

Abstract
Three cDNAs, termed EpC1, TPxEg and EgG5, were isolated by immunoscreening from an Echinococcus granulosus cDNA library. The recombinant phages exhibited strong reactivity with sera from humans with confirmed cystic echinococcosis (CE) and with sera from mice infected with E. granulosus oncospheres. The cDNAs were subcloned into a pET vector, expressed as fusion proteins tagged with GST and affinity purified against the GST tag. Of the three recombinant proteins, EpC1 achieved the highest performance for serodiagnosis of CE in Western blot analysis using a panel of clinically defined human sera to initially address the sensitivity and specificity of the molecules. The protein yielded an overall sensitivity of 92.2% and specificity of 95.6%, levels unprecedented taking into account the large panel of 896 human sera that were tested. The strategy used may also prove suitable for improved immunodiagnosis of other parasitic infections.
Keywords: Echinococcus; Recombinant proteins; Blotting, Western
Introduction
Parasites, especially helminth worms, generally produce longstanding chronic infections. The elevated levels of serum antibodies that are generated during the course of infection can provide a sound basis for the development of sensitive and specific serodiagnostic methods for parasite detection. Nevertheless, serodiagnosis of a number of helminthic infections, such as cystic echinococcosis (CE), remains a challenge. The major problem is that crude or secretory/excretory proteins from the targeted parasite are often cross-reactive with sera from humans and animals infected with other parasites; this results in low specificity in the performance of serodiagnostic assays.
CE is a near-cosmopolitan zoonosis caused by larval stages of the dog tapeworm (cestode) Echinococcus granulosus (family Taeniidae; genus Echinococcus). Larval infection (also termed hydatid disease; hydatidosis) is characterized by long term growth of metacestode (hydatid) cysts in the internal organs (mainly the liver and lungs) of humans and herbivorous (sheep, cattle, pigs goats, camels) intermediate hosts. Definitive hosts, such as dogs, wolves and foxes, harbour the adult tapeworms in their intestines. Periodically these worms release segments and eggs that are passed in the faeces into the external environment. Eggs are ingested by humans and ungulates and the oncosphere larvae that are released penetrate the gut and are transported through blood or lymph to target organs (generally the liver or lungs). There they develop into hydatid cysts. Carnivores are infected by eating ungulate offal harbouring hydatid cysts containing viable brood capsules and protoscoleces.
Early diagnosis of CE can provide significant improvements in the quality of the management and treatment of the disease. Generally, the early phases of infection are asymptomatic, so methods that are relatively easy to use and that are cheap are required for large scale screening of populations at high risk (1). Immunodiagnosis is considered to be a useful approach that can also confirm clinical findings based on the identification of cyst structures by imaging techniques such as ultrasonography (2-6).
E. granulosus hydatid cyst fluid (HCF) antigens are the usual source of antigenic material for immunodiagnostic assays (6). However, there are difficulties associated with the currently available tests, related to their lack of sensitivity and specificity, and problems with the standardization of their use (7). Antibody cross-reactivity with antigens from other parasites, notably other taeniid cestodes, is a major problem when using HCF antigens in CE immunodiagnosis. It has been suggested that CE serology may be improved by the use of recombinant proteins, combining several defined antigens (including synthetic peptides) and the design of new E. granulosus-specific peptides that react with otherwise false-negative sera (6). We recently reported a novel DNA sequence from E. granulosus (termed EpC1) that was isolated from a protoscolex (larval) cDNA library by immunoscreening (8). The sequence was expressed in bacteria, purified and the value of the recombinant protein in diagnosis of human CE assessed. Here we report further on the strategy that we used to isolate this and other E. granulosus cDNAs, their expression and purification, and evaluation of their serodiagnostic performance.
Materials and Methods
Parasites
Sheep hydatid cysts were obtained from a slaughterhouse in Urumqi, Xinjiang, PR China. Hydatid cyst fluid (HCF) was collected from fertile cysts with a syringe and used for crude antigen preparation (see below). Then, brood capsules and protoscoleces were aspirated and washed 10 times with phosphate buffered saline (PBS). The brood capsules and protoscoleces were aliquoted into tubes and stored in liquid nitrogen until used.
Activated oncospheres for infection of mice
Mature adult worms were collected from dogs experimentally infected with fresh protoscoleces at Xinjiang Veterinary Research Institute, Urumqi. Eggs were released by homogenizing the worms in an electric blender. The homogenate was passed through a 132 mm sieve and the sheared worm material was discarded after thoroughly rinsing the worm tissues through the sieve. The eggs were further washed and retained on a 20 mm mesh. The washed eggs were stored in PBS containing 1000 i.u./ml benzyl penicillin and 1000 mg/ml streptomycin sulphate at 4°C (9).
Eggs were incubated in 50 ml screw-capped tubes at 37°C for 45 min in a sterile solution of 1% (w/v) pepsin (Sigma, St. Louis, MO, 1:2500) and 1% (v/v) HCl in 0.85% NaCl (w/v). After centrifugation (500 × g, 5 min), the pepsin solution was decanted. The eggs were washed once with PBS and incubated in a sterile solution of 1% (w/v) pancreatin (Sigma, 4 × U.S. Pharmacopeia.), 1% (w/v) NaHCO3 and 5% (v/v) sterile sheep bile. The oncospheres were checked every 2 min with a microscope until all the oncospheres had been released from embryonic membranes. The oncospheres were pelleted by centrifugation (1,000 × g, 5 min). The supernatant was discarded and the oncospheres were washed twice with Hank’s buffer.
The oncospheres were further purified by density-gradient separation with 100% Percoll (Sigma) (10). After washing 3 times with PBS, the supernatant was discarded and the oncospheres were used directly to infect mice.
Infection of mice and preparation of mouse infection sera (MIS)
Twenty-six (15 females, 11 males, 6-8 weeks old) Chinese Kunming White (CKW) mice were used for challenge infection with hatched/activated oncospheres (AO) (9) and production of antisera. The mice were infected with 2,500 AO injected intraperitoneally (i.p.), 500 AO injected intravenously (i.v.) or by a combination of both routes. The mice were sacrificed using CO2 at 26 weeks after the first infection and the fluid-filled cysts (none had protoscoleces) present in the peritoneal and/or thoracic cavities were individually counted and measured for size. One individual was found to be uninfected. Blood samples were collected by heart puncture from the other 25 mice and sera were separated from clots by centrifugation at 12,000 × g for 3 min after placing the samples at 4°C for overnight. The sera were stored at -20°C until use.
Preparation of hyper immune rabbit serum (HIRS) raised against HCF
HCF was collected from fertile hydatid cysts as described above and dialyzed against PBS (10 × the volume of HCF) for 48 h at 4°C, with 8 changes of PBS. The HCF was concentrated ten-fold using the Tangential Flow Filtration (TFF) system (Millipore, Bedford, MA) and kept at -80°C until used.
HIRS was produced by immunization of an outbred rabbit (Lop Hybrid) with HCF antigens. HCF proteins (400 mg) were mixed with an equal volume of Freund’s complete adjuvant (Difco Laboratoies, Detroit, MI) for the first immunization given subcutaneously, and mixed with incomplete adjuvant for the second intramuscular immunization 3 weeks after the first injection. Two subsequent immunizations were carried out as for the second but at fourteen day intervals. The rabbit was ear-bled and the blood was clotted at room temperature for 30 min and then at 4°C for 4 h. The clot was separated from the serum by centrifugation at 3,000 × g for 10 min, and the serum stored in 1 ml aliquots at -20°C after being pre-adsorbed against E. coli lysate following a protocol described in (11).
RNA extraction
Total RNA was extracted from protoscoleces of E. granulosus freshly isolated from sheep hydatid cysts using TRIZOL Reagent (Lifetechnologies, Rockville, MD). Messenger RNA was extracted from 2 mg of total RNA using an Oligotex mRNA Mini Kit (Qiagen, Hilden, Germany). The mRNA was then precipitated by adding 1/10 vol of 3 M sodium acetate (pH 5.2), and 0.8 vol of isopropanol. The tube was kept at -20°C overnight, then centrifuged at 12,000 × g for 30 min at 4°C. The pellet was washed with 70% (v/v) ethanol, air-dried at room temperature 10-15 min, and dissolved in 20 ml DEPC-treated water. The resulting mRNA was used for constructing the cDNA library.
Construction of a protoscolex cDNA library
The cDNA library was constructed using the ZAP Express® cDNA Synthesis Kit (Stratagene, La Jolla, CA). Briefly, frst strand cDNA was synthesized commencing with 5 mg (in 25 ml of water) of poly(A)+ RNA in a final reaction volume of 50 ml containing 5 ml of 10 × first-strand buffer, 3 ml of first-strand methyl nucleotide mixture, 2 ml of EcoR I adapters (1.4 mg/ml), 12.5 ml of DEPC-treated water, and 1ml of RNase Block Ribonuclease Inhibitor (40 U/ml) (Stratagene). The reaction was incubated to anneal the primer to the template for 10 min at room temperature. 1.5 ml of MMLV-RT (50 U/ml) (Stratagene) was added to the first-strand synthesis reaction. Then, the reaction was further incubated at 37°C for 1 h.
Second-strand cDNA synthesis was performed in a reaction volume of 200 ml containing 50 ml of the first-strand synthesis reaction, 20 ml of 10 × second-strand buffer, 6 ml of second-strand dNTP mixture, 109 ml of sterile distilled water, 2 ml of [α-32P] dATP (800 Ci/mmol, Amersham Pharmacia Biotech. Inc., Piscatway, NJ), 2 ml of RNase H (1.5 U/ml) and 11 ml of DNA polymerase I (9.0 U/ml ) (all supplied by Stratagene). The mixture was incubated for 2.5 h at 16°C.
Blunting the cDNA termini was carried out by adding 23 ml of blunting dNTP mixture and 2 ml of cloned pfu DNA polymerase (2.5 U/ml) to the second-strand synthesis reaction and incubating the reaction at 72°C for 30 min. The reaction was extracted by phenol-chloroform (1:1 (v/v), pH 7.5) and then chloroform. The cDNAs were precipitated by adding 1/10 volume of 3 M sodium acetate (pH 5.2), 2.5 volumes of 100% (v/v) cold ethanol at -20°C overnight and microcentrifugation at maximum speed for 60 min at 4°C. The pellet was washed with 70% (v/v) ethanol. The cDNA pellet was resuspend in 9 ml of EcoR I adapters (0.4 mg/ml) (Stratagene) and incubated at 4°C for 30 min. Seven ml of the cDNA solution containing EcoR I adapters was transferred into a fresh tube followed by adding 1 ml of 10 × ligase buffer, 1 ml of 10 mM rATP and 1 ml of T4 DNA ligase (4U/ml) (all supplied by Stratagene). The reaction was incubated overnight at 8°C to ligate the adapters to cDNA. Then the tube was heated in a 70°C water bath for 30 min and then the reaction was cooled at RT for 5 min.
The cDNA containing EcoR I ends was phosphorylated by adding 1 ml of 10 × ligase buffer, 2 ml of 10 mM rATP, 6 ml of sterile water and 1 ml of T4 polynucleotide kinase (all supplied by Stratagene), and incubating at 37°C for 30 min. The kinase was inactivated by heating at 70°C for 30 min, then the reaction allowed to equilibrate to RT for 5 min. 3' termini was cut by Xho I restriction enzyme as the Oligo(dT) linker-primer containing the enzyme cutting site with adding 28 ml of Xho I buffer supplement and 3 ml of Xho I (40 U/ml) (all supplied by Stratagene).Then the reaction was incubated at 37°C for 90 min, the cDNA was precipitated by adding 1/10 volume of 10 × STE buffer and 2.5 volume of 100% (v/v) ethanol overnight at -20°C and the reaction mix centrifuged at maximum speed for 60 min at 4°C. The pellet was resuspended in 14 ml of 1 × STE buffer and 3.5 ml of the column loading dye.
In order to obtain larger fragments, the cDNA was fractionated through a drip column containing Sepharose CL-2B gel filtration medium. The cDNA was run and eluted through the column by adding 1 × STE buffer and fractionated by the size of the cDNA. Larger fragments run faster than the smaller fragments. The size of cDNAs in each fraction was inspected by running 1 ml of each fraction (100 ml volume) on an agarose gel and exposing to an X-Ray film (Fig. 2). Fractions containing cDNAs more than 500 bp in size were combined, then extracted with phenol-chloroform, chloroform, and precipitated with ethanol. The pellet was suspended in 10 ml of water and stored at -20°C until the next stage.
Fig. 2. Size of cDNA synthesised from mRNA isolated from protoscoleces of E. granulosus.
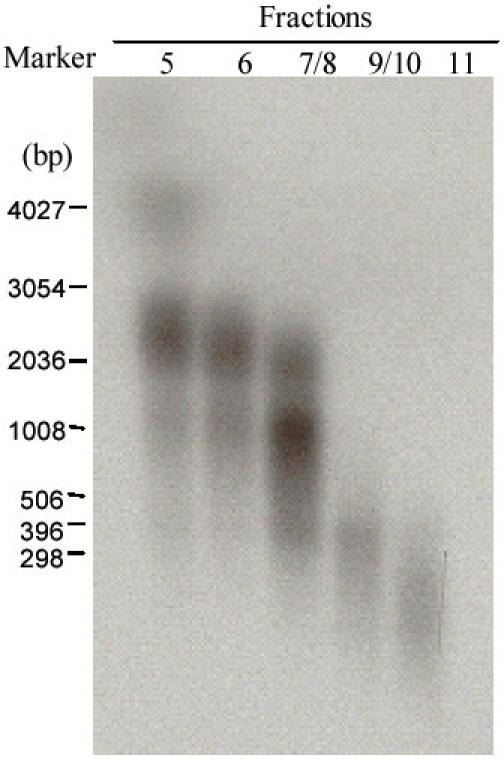
After the second-strand cDNA was synthesised by adding 2μl of [α-32P] dATP to the reaction, the cDNA was fractioned through a CL-2B column after being linked with EcoR I adapters and digested with Xho I to create an Xho I cut end. Fractions 5 to 11 (1μl of elution) were run on an agarose gel and exposed for 4 h to an X-ray film before being developed. Standard RNA markers, in nucleotides are shown on the left hand of the figure.
The cDNAs were ligated into the ZAP Express vector (Stratagene) in a final volume of 5 ml containing 1 ml of the ZAP Express vector (1 mg/ml), 2 ml of cDNA (about 200 ng), 0.5 ml of 10 × ligase buffer, 0.5 ml of 10 mM rATP (pH 7.5), 0.5 ml of water and 0.5 ml of T4 DNA ligase (4 U/ml) (all supplied by Stratagene). The reaction mixture was incubated overnight at 12°C.
Packaging of the cDNA library was performed by adding 1 ml of the ligated cDNA to 25 ml of packaging extract and incubation at 22°C for 2 h. Then 500 ml of salt and magnesium (SM) buffer and 20 ml of chloroform were added. After a brief microfuge spin, the supernatant containing the phage was ready to be titered and storing at 4°C for screening and amplifying.
A small amount (1 ml) of stock lambda phage was serially diluted with SM buffer. One ml of the dilution was added to a tube containing 200 ml of host XL1-Blue cells cultured to an OD600nm of 1.0 and diluted in 10 mM MgSO4 to an OD600nm of 0.5. After the tube was incubated at 37°C for 15 min, 3 ml of NZY top agar (melted and cooled to 48°C), 15 ml of 0.5 M IPTG (in water), and 50 ml of X-gal {250 mg/ml [in N,N'-dimethyl-formamide (DMF)]} were added. The mixture was plated on the top of a NZY agar plate. The plate was cooled at RT for 10 min and then inverted and incubated at 37°C. The phage plaques could be seen visible after 6-8 h. The recombinants that were in a white (clear) background and non recombinants that were in a blue background were counted. The integrity of the library [in plaque-forming units (pfu) per milliliter (pfu/ml)] was calculated using the formula:
[Number of plaques (pfu) × dilution factor/volume plated (ml)] × 1000 ml / ml.
Immunoscreening the cDNA library
The protoscolex cDNA library was immunoscreened using the hyper immune rabbit serum (HIRS) and a pool of the mouse infection sera (MIS). Briefly, XL1-blue host cells (Stratagene) were incubated with 1 ml of an optional dilution of the library, and plated on NZY agar plates. The agar plates were incubated at 42°C for 4.5 h, transferred to a 37°C incubator and overlaid with 10 mM IPTG-nitrocellulose membranes. The membranes were orientated on the plates using a code of punctures that penetrated the membrane and agar surface, and the plates were incubated at 37°C for a further 4 h. Each membrane was removed, air dried, and 1 ml of HCF (2 mg/ml of proteins) was spotted at one point of the filter and acted as control for a positive reaction before the membrane was washed in PBST (3 × 15 min) and blocked overnight at 4°C in 5% (w/v) skim milk in PBS. Following the blocking step, all incubation steps were carried out at room temperature on a moving platform with the membranes being washed thoroughly in 3 × 15 min washes after each step. The membranes were washed in PBST, incubated for 2 h with HIRS or MIS (diluted to 1 in 100), and further washed 3 × 10 min in PBST. The secondary antibody of horseradish peroxidase conjugated sheep anti-mouse Ig (Silenus) or goat anti-rabbit IgG (Bio-Rad) was diluted 1:2000 in 5% (w/v) skim milk in PBST, and incubated with the membranes for 2 h. The membranes were developed in 45 ml of PBS containing 30 ml of 30% (v/v) hydrogen peroxide and 5ml of the chromogenic substrate 4-chloro-1-naphthol (60 mg stock solution of 4-chloro-1-naphthol in 20 ml of ice cold methanol) after three washes in PBST and a final wash in PBS only. Fusion proteins adsorbed to the membranes that were recognized by the MIS or HIRS appeared as pink to purple coloured spots, with colour intensity indicating immunoreactivity. Positive plaques were identified by aligning the membrane with the agar plate, and plaques of interest were removed using a sterile Pasteur pipette and transferred to 500 ml of sterile SM buffer. Twenty μl chloroform was added to the phage stock to lyse the host cells, and the stock was stored at 4°C.
Preparation of plasmid DNA by single-clone excision
Automatic excision is a process where purified phage particles are converted to phagemid (plasmid) by in vito excision using a suitable helper phage. A single XL1-blue colony was incubated overnight at 30°C in NZY broth (one liter contained 5g NaCl, 2g MgSO4·7H2O, 5g yeast extract, 10g casein hydrolysate)) containing 0.2% (w/v) maltose. A single XLOLR (strain) E. coli (Stratagene) colony was incubated overnight at 30°C in NZY broth. The cells were pelleted (1,000 × g, 10 min, RT) and resuspended at an OD600nm of 1.0 in 10 mM MgSO4. Washed cell (200 ml of XL1-blue MRF cells) were transferred to a sterile Falcon 2059 polypropylene tube containing 250 ml of purified phage stock (containing >1 × 105 phage particles) and 1 ml of the ExAssist helper phage (>1 × 106 pfu/ml) (Stratagene). The tube was incubated at 37°C for 15 min, 3 ml NZY broth added and incubated for a further 3 h at 37°C with shaking (100 rpm). The cells were lysed by heating at 68°C for 20 min and then cellular debris was pelleted (1,000 × g, 15 min). The supernatant (10 ml) was incubated with 200 ml of washed cells (XLOLR cells) at 37°C for 15 min, then 300 ml of NZY broth was added and incubated at 37°C for 45min. One hundred ml of infected cells were plated (spread) onto LB agar plates containing 50 mg/ml of Kanamycin and incubated at 37°C for overnight. Single colonies were incubated overnight in 10 ml of LB broth containing kanamycin (50 mg/ml) and plasmid DNA was extracted from the cells the following day.
Colony immunoassay for initial screening with mouse and human sera
The initial tests for each of the positive clones were performed in a colony immunoassay. Plaques formed by cloned phage infected host bacterial cells were transferred and lifted to a sheet of nitrocellulose membrane. The sheet was cut into strips and each of the strips was probed using individual mouse infection serum and that from a patient with clinically diagnosed CE. The reaction procedures including the colour development were the same as those used for immunoscreening the cDNA library.
Sub-cloning, expression and purification of recombinant proteins
Positive clones with diagnostic sensitivity in the initial serum tests were sub-cloned into the BamH I (New England BioLabs, Beverly, MA) and Not I (NEB) site of the pET-41b(+) expression vector (Novagen, Inc. Madison, WI). The GST fusion proteins were expressed in E. coli BL21(DE3) cells induced by 0.2 mM IPTG and affinity purified by GST∙Bind™ Kits (Novagen) under native conditions according to the manufacturer’s instructions. Recombinant GST was obtained by expressing plasmid pET-41b(+) vector in BL21(DE3). The vector contained GST and S- and 2 × His Tags to facilitate purification. The bacterial cells were cultured at 37°C, soluble protein production was induced by 0.2 mM IPTG, and purified by the same GST affinity purification kit (for details, see Protocol 1).
SDS-PAGE and immunoblot analysis
Affinity purified GST fusion proteins or concentrated HCF were electrophoresed on 12% (w/v) SDS-PAGE gels under reducing conditions and transferred onto nitrocellulose membrane. The membrane was then cut into strips. Each strip contained about 0.3 μg recombinant fusion protein or 15 μg HCF proteins. After blotting with 5% (w/v) skim milk, the strips were incubated with mouse or human test sera (diluted 1:100) for 1 h at 37°C. After 3 washes with PBST, the strips were incubated with sheep anti-mouse whole Ig (Silenus, Amrad Operations Pty Ltd, Melbourne, Australia) or goat anti-human IgG (Sigma) conjugate. The strips were visualized for colour after development in 4-chloro-1-naphanol substrate solution for 15 min at room temperature. Further details are provided in Protocol 2.
Patients and sera
Serum samples from patients with surgically confirmed CE were collected from different geographical areas and used to assess the diagnostic sensitivity of the selected proteins (Table 3). Serum samples used for assessing their diagnostic specificity were from patients with other proven parasitic infections, including alveolar echinococcosis (AE), neurocysticercosis and schistosomiasis (Table 3). All sera were stored at -20°C until used. Full clinical details and geographical origins of the sera can be found in Li et al. (8).
Table 3. Diagnostic performance, by immunoblotting, of the test proteins screened against human sera clinically confirmed with an infection or other clinical presentation.
| Number of positive sera/total sera tested (% positivity rate) | ||||||
| CE | AE | NCC | Schist | Others | Healthy | |
| GST | 5/116 (4.3) | 2/89 (2.2) | 7/172 (4.1) | 1/78 (1.3) | 2/153 (1.3) | 1/70 (1.4) |
| HCF-AgB | 98/116 (84.5) | 7/9 (77.8) | 14/100 (14.0) | 5/50 (10.0) | 5/119 (4.2) | 3/70 (4.3) |
| EpC1-GST | 107/116 (92.2) | 4/89 (4.5) | 16/172 (9.3) | 1/78 (1.3) | 1/153 (0.65) | 3/70 (4.3) |
| EgTPx-GST | 39/100 (39.0) | 50/89 (56.2) | 14/72 (19.4) | 0/28 (0) | 5/29 (17.2) | 4/20 (20.0) |
| EgG5-GST | 14/23 (60.9) | 9/9 (100) | 32/72 (44.4) | 0/28 (0) | 5/29 (17.2) | 3/20 (15.0) |
Results
Quality of sera for screening the cDNA library
Before screening the cDNA library, the reactivities of the MIS and HIRS were tested with HCF antigens by Western blot and these reactions were also compared with other sources of sera. Native HCF antigens were run on SDS-PAGE, then transferred onto a nitrocellulose membrane. The stripped membranes were then probed with HIRS, MIS, serum pools from mice infected with schistosomiasis and malaria, and serum pools from humans with confirmed CE, AE, NCC and schistosomiasis. The MIS and HIRS recognized a similar pattern of HCF antigens (Fig. 1); more than 12 HCF bands of molecular sizes ranging from 8 to 200 kDa, including the strongly reacting 8 kDa sub-unit of antigen B (12) (Fig. 1, arrowed), were recognized. Pools of sera from human subjects with confirmed CE, AE and NCC reacted with 6 or more HCF components (Fig. 1).
Fig. 1. Western blot analysis showing the recognition of hydatid cyst fluid (HCF) antigens of Echinococcus granulosus by different sources of sera.

Lane M, protein marker; Lane 1, pooled sera from normal mice; Lane 2, pooled sera from mice experimentally infected with oncospheres of E. granulosus; Lane 3, pooled sera from mice infected with Plasmodium yoelii; Lane 4, pooled sera from mice infected with Schistosoma japonicum; Lane 5, pooled normal human sera; Lane 6, pooled sera from humans with proven cystic hydatid disease infection; Lane 7, pooled sera from humans with proven alveolar echinococcosis infection; Lane 8, pooled sera from humans with proven neurocysticercosis infection; Lane 9, hyperimmune rabbit serum prepared against hydatid cyst fluid antigens. HCF antigen B (AgB) is arrowed. Protein standard markers are shown on the left hand of the figure.
Construction and titering of the protoscolex cDNA library
In order to obtain good quality cDNA, a column was used to fractionate the cDNA after the second strand synthesis. cDNAs in fractions with sizes greater than 0.5 kb (Fractions 5, 6, 7/8 in Fig. 2) were combined and ligated to the λZAP phage vector. Clones were randomly selected and amplified by PCR. The majority of cDNA inserts were more than 1 kb (1-3.0 kb) in size (Fig. 3). The titre of the unamplified protoscolex cDNA library was 2.31 × 106 plaque forming units (pfu) per ml. An estimated 99.5% of the phage in the cDNA library were recombinants.
Fig. 3. The cDNA insert sizes of randomly selected clones from the protoscolex cDNA library.

The cDNA insert sizes were determined by PCR using T3 and T7 primers with the PCR products being electrophorezed on a 1% (w/v) agarose gel. The size of the DNA fragments (Lanes 1-21) were compared with standard DNA ladder (Gibco BRL) (Lane M).
Screening the cDNA library
To identify genes encoding putative immunodiagnostic antigens, we first used HIRS to immunoscreen 1.65 × 105 pfu of the unamplified E. granulosus protoscolex expression cDNA library. Ninety six clones were isolated. Four clones that showed strong signals were sequenced (Table 1). All of the four proteins were expressed in the phage vector and then lifted to nitrocellulose membrane from plate plaques. None of the proteins were recognized by MIS or a pool of human CE sera (data not shown).
Table 1. Clones isolated from the E. granulosus protoscolex cDNA library by immunoscreening using a pool of sera from mice experimentally infected with oncospheresa and a hyper-immune rabbit serum raised against hydatid cyst fluidb.
| Protein | Number of representatives | Estimated cDNA insert size (kb) |
| aEgTPx | 5 | 0.661-0.662 |
| aEpC1 | 1 | 0.42 |
| aEpG5 | 5 | 1.8-2.0 |
| bEgP1 | 4 | 1.3-1.8 |
| bEgP3 | 5 | 1.2-1.4 |
| bEgP5 | 3 | 2.1-2.4 |
| bEgP6 | 2 | 2.0-2.2 |
MIS was then used to immunoscreen the E. granulosus cDNA library. Ninety immunogenic clones were isolated by screening 1.6 × 104 recombinant phages from the unamplified cDNA library. Among these, 11 strongly positive clones were excized into plasmid vectors and sequenced. Sequencing analysis showed that they belonged to three sequence groups, termed EpC1, EgTpx and EgG5 (GenBank accession numbers, AF481884, AF478688 and Z29489, respectively) (Table 1). Clone EpC1 is a novel E. granulosus gene containing a putative ORF of 231 bp coding for a protein with a theoretical molecular mass of 8.5 kDa. Clone EgTPx contained a putative ORF of 582 bp coding for a protein with a theoretical molecular mass of 21.4 kDa. Clone EgG5 contained a putative ORF of 1680 bp coding for a protein with a theoretical molecular mass of 65.17 kDa. The three antigens were subsequently expressed as GST-fusion proteins, purified by glutathione-affinity chromatography and assessed for diagnostic performance.
Recognition of cDNA clones by mouse and human antisera
Phage-expressed proteins were transferred onto nitrocellulose strips and then processed by dot Western blotting (Fig. 4). Phage plaque dot ELISAs showed that 84% (21 out of 25) mouse infection sera reacted with EpC1, 11/25 (44%) with EgG5, 18/25 (72%) with EgTPx and 23/25 (92%) with the mixture of EpC1, EgG5 and EgTPx. 75.6% (34/45) human sera reacted with the mixture of EpC1, EgG5 and EgTPx (data not shown). These results indicated that the proteins were recognized by both mouse and human infection sera and the reactivities of the mixed phage expressed proteins were superior to the individual proteins. For diagnostic proposes, the use of natural infection serum to screen the cDNA library was considered more appropriate than a serum prepared against HCF antigens.
Fig. 4. Conventional Western blot (Panel I) of hydatid cyst fluid antigen B (arrowed) and plaque-dot blots of recombinant phages (Panel II) using individual sera from 26 mice (numbered at the top of Panel I) infected with oncospheres of E. granulosus.
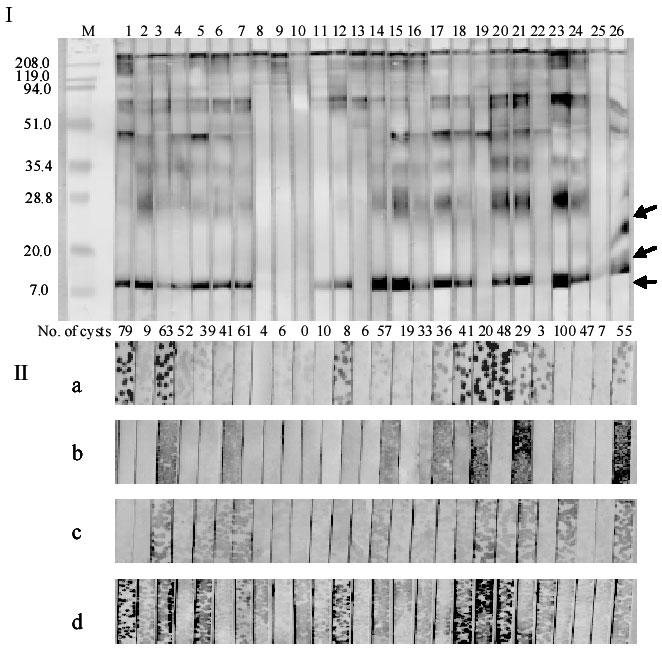
Recombinant phage vectors were transfected into XL1-Blue bacterial cells (Stratagene), then plated on top of LB plates with top agar. After growing the transfected cells for 5 h at 42°C, a nitrocellulose membrane was placed on top of the agar. Then the plate was incubated at 37°C for a further 4 h. The membrane was washed three times with PBST and cut into strips. After blocking in 5% (w/v) skim milk in PBS, the strips were reacted with the individual mouse sera and then sheep anti-mouse IgG conjugate (full details are described same as Western blotting in Protocol 2). The strips were developed with 4-chloro-1-naphthol substrate. Recombinant phage are EgTPx (a), EgG5 (b), EpC1 (c), and a mixture of EgTPx, EgG5 and EpC1 (d). The numbers of hydatid cysts resulting from oncospheral infection in each of the mice are shown between Panels I and Panels II. Protein standard markers (M) (in kDa) are shown on the left side of Panel I.
Cloning and expression of cDNAs
Having shown the potential serodiagnostic value of their expression products, the EpC1, EgTPx, EgG5 cDNAs were subcloned into the pET-41b(+) vector and expressed as GST fusion proteins induced with 0.2 mM IPTG for 3 h at 37°C. The soluble fusion proteins that resulted were affinity purified. Fig. 5a shows the expression and purification of the recombinant proteins on a Coomassie-stained 12% SDS-gel, run under reducing conditions. The affinity purified fusion proteins were clearly recognized by a pool of sera from CE patients (Fig. 5b) and MIS (Fig. 5c).
Fig. 5. Expression, purification (Panel A, SDS-PAGE stained by Coomassie blue) and Western blotting of recombinant proteins for diagnosis of cystic echinococcosis (CE) with a pool of sera from patients confirmed with CE (Panel B), and a pool of sera from mice experimentally infected with oncospheres of E. granulosus (Panel C).
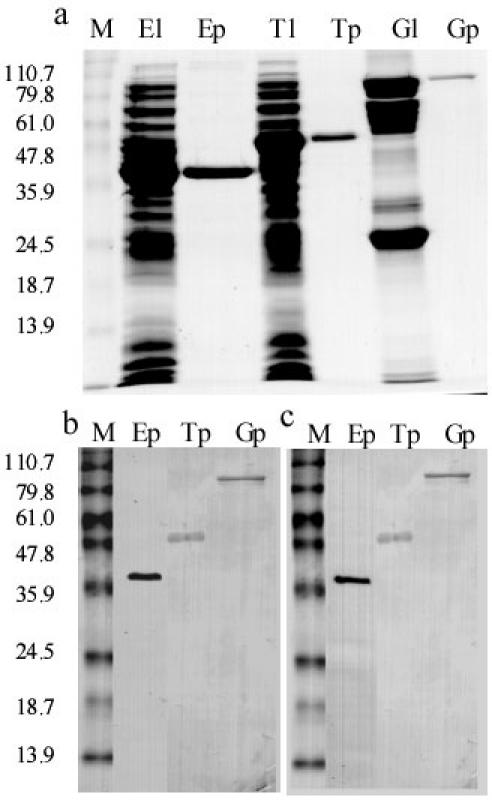
Lane El, bacterial lysate containing EpC1-GST; Lane Ep, affinity purified EpC1-GST; Lane Tl, bacterial lysate containing EgTPx-GST; Lane Tp, affinity purified EgTPx-GST; Lane Gl, bacterial lysate containing EgG5-GST; Lane Gp, affinity purified EgG5-GST. Protein standard markers (in kDa) are shown on the left side of the panels.
Diagnostic performance of rEpC1, EgTPx and EgG5 using immunoblotting
Immunoblotting (IB) was used to test the diagnostic sensitivity of rEpC1-GST, rEgTPx-GST, rEgG5-GST and HCF antigens against 25 individual MIS sera (mice infected with E. granulosus oncospheres) (Table 2). Of these, 23 (92%) recognized rEpC1-GST, whereas 22 (88%) of the sera reacted with HCF AgB. Sensitivity was much lower with rEgTPx-GST (64%) and rEgG5-GST (40%). Then, the same proteins were tested in an IgG-IB with individual sera from patients with clinically diagnosed CE, patients who had other helminthic infections, patients with other liver diseases and subjects who were clinically normal. The overall results are summarized in Table 3 and show clearly that EpC1 was the most diagnostically useful for detecting CE infection. In total, the EpC1 diagnostic antigen was tested on 896 sera including sera from 324 CE, 172 NCC, 89 AE, 241 other infections/liver cancer cases and 70 confirmed negative controls. The overall sensitivity obtained with EpC1 was 92.2% and specificity was 95.6% (8).
Table 2. Western blot analysis of the three selected antigens, HCF-AgB and GST using 25 sera from mice experimentally infected with oncospheres of E. granulosus.
| Protein | Number of positively reacting sera | % |
| HCF-AgB | 22 | 88 |
| GST | 0 | 0 |
| EpC1-GST | 23 | 92 |
| EgTPx-GST | 16 | 64 |
| EgG5-GST | 10 | 40 |
Discussion
Sufficient quantity and high quality mRNA was crucial for construction of the E. granulosus cDNA library. Accordingly, E. granulosus protoscoleces were aspirated from hydatid cysts within 2 h after slaughter of infected sheep. After a final wash with PBS, the protoscoleces were placed into a liquid nitrogen container directly and stored in the container until use. The steps for RNA extraction were undertaken under very cold conditions before adding the TRIZOL Reagent. The mortar and pestle for grinding the protoscolex tissue were pre-cooled with liquid nitrogen. The tissue was ground by continuously adding liquid nitrogen to keep the mortar cool. Once the tissue was powdered, the powder was poured into a tube containing TRIZOL Reagent. Careful fractionation of cDNA was also important for constructing the high quality cDNA library containing large fragments or full length cDNA sequences.
Several different types of pooled sera were tested with crude hydatid cyst antigens by Western blot (Fig. 1) in order to determine the optimum serum for screening the E. granulosus cDNA library. The results showed that many HCF antigens could be recognized by MIS and HIRS, and by human infection sera. The CKW mouse, which is widely used in China for medical science research, is a good experimental animal model for the primary infection of hydatid disease as shown in previous studies (9). 25 out of 26 mice were successfully infected with E. granulosus oncospheres producing from 4 to 100 cysts (8). The mouse sera were also individually probed against HCF antigens in Western Blot analysis which showed that most of the sera (22/25) were able to strongly recognize HCF antigens (8).
HIRS was used initially to screen the cDNA library. 1.65 × 105 plaques were probed with HIRS, which was more than 10 times the number of plaques (1.6 × 104) screened by MIS. Almost the same numbers of positive clones (96 vs 90) were obtained with the two screens, which indicated that infection serum was more sensitive for this purpose than the hyper immune serum. The clones strongly reacting with HIRS belonged to four groups designated as EgP1, EgP3, EgP5 and EgP6 (Table 1). EgP1 is a partial severin sequence and the others are novel genes. EgP6 has a sequence with 55 amino acid repeated 7 times. The repeat sequence has no motif similarity in the GenBank™ database. The dot-ELISA using phage plaques showed that those recombinant proteins were not recognized by infection sera from mice or humans. This was further evidence that infection serum is preferable to hyper immune serum in screening a cDNA library for isolating genes encoding putatively diagnostic antigens. We did not undertake any further expression or serological analysis of these four groups of clones.
The most frequent cross-reactions in immunodiagnostic assays for cystic echinococcosis are due to E. multilocularis and T. Solium (13). Antigen 5, one of the two major antigenic lipoprotein components in HCF, cross-reacts with antibodies in sera from subjects infected with E. multilocularis, E. vogeli, T. solium and other parasites (14). The other dominant lipoprotein, antigen B (15, 16) is more specific although cross reactivity occurs with serum antibodies in patients infected with E. multilocularis and E. Vogeli (17), and T. Solium (14). In our study, a pool of sera from 9 AE patients also showed reactivity with antigen B-like proteins in HCF (Fig. 1).
We used immunoblotting (IB) as the basis of our serodiagnostic method due to its simplicity and sensitivity (18-20). Furthermore, IB directly shows the reactivity of target protein with a positive serum sample. We compared rEpC1-GST with HCF antigen B to evaluate its diagnostic performance. Antigen B, banding at approximately 8 kDa in SDS-PAGE, was used for comparative purposes as it has been shown to be a useful marker for diagnosis of CE (21-23). The overall sensitivity (92.2%) and specificity (95.6%) obtained with rEpC1-GST indicate that this protein has probably achieved a better level of diagnostic performance than other reported reagents, particularly when the large number of serum samples tested are taken into consideration. We are currently evaluating the performance of rEpC1 in an ELISA format.
Although the GST tag present on the rEpC1-GST fusion protein was derived from S. japonicum, only one of 88 sera from patients with schistosomiasis (20 confirmed S. japonicum cases; 68 confirmed S. mansoni cases) reacted with rEpC1-GST (8). The reasons for this lack of reactivity are unclear but it may be due to the fact that the E. coli expressed GST did not contain conformational epitopes required for recognition by the schistosomiasis infection sera or the antigenicity of GST was too weak to be recognized. The rEpC1-GST fusion protein was digested with enterokinase to remove GST using a recombinant enterokinase kit (Novagen). Forty-five sera from confirmed CE cases were used to compare the reactivity of rEpC1 with rEpC1-GST by IB. Sensitivity, in terms of positively reacting sera, was similar with both antigens but weaker signals were obtained with rEpC1 (data not shown). The EpC1 cDNA was also subcloned into the pCAL-n vector (Stratagene) to express rEpC1 as a fusion protein with calmodulin binding protein (rEpC1-CBP). Again weaker signals were obtained (data not shown) indicating that the presence of GST enhanced the sero-reactivity of rEpC1. This may have been due to the GST presenting a confirmational structure with the epitope(s) of EpC1, which resulted in an increase in the signal.
CE is characterized by the triggering of an intense humoral response with a corresponding rise in the titers of specific antibodies (6). Such a response is encouraging for the development of a CE serodiagnostic assay but available antigens do not provide an appropriate level of sensitivity or specificity. Furthermore, due to the lack of standardization of these antigens and a lack of concordance among the various tests available for CE serology, a diagnostic assay for routine laboratory diagnosis of hydatidosis is not yet available. A rapid, simple antibody-based test incorporating a single defined recombinant E. granulosus antigen is required urgently for diagnosis of early CE infection in humans, ensuring early treatment, and for discrimination from AE infection. We have described the cloning, expression and purification of a small panel of E. granulosus antigenic components resulting in the identification of a suitable molecule, rEpC1-GST that can provide the requisite levels of specificity and sensitivity, and applied it in a simple immunoblot format for CE diagnosis. We plan to extend the diagnostic potential of the test by improving its specificity without affecting sensitivity, thereby supporting its utility for routine application in the clinic, laboratory and in the field for community surveillance surveys that monitor CE control programs. Further, as there is no assay currently available for serodiagnosis of CE in livestock animals that are responsible for the transmission of E. granulosus to humans, our objective of developing a rapid test, based on the EpC1 protein, is timely. The assay would allow the monitoring of animals imported into countries free from hydatidosis and as an aid to countries where control schemes for the disease are in operation. Given the diagnostic properties of EpC1 and the fact that large quantities of the protein in pure form can be produced, the antigen should be readily amenable to any commercial protocols currently available for antibody detection applicable both clinically and to detect infected livestock. We plan future simplification of the test to a dipstick dye assay format that will be rapid, cheap and not requiring any equipment. Such assays have been developed for the detection of antibodies against a range of infectious agents including Schistosoma (24) and Leishmania (25).
The strategy we used for isolation of E. granulosus cDNAs expressing proteins with serodiagnostic value and their application in antibody-based detection assays may also prove suitable for immunodiagnosis of other parasitic infections.
Supplemental Information
Other areas of active research undertaken by the authors can be found at http://www.qimr.edu.au/research/labs/donm/index.html.
Acknowledgments
The work was supported by the Queensland Institute of Medical Research Development fund (JL), Wellcome Trust (GR057212) (WBZ and DPM), The National Health and Medical Research Council of Australia (DPM), Australian International Postgraduate Research (IPRS) and University of Queensland International Postgraduate Research (UQIPRS) Scholarships to WBZ. The rabbit anti-serum used in this study was kindly provided by Dr. Lihua Zhang, QIMR. We thank Professor Akira Ito, Dr Marianna Wilson, and Dr Peter Schantz for technical assistance and advice. We also thank Dr Irving Kagan for his expert advice and provision of excellent laboratory facilities.
Abbreviations
- CE
cystic echinococcosis
- AE
alveolar echinococcosis
- NCC
neurocysticercosis
- Schist
schistosomiasis
- Others
other infections/clinical presentations
- Healthy
uninfected controls
Appendix
Protocols
EpC1-GST fusion protein expression
Pick a colony from a plate using a tooth pick and inoculate into 30 ml LB plus kanamycin antibiotic at a final concentration of 30 μg/ml. Culture overnight at 37°C.
Dilute the culture 10 times to 300 ml LB containing kanamycin at a final concentration of 30 μg/ml and further grow the bacteria by measuring the optical density at 600nm (OD600).
Induce recombinant protein expression by adding isopropyl bD-thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM when the OD600 reaches 0.6. Further culture for 4-5 h and harvest cells by centrifugation for 30 minutes at 5000 rpm. Use the GS-3 rotor in a Sorvall (or the JA-10 in a Beckman) centrifuge.
Discard the supernatant and resuspend the cell pellet with GST binding buffer (25 mM Tris pH 7.5, 150 mM NaCl, 10 mM DTT, 1 mM EDTA and 1 mm PMSF). Use about 10 ml of GST binding buffer per g of wet cells.
After three frozen-thaw cycles, ultrasonicate for 3 min using the standard large attachment head (90% output and 50% duty cycle). Repeat 3 times. Cool on ice for 3 min after each sonication.
Centrifuge for 30 min at 12,000g at 4°C.
Load and run 10 ml of supernatant on a SDS-PAGE gel to estimate the expression rate of the fusion protein. Measure the concentration of protein in the lysate.
Dilute the lysate to 1 mg/ml of total protein with GST binding buffer and load onto a gluthathione sepharose column (Novagen). Normally 2 ml of gluthatione sepharose (50% slurry) can bind 10 mg of recombinant protein. Calculate the volume of lysate needed and keep the sample at 4°C when passing through the gluthathione sepharose column.
Wash the column by flowing through with GST binding buffer at 4°C until no protein can be detected using the Protein Assay Dye Reagent (Bio-Rad) (26). We use the mini measure method: 5 μl of flow sample is added to 250 μl of diluted (1 in 5 dilution) Protein Assay Dye Reagent in a 96-wall plate. No color change indicates no protein remains in the flow sample.
EpC1-GST fusion protein is eluted by adding elution buffer (50 mM Tris-HCl (pH 8.0) with 10 mM reduced glutathione) at a flow rate of 1 ml/min until no protein remains on testing with the protein assay dye reagent.
Measure the protein concentration in the eluted sample, aliquot into tubes and store at -70°C until use.
SDS-PAGE of recombinant proteins and Western blotting (immunoblotting)
SDS-PAGE
We use 12% (w/v) SDS-PAGE gels with the Bio-Rad Protean Minigel system. Full details for preparation of the SDS-PAGE gels can be found in Chapter 10 of reference 27. We routinely run whole-well gels, loading 10µg protein per gel. Then, the protein is transferred onto nitrocellulose membrane for Western blotting.
Western blotting (immunoblotting)
Once recombinant proteins are separated using SDS-PAGE, place a nitrocellulose membrane on the gel and, using electrophoresis, transfer the protein bands onto the nitrocellulose membrane (27).
Block the membrane with 5% (w/v) skim milk in PBS for 1 h at 37°C and cut the membrane into strips 2.5 mm wide with 32 strips containing 0.3 μg protein per strip.
Incubate the nitrocellulose membrane with human or animal test serum. The test serum is normally diluted 1 in 100 with 5% (w/v) skim milk in PBST (PBS containing 0.05% (v/v) Tween-20). Incubation can be undertaken at 37°C, or alternatively, over-night at 4°C on a rocking platform.
Incubate the nitrocellulose membrane with an antibody-enzyme conjugate (secondary antibody) after washing 3 × 15 min on a rocking platform in PBST. We routinely use sheep anti-human IgG (Sigma), or sheep anti-mouse IgG (Silenus, AMRAD, Melbourne, Australia) conjugate diluted 1 in 1000 with 5% (w/v) skim milk in PBST.
The location of the antibody-binding band is revealed by incubating the membrane with a colorless substrate* at room temperature for 15 min. The reaction is stopped with three washes with water.
A purple or dark purple colour occurring at the position of the EpC1-GST band represents a positive reaction.
* - For stock solution, dissolve 60 mg of 4-chloro-1-naphthol in 20 ml cold methanol and the solution can be stored at -20°C for a month. For the working solution, dilute the stock solution 1 in 10 with PBS and add 6 μl of hydrogen peroxide (30% (v/v) solution) per 10 ml of the working solution.
References
- McManus DP, Zhang WB, Li J, Bartley PB. Echinococcosis. Lancet. 2003;362(10):1295–1304. doi: 10.1016/S0140-6736(03)14573-4. [DOI] [PubMed] [Google Scholar]
- Rickard MD, Lightowlers M. Immunodiagnosis of hydatid disease. London, UK: George Allen & Unwin (Publishers) Ltd., 1986.
- Gottstein B. Molecular and immunological diagnosis of echinococcosis. Clin Microbiol Rev. 1992;5(3):248–261. doi: 10.1128/cmr.5.3.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightowlers MW, Gottstein B. Echinococcosis/hydatidosis: antigens, immunological and molecular diagnosis. Wallingford, Oxon, UK: CAB International, 1995.
- Poretti D, Felleisen E, Grimm F, et al. Differential immunodiagnosis between cystic hydatid disease and other cross-reactive pathologies. Am J Trop Med Hyg. 1999;60(2):193–198. doi: 10.4269/ajtmh.1999.60.193. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li J, McManus DP. Concepts in immunology and diagnosis of hydatid disease. Clin Microbiol Rev. 2003;16(1):18–36. doi: 10.1128/CMR.16.1.18-36.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babba H, Messedi A, Masmoudi S, et al. Diagnosis of human hydatidosis: comparison between imagery and six serologic techniques. Am J Trop Med Hyg. 1994;50(1):64–68. doi: 10.4269/ajtmh.1994.50.64. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang WB, Wilson M, Ito A, McManus DP. A novel recombinant antigen for immunodiagnosis of human cystic echinococcosis. J Infect Dis. 2003;188(12):1552–1561. doi: 10.1086/379976. [DOI] [PubMed] [Google Scholar]
- Zhang W, You H, Zhang Z, Turson G, Hasyet A, McManus DP. Further studies on an intermediate host murine model showing that a primary Echinococcus granulosus infection is protective against subsequent oncospheral challenge. Parasitol Int. 2001;50(4):279–283. doi: 10.1016/s1383-5769(01)00086-1. [DOI] [PubMed] [Google Scholar]
- Rajasekariah GR, Rickard MD, Mitchell GF. Density-gradient separation of Taenia pisiformis oncospheres. J Parasitol. 1980;66(2):355–356. [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning. A laboratory manual. 3rd ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 2001.
- Lightowlers MW, Liu DY, Haralambous A, Rickard MD. Subunit composition and specificity of the major cyst fluid antigens of Echinococcus granulosus. Mol Biochem Parasitol. 1989;37(2):171–182. doi: 10.1016/0166-6851(89)90149-7. [DOI] [PubMed] [Google Scholar]
- Schantz PM, Shanks D, Wilson M. Serologic cross-reactions with sera from patients with echinococcosis and cysticercosis. Am J Trop Med Hyg. 1980;29(4):609–612. doi: 10.4269/ajtmh.1980.29.609. [DOI] [PubMed] [Google Scholar]
- Leggatt GR, Yang W, McManus DP. Serological evaluation of the 12 kDa subunit of antigen B in Echinococcus granulosus cyst fluid by immunoblot analysis. Trans R Soc Trop Med Hyg. 1992;86(2):189–192. doi: 10.1016/0035-9203(92)90566-u. [DOI] [PubMed] [Google Scholar]
- Ito A, Ma L, Schantz PM, et al. Differential serodiagnosis for cystic and alveolar echinococcosis using fractions of Echinococcus granulosus cyst fluid (antigen B) and E. multilocularis protoscolex (EM18). Am J Trop Med Hyg. 1999;60(2):188–192. doi: 10.4269/ajtmh.1999.60.188. [DOI] [PubMed] [Google Scholar]
- Mamuti W, Yamasaki H, Sako Y, et al. Molecular cloning, expression, and serological evaluation of an 8-kilodalton subunit of antigen B from Echinococcus multilocularis. J Clin Microbiol. 2004;42(3):1082–1088. doi: 10.1128/JCM.42.3.1082-1088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JC, McManus DP. Specific and cross-reactive antigens of Echinococcus granulosus hydatid cyst fluid. Mol Biochem Parasitol. 1987;25(2):143–154. doi: 10.1016/0166-6851(87)90003-X. [DOI] [PubMed] [Google Scholar]
- Siracusano A, Ioppolo S, Notargiacomo S, et al. Detection of antibodies against Echinococcus granulosus major antigens and their subunits by immunoblotting. Trans R Soc Trop Med Hyg. 1991;85(2):239–243. doi: 10.1016/0035-9203(91)90039-2. [DOI] [PubMed] [Google Scholar]
- Ioppolo S, Notargiacomo S, Profumo E, et al. Immunological responses to antigen B from Echinococcus granulosus cyst fluid in hydatid patients. Parasite Immunol. 1996;18(11):571–578. doi: 10.1046/j.1365-3024.1996.d01-31.x. [DOI] [PubMed] [Google Scholar]
- Ortona E, Rigano R, Margutti P, et al. Native and recombinant antigens in the immunodiagnosis of human cystic echinococcosis. Parasite Immunol. 2000;22(11):553–559. doi: 10.1046/j.1365-3024.2000.00336.x. [DOI] [PubMed] [Google Scholar]
- Matossian RM, Kane GJ, Chantler SM, Batty I, Sarhadian H. The specific immunoglobulin in hydatid disease. Immunology. 1972;22(3):423–430. [PMC free article] [PubMed] [Google Scholar]
- Kanwar JR, Kaushik SP, Sawhney IM, Kamboj MS, Mehta SK, Vinayak VK. Specific antibodies in serum of patients with hydatidosis recognised by immunoblotting. J Med Microbiol. 1992;36(1):46–51. doi: 10.1099/00222615-36-1-46. [DOI] [PubMed] [Google Scholar]
- Maddison SE, Slemenda SB, Schantz PM, Fried JA, Wilson M, Tsang VC. A specific diagnostic antigen of Echinococcus granulosus with an apparent molecular weight of 8 kDA. Am J Trop Med Hyg. 1989;40(4):377–383. doi: 10.4269/ajtmh.1989.40.377. [DOI] [PubMed] [Google Scholar]
- Zhu Y, He W, Liang Y, et al. Development of a rapid, simple dipstick dye immunoassay for schistosomiasis diagnosis. J Immunol Methods. 2002;266(1-2):1–5. doi: 10.1016/S0022-1759(02)00086-8. [DOI] [PubMed] [Google Scholar]
- Schallig HD, Cardoso L, Hommers M, et al. Development of a dipstick assay for detection of Leishmania-specific canine antibodies. J Clin Microbiol. 2004;42(1):193–197. doi: 10.1128/JCM.42.1.193-197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moor DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. Volume 1. Analysis of Proteins. New York: John Wiley & Sons, Inc.; 1994. P. 10.0.01-10.8.17.