained significant after Bonferroni correction of 940 tests.
We performed additional analyses in a subset of men with complete information on baseline occupation and development of competing diagnoses of cancer, cardiovascular disease, asthma and/or COPD, and hospitalization for pneumonia during follow-up. In general, results were similar to the original analysis, although p values were not as small given the smaller sample size. Two SNPs (rs4849384 in DPP10 and rs17170012 in NPSR1) were significantly associated with lung function decline in TC and RC (Supplementary Table S6). Likely because this was a smaller sample (TC: n = 452; RC: n = 383), the results in Supplementary Table S6 would not survive adjustment for multiple testing.
Discussion
We conducted a candidate gene analysis and report genetic variants influencing lung function decline in a general population sample of men that was initially recruited to study healthy aging. Findings of genetic association with lung function decline in general populations have been reported using longitudinal data (14,15), yet the number of candidate genes and their SNPs investigated were limited in these earlier studies. One recent study has investigated genetic predictors of lung function decline in an elderly cohort (26), and another recent study investigated genetic predictors of lung function decline in active smokers (27). However, these two recent studies had less than 10 years of follow-up, and candidate genes in these more recent studies focused on antioxidant genes and smoking-related genes. To date, only one genome-wide study of lung function decline has been published and that study had no findings that reached genome-wide statistical significance (28). However, in that study, the longest mean follow-up time was only 14.6 (±7.2 years), and most of the cohorts only had two lung function measures, and there was likely significant heterogeneity in the studies that precluded more robust results. The strength of our study derives from the availability of multiple, repeated lung function and predictor information gathered over a mean of 25 years in one cohort, allowing us to account for changes in exposures (ie, age and smoking) and physical characteristics (ie, height) over time.
We found DPP10, NPSR1, and ADAM33 to be associated with lung function decline and these genes have all been shown to be associated with asthma in multiple populations; ADAM33 was implicated as an asthma gene in 17 populations, DPP10 in 4, and NPSR1 (G-protein receptor 154) in 9, as summarized in Michel and colleagues (29). The four associated variants of DPP10, which encodes dipeptidyl peptidase X, all reside between exons 1 and 2, an area where alternate splicing occurs to encode multiple isoforms of different length and where association with asthma has been observed (30). For example, a variant located in intron 1 (rs13011555) had been found to be associated with FEV1 in Caucasian adults (The British 1958 Birth Cohort) (31). We did not genotype this specific variant in our study, and none of the SNPs that we did genotype tags this variant, based on data from HapMap (http://hapmap.ncbi.nlm.nih.gov/). The significant yet small effect sizes estimated in our study and the British 1958 Birth Cohort suggest that DPP10 is one of the many genes influencing lung function. DPP10 belongs to the dipeptidyl peptidase family and it is a structural homology of DPP4, whose function as a serine protease has been better characterized (32,33). In a rat model of asthma, DPP10 was found to be expressed in the bronchi, trachea, and leukocytes and elevated in asthmatic as compared with control animals (34). However, the function of DPP10 in lung function remains unclear. The limited knowledge of its function is its association with voltage-gated potassium channels and that it plays a role in current kinetics (35). Hence, one can speculate that DPP10 may influence lung function by regulating the electrophysiological properties of excitable cells and altering neuronal regulation of the lungs (36).
ADAM33 is a member of the metalloprotease-disintegrin family that proteolytically removes extracellular domains of various transmembrane proteins to induce shedding of cell surface ligands and downstream signaling pathways, mediate cell–cell and cell–matrix interactions (37). Polymorphisms in this gene have been associated with faster rates of lung function decline in both asthmatic participants (38) and general population participants (15). In the airways, the expression pattern of ADAM33 differed between asthmatic and control individuals (39), with almost complete absence of expression in the smooth muscle of control participants and weak expression in the epithelium as compared with asthmatic participants.
In addition to ADAM33 and DPP10, NPSR1, also known as G-protein receptor 154 is the third asthma gene discovered through positional cloning (40,41) and found to be associated with rates of FEV1 decline in our study. NPSR1 is a seven-transmembrane G-protein–coupled receptor and the expression pattern of NPSR1 in the lungs has been inconsistently reported. Although immunocytochemistry staining of bronchial biopsy tissue of asthmatic and nonasthmatic participants has shown that NPSR1 protein is expressed in bronchial epithelium cells of asthmatic and not in control participants (42), messenger RNA expression of NPSR1 was low to absent for various human airway-derived epithelial cells, smooth muscle cells. and fibroblasts (43). In another study, messenger RNA expression of NPSR1 has been found to be elevated in the ciliated cells of the airway epithelium of asthmatic participants compared with controls (41). Polymorphisms of NPSR1 have been found to be associated with asthma or related phenotypes (eg, IgE, airway responsiveness) in several populations (41,44,45). Interestingly, however, in two murine studies of experimental asthma using NPSR1-deficient mice, there was no impact of NPSR1 on airway inflammation, airway hyperresponsiveness, and the development of asthma (43,46). Yet NPSR1 appeared to influence respiratory function in mice (46). When treated with neuropeptide S intracerebroventricularly, wild-type mice had increased respiratory frequency and decreased tidal volume from baseline as compared with NPSR1-deficient mice. A total of two NPSR1 variants was found to be associated with rates of FEV1 decline in this study, after adjustment for multiple testing. Homozygotes of the rare alleles for all variants confer a slower rate of FEV1 decline. The associated variants all reside in introns and have unknown biological functions to date. Variant rs323917 has previously been shown to be associated with airway hyperresponsiveness, with the rare allele associated with increased airway hyperresponsiveness (44). Increased airway hyperresponsiveness has been recognized as a risk factor for longitudinal lung function decline in several cohorts (47–49), including this cohort (48).
In addition to rates of FEV1 decline, two of the seven variants also showed significant association with rates of FVC decline, suggesting that the mechanisms which DPP10 and NPSR1 operate under, affect rates of lung function decline potentially through both airway caliber and lung volume.
It is of interest to note that the associations that survived multiple-testing adjustment (Table 2) and other top associations in Supplementary Table S4 were those in recessive models of the candidate genes. For the SNPs in Table 2, aside from SNP rs17636812 in DPP10, where the recessive genotype category in TC and RC combined was comprised of 42 men, all other recessive genotype categories for the other SNPs were comprised of 10 participants or less. The additive genetic models of these SNPs did not reach statistical significance. Thus, it appears that the genetic effects of these genes are only expressed in a small subset of populations that are homozygous for the particular SNP. Additionally, this suggests that factors other than genetics are important predictors of lung function decline, and is consistent with the fact that the only genome-wide association study on lung function decline (28) did not find any statistically significant results. It is possible that gene-by-environment interaction analyses might yield more results.
This study has its limitations. Because the cohort is composed of Caucasian men, associations detected in this cohort may not be generalizable to women and non-Caucasians. In addition, because the mean age of this cohort at baseline was 41 years, genetic factors which we have identified to influence lung function decline may not be the same as those that influence growth in younger populations. Because genotyping for this project began several years ago, we were unable to include variants from genes that have recently been associated with asthma (eg, ORMDL3, PDE4D), COPD (CHRNA 3/5), or lung function (GSTO2 and IL6R). Nevertheless, our findings that genetic variants of genes involved in asthma and COPD pathogenesis are associated with lung function decline in normal aging participants suggest that similar genetic mechanisms underlie lung function decline in both disease and normal aging processes. Although longitudinal studies are generally thought to provide more accurate estimates of lung function decline, these types of studies also have problems such as learning effects, loss to follow-up, variability over time of spirometers and technicians. We have attempted to minimize these effects where possible. Although spirometry was performed in a standardized manner from the inception of the study in the 1960s (17), standardization was modified beginning in 1984 to comply with the recommendations from the American Thoracic Society (18), and a different spirometer was used. We accounted for this change by creating a variable that identified the method and adjusted this in our analyses. Although the standardization method itself was a significant determinant of lung function decline, it did not affect the observed associations between the SNPs and lung function decline, whether the variable was in the model or not. Additionally, we used standardized protocols for measuring spirometry to minimize variability between technicians.
Another limitation is the comprehensiveness of coverage of the genome. We took a candidate gene approach in this study, thus, this was only limited to genes that have known associations to either asthma or COPD. Other genes that have not been associated with these two disorders may have effects on lung function decline. Although a genome-wide association study may have given greater coverage of the genome, other current methods, such as exome sequencing, would give more comprehensive coverage and result in more novel findings in future studies.
In summary, we have found that variants in seven asthma and COPD candidate genes were determinants of lung function decline over a mean of 25 years in a cohort of men. Our results suggest that mechanisms involved in the development of asthma and COPD are operating in the normal process of decline in lung function seen with aging.
Supplementary Material
Supplementary material can be found at: http://biomedgerontology.oxfordjournals.org/
Funding
This work was funded by the National Institutes of Health (AG 027014); Dr. A.H.P. was supported by a grant from The Croucher Foundation; Dr. D.S. was supported by a VA Research Career Scientist award; The VA Normative Aging Study is supported by the Cooperative Studies Program/Epidemiology Research and Information Center of the U.S. Department of Veterans Affairs, and is a component of the Massachusetts Veterans Epidemiology Research and Information Center, Boston, Massachusetts.
References
- 1. Kerstjens HA, Rijcken B, Schouten JP, Postma DS. Decline of FEV1 by age and smoking status: facts, figures, and fallacies. Thorax. 1997;52:820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127:725–734 [DOI] [PubMed] [Google Scholar]
- 3. Tager IB, Segal MR, Speizer FE, Weiss ST. The natural history of forced expiratory volumes. Effect of cigarette smoking and respiratory symptoms. Am Rev Respir Dis. 1988;138:837–849 [DOI] [PubMed] [Google Scholar]
- 4. Anstey KJ, Windsor TD, Jorm AF, Christensen H, Rodgers B. Association of pulmonary function with cognitive performance in early, middle and late adulthood. Gerontology. 2004;50(4):230–234 [DOI] [PubMed] [Google Scholar]
- 5. Mannino DM, Buist AS, Petty TL, Enright PL, Redd SC. Lung function and mortality in the United States: data from the First National Health and Nutrition Examination Survey follow up study. Thorax. 2003;58:388–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weuve J, Glymour MM, Hu H, et al. Forced expiratory volume in 1 second and cognitive aging in men. J Am Geriatr Soc. 2011;59:1283–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannino DM, Gagnon RC, Petty TL, Lydick E. Obstructive lung disease and low lung function in adults in the United States: data from the National Health and Nutrition Examination Survey, 1988–1994. Arch Intern Med. 2000;160(11):1683–1689 [DOI] [PubMed] [Google Scholar]
- 8. Givelber RJ, Couropmitree NN, Gottlieb DJ, et al. Segregation analysis of pulmonary function among families in the Framingham Study. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1445–1451 [DOI] [PubMed] [Google Scholar]
- 9. Wilk JB, Djousse L, Arnett DK, et al. Evidence for major genes influencing pulmonary function in the NHLBI family heart study. Genet Epidemiol. 2000;19:81–94 [DOI] [PubMed] [Google Scholar]
- 10. Joost O, Wilk JB, Cupples LA, et al. Genetic loci influencing lung function: a genome-wide scan in the Framingham Study. Am J Respir Crit Care Med. 2002;165:795–799 [DOI] [PubMed] [Google Scholar]
- 11. Wilk JB, DeStefano AL, Arnett DK, et al. A genome-wide scan of pulmonary function measures in the National Heart, Lung, and Blood Institute Family Heart Study. Am J Respir Crit Care Med. 2003;167:1528–1533 [DOI] [PubMed] [Google Scholar]
- 12. Wilk JB, DeStefano AL, Joost O, et al. Linkage and association with pulmonary function measures on chromosome 6q27 in the Framingham Heart Study. Hum Mol Genet. 2003;12:2745–2751 [DOI] [PubMed] [Google Scholar]
- 13. Guénégou A, Leynaert B, Bénessiano J, et al. Association of lung function decline with the heme oxygenase-1 gene promoter microsatellite polymorphism in a general population sample. Results from the European Community Respiratory Health Survey (ECRHS), France. J Med Genet. 2006;43:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Imboden M, Downs SH, Senn O, et al. ; SAPALDIA Team. Glutathione S-transferase genotypes modify lung function decline in the general population: SAPALDIA cohort study. Respir Res. 2007;8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Diemen CC, Postma DS, Vonk JM, Bruinenberg M, Schouten JP, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. Am J Respir Crit Care Med. 2005;172:329–333 [DOI] [PubMed] [Google Scholar]
- 16. Bell B, Rose DL, Damon H. The Normative Aging Study: an interdisciplinary and longitudinal study of health and aging. Aging Hum Devel. 1972;3:5–17 [Google Scholar]
- 17. Kory RC, Callahan R, Boren HG, Syner JC. The Veterans Administration-Army cooperative study of pulmonary function. I. Clinical spirometry in normal men. Am J Med. 1961;30:243–258 [DOI] [PubMed] [Google Scholar]
- 18. ATS statement--Snowbird workshop on standardization of spirometry. Am Rev Respir Dis. 1979;119(5):831–838 [DOI] [PubMed] [Google Scholar]
- 19. Standardization of spirometry--1987 update. Statement of the American Thoracic Society. Am Rev Respir Dis. 1987;136(5):1285–1298 [DOI] [PubMed] [Google Scholar]
- 20. Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med. 1995;152(3):1107–1136 [DOI] [PubMed] [Google Scholar]
- 21. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159:179–187 [DOI] [PubMed] [Google Scholar]
- 22. Lee LG, Connell CR, Bloch W. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 1993;21:3761–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–372 [PubMed] [Google Scholar]
- 24. Fisher RA. Statistical Methods for Research Workers. 11th ed New York: Hafner; 1950 [Google Scholar]
- 25. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang W, Bentley AR, Kritchevsky SB, et al. ; Health ABC study. Genetic variation in antioxidant enzymes, cigarette smoking, and longitudinal change in lung function. Free Radic Biol Med. 2013;63:304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohamed Hoesein FA, Wauters E, Janssens W, et al. Variants in the 15q24/25 locus associate with lung function decline in active smokers. PLoS One. 2013;8:e53219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imboden M, Bouzigon E, Curjuric I, et al. Genome-wide association study of lung function decline in adults with and without asthma. J Allergy Clin Immunol. 2012;129:1218–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Michel S, Liang L, Depner M, et al. Unifying candidate gene and GWAS Approaches in Asthma. PLoS One. 2010;5:e13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Allen M, Heinzmann A, Noguchi E, et al. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat Genet. 2003;35:258–263 [DOI] [PubMed] [Google Scholar]
- 31. Blakey JD, Sayers I, Ring SM, Strachan DP, Hall IP. Positionally cloned asthma susceptibility gene polymorphisms and disease risk in the British 1958 Birth Cohort. Thorax. 2009;64(5):381–387 [DOI] [PubMed] [Google Scholar]
- 32. Engel M, Hoffmann T, Wagner L, et al. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci USA. 2003;100:5063–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261:466–469 [DOI] [PubMed] [Google Scholar]
- 34. Schade J, Stephan M, Schmiedl A, et al. Regulation of expression and function of dipeptidyl peptidase 4 (DP4), DP8/9, and DP10 in allergic responses of the lung in rats. J Histochem Cytochem. 2008;56:147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cotella D, Radicke S, Cipriani V, et al. N-glycosylation of the mammalian dipeptidyl aminopeptidase-like protein 10 (DPP10) regulates trafficking and interaction with Kv4 channels. Int J Biochem Cell Biol. 2012;44:876–885 [DOI] [PubMed] [Google Scholar]
- 36. Ren X, Hayashi Y, Yoshimura N, Takimoto K. Transmembrane interaction mediates complex formation between peptidase homologues and Kv4 channels. Mol Cell Neurosci. 2005;29:320–332 [DOI] [PubMed] [Google Scholar]
- 37. van Goor H, Melenhorst WB, Turner AJ, Holgate ST. Adamalysins in biology and disease. J Pathol. 2009;219:277–286 [DOI] [PubMed] [Google Scholar]
- 38. Jongepier H, Boezen HM, Dijkstra A, et al. Polymorphisms of the ADAM33 gene are associated with accelerated lung function decline in asthma. Clin Exp Allergy. 2004;34:757–760 [DOI] [PubMed] [Google Scholar]
- 39. Foley SC, Mogas AK, Olivenstein R, et al. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J Allergy Clin Immunol. 2007;119:863–871 [DOI] [PubMed] [Google Scholar]
- 40. Castro-Giner F, de Cid R, Gonzalez JR, et al. Positionally cloned genes and age-specific effects in asthma and atopy: an international population-based cohort study (ECRHS). Thorax. 2010;65:124–131 [DOI] [PubMed] [Google Scholar]
- 41. Laitinen T, Polvi A, Rydman P, et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–304 [DOI] [PubMed] [Google Scholar]
- 42. Orsmark-Pietras C, Melén E, Vendelin J, et al. ; PARSIFAL Genetics Study Group. Biological and genetic interaction between tenascin C and neuropeptide S receptor 1 in allergic diseases. Hum Mol Genet. 2008;17:1673–1682 [DOI] [PubMed] [Google Scholar]
- 43. Allen IC, Pace AJ, Jania LA, et al. Expression and function of NPSR1/GPRA in the lung before and after induction of asthma-like disease. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1005–L1017 [DOI] [PubMed] [Google Scholar]
- 44. Feng Y, Hong X, Wang L, et al. G protein-coupled receptor 154 gene polymorphism is associated with airway hyperresponsiveness to methacholine in a Chinese population. J Allergy Clin Immunol. 2006;117:612–617 [DOI] [PubMed] [Google Scholar]
- 45. Melén E, Bruce S, Doekes G, et al. ; PARSIFAL Genetics Study Group. Haplotypes of G protein-coupled receptor 154 are associated with childhood allergy and asthma. Am J Respir Crit Care Med. 2005;171:1089–1095 [DOI] [PubMed] [Google Scholar]
- 46. Zhu H, Perkins C, Mingler MK, Finkelman FD, Rothenberg ME. The role of neuropeptide S and neuropeptide S receptor 1 in regulation of respiratory function in mice. Peptides. 2011;32:818–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hodgins P, Henneberger PK, Wang ML, Petsonk EL. Bronchial responsiveness and five-year FEV1 decline: a study in miners and nonminers. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1390–1396 [DOI] [PubMed] [Google Scholar]
- 48. O’Connor GT, Sparrow D, Weiss ST. A prospective longitudinal study of methacholine airway responsiveness as a predictor of pulmonary-function decline: the Normative Aging Study. Am J Respir Crit Care Med. 1995;152:87–92 [DOI] [PubMed] [Google Scholar]
- 49. Rijcken B, Schouten JP, Xu X, Rosner B, Weiss ST. Airway hyperresponsiveness to histamine associated with accelerated decline in FEV1. Am J Respir Crit Care Med. 1995;151:1377–1382 [DOI] [PubMed] [Google Scholar]