

Abstract
Inhibitors of the DNA damage checkpoint kinase, Chk1, are highly effective as chemo- and radio-sensitizers in preclinical studies but are not well-tolerated by patients. We exploited the promiscuous nature of kinase inhibitors to screen 9 clinically relevant kinase inhibitors for their ability to sensitize pancreatic cancer cells to a sub-lethal concentration of gemcitabine. Bosutinib, dovitinib, and BEZ-235 were identified as sensitizers that abrogated the DNA damage checkpoint. We further characterized bosutinib, an FDA-approved Src/Abl inhibitor approved for chronic myelogenous leukemia. Unbeknownst to us, we used an isomer (Bos-I) that was unknowingly synthesized and sold to the research community as “authentic” bosutinib. In vitro and cell-based assays showed that both the authentic bosutinib and Bos-I inhibited DNA damage checkpoint kinases Chk1 and Wee1, with Bos-I showing greater potency. Imaging data showed that Bos-I forced cells to override gemcitabine-induced DNA damage checkpoint arrest and destabilized stalled replication forks. These inhibitors enhanced sensitivity to the DNA damaging agents’ gemcitabine, cisplatin, and doxorubicin in pancreatic cancer cell lines. The in vivo efficacy of Bos-I was validated using cells derived directly from a pancreatic cancer patient’s tumor. Notably, the xenograft studies showed that the combination of gemcitabine and Bos-I was significantly more effective in suppressing tumor growth than either agent alone. Finally, we show that the gatekeeper residue in Wee1 dictates its sensitivity to the 2 compounds. Our strategy to screen clinically relevant kinase inhibitors for off-target effects on cell cycle checkpoints is a promising approach to re-purpose drugs as chemosensitizers.
Keywords: Checkpoint override, DNA damage, Kinase inhibitors, Mitosis, drug repurposing
Introduction
The activation of cell cycle checkpoints in response to genotoxic insults plays a critical role in preventing cells from entering mitosis with either damaged or unreplicated DNA. Loss of p53 functions in many cancer cells abrogates their G1 DNA damage checkpoint resulting in their reliance on the S and G2 checkpoints that are mediated by ATM/ATR and Chk1/Chk2 axis.1,2 These checkpoints are believed to contribute to the ability of tumor cells to survive a variety of genotoxic drugs, as disruption of these failsafe mechanisms (checkpoint override) enhance drug killing by forcing them into a catastrophic mitosis that kills them.3-5 The development of checkpoint inhibitors as chemosensitizers, such as those that target Chk1 kinase,6 has been validated in pre-clinical studies.7-9 Unfortunately, the potential clinical benefits of Chk1 inhibition have never been realized owing to toxicities reported in clinical trials (reviewed in ref. 10 and references therein). It is unclear whether the toxicity issues are due to potent Chk1 inhibition in vivo (on-target) or due to off-target effects. Recent preclinical studies of various ATR11 and Wee18 inhibitors have validated their chemosensitization properties. This new generation of sensitizers is currently being tested in clinical trials. The results from such clinical trials will be informative in evaluating whether on-target activity or off-target activity is responsible for unacceptable side-effects. Thus, until checkpoint inhibitors that have limited side-effects and can be tolerated by patients are discovered, the concept of chemosensitization by overriding DNA damage checkpoints as a bona fide therapeutic strategy remains untested.
Large scale efforts that examined the selectivity of kinase inhibitors has revealed complex and unanticipated interactions that extend beyond the target that they were designed to inhibit.12,13 Recently, pazopanib, an FDA-approved multi-targeted receptor tyrosine kinase inhibitor for use against soft-tissue sarcoma was shown to inhibit aurora kinase A in an off-target manner. Importantly, treatment with combinations of pazopanib and taxol enhanced killing and improved clinical outcome.14 Additionally, imatinib, which is used to inhibit BCR-Abl in CML patients, has off-target activity toward c-Kit and subsequently displays clinical efficacy in patients with gastrointestinal stromal tumors.15 These and other examples validate the concept of re-purposing clinically relevant kinase inhibitors by exploiting off-target activities. The concept of drug repurposing is an increasingly attractive approach for drug discovery as it can save significant amounts of time and money and, just as importantly, can fast-track drugs into the clinic (reviewed in ref. 16). We therefore wanted to exploit the promiscuous nature of kinase inhibitors to screen existing drugs for off target chemosensitization activity. Here we report the re-purposing of clinically relevant compounds with novel checkpoint inhibitory activities that maybe tested in the clinic as chemosensitizers. We used pancreatic cancer cells for our screen given the desperate need to improve treatment outcome of this deadly cancer.
Results
Identification of clinically relevant kinase inhibitors that sensitize cells to gemcitabine
We screened a panel of 9 clinically relevant kinase inhibitors, covering a variety of primary targets (Table 1), to test for their ability to enhance the growth inhibitory effect of gemcitabine of PANC1 cells. Cells were treated with gemcitabine, a nucleoside analog, for 24 h to induce replicative stress and impair cell cycle progression through S phase (Fig. S1A). Kinase inhibitors (all at 1 μM final) were added and cell proliferation was measured 48 h later (Fig. 1A). UCN-01, a Chk1 inhibitor, was included as a positive control for gemcitabine sensitization, as reported,17 and was effective in this screen (P < 0.00001 compared with gemcitabine alone). Our screen identified dovitinib (P = 0.004), bosutinib (P < 0.0001), and BEZ-235 (P < 0.0001) as compounds that significantly enhance gemcitabine-mediated growth suppression. BEZ-235 was designed as an mTOR/PI3K inhibitor but was recently shown to also inhibit the ATR/ATM/DNA PKcs checkpoint kinases that are members of the PI3K family.18,19 Bosutinib and dovitinib are Src/Abl and multi-receptor tyrosine kinase (RTK) inhibitors, respectively, that are not known to exhibit chemosensitization activity. We validated the results from the short-term cell proliferation assay with long-term clonogenic survival studies. Cells were either treated with 10 nM gemcitabine for 24 h followed by the addition of kinase inhibitors (all at 1 μM except for UCN-01 which was 100 nM) for 3 h before drugs were washed out and clonogenic survival assessed 10 d later. Both bosutinib and dovitinib reduced survival (P = 0.01, P = 0.05, respectively) as did UCN-01 (P < 0.005) (Fig. 1B). However, BEZ-235 alone was found to greatly reduce colony formation and thus we were unable to demonstrate drug sensitization in the clonogenic assay (Fig. S1B). Since bosutinib gave the greatest sensitization, we characterized its activity further. To confirm the reduction in cell proliferation, as determined by the MTS assay, was due to the induction of apoptosis we quantified the percentage of Annexin V positive cells following treatments. PANC1 cells were treated with gemcitabine at either 10 nM for 24 h or with 2 μM for 2 h followed by 22 h in drug-free media. As shown in Figure 1C, the addition of UCN-01 or bosutinib to gemcitabine-treated cells resulted in a significant increase in apoptosis.
Table 1. A list of kinase inhibitors used in this study, their current clinical status and their primary intended targets.
| Agent (trade name) | Clinical status | Intended primary targets |
|---|---|---|
| Bosutinib (Bosulif) | FDA approved | Src, Bcr-Abl |
| Dasatinib (Sprycel) | FDA approved | Src, Bcr-Abl, |
| Dovitinib | Experimental/Clinical trials | Flt3, KIT, FGFR, VEGFR, PDGFR |
| Erlotinib (Tarvceva) | FDA approved | EGFR |
| Gefitinib (Iressa) | FDA approved | EGFR |
| Imatinib (Gleevec) | FDA approved | Bcr-Abl, KIT, |
| Lapatinib (Tykerb) | FDA approved | EGFR/Her2 |
| Sunitinib (Sutent) | FDA approved | PDGFR, VEGFR, KIT, RET, CSF-1R, Flt3 |
| BEZ235 | Experimental/Clinical trials | PI3-K, mTOR |
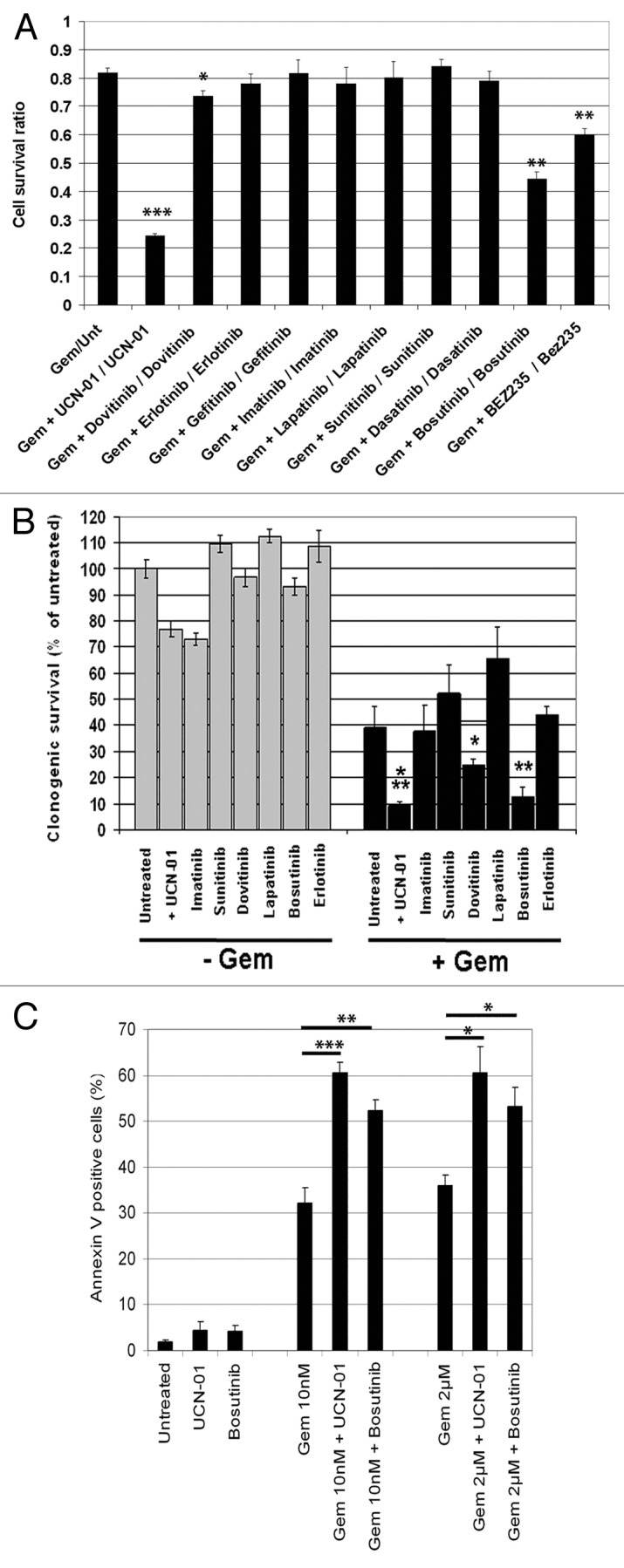
Figure 1. Identification of clinically relevant kinase inhibitors that sensitize cells to gemcitabine. (A) PANC1 cells were treated in triplicate with gemcitabine (10 nM) for 24 h before the addition of kinase inhibitors (all 1 µM) or UCN-01 (100 nM). Viability was assessed 48 h later using MTS assay. Assays were conducted at least 3 times and data are presented as the mean ± SD ***P < 0.00001, **P ≤ 0.0001, and *P < 0.005 compared with gemcitabine/untreated cells, respectively. (B) Clonogenic assays of PANC1 cells treated with or without gemcitabine (10 nM) for 24 h followed by a 3 h treatment with the indicated kinase inhibitors (1 µM) or UCN-01 (100 nM). All drugs were washed out and surviving colonies determined 7–10 d later. Assays were conducted in duplicate a minimum of 3 times and data are presented as the mean ± SD ***P < 0.005, **P < 0.01, and *P < 0.05. (C) PANC1 cells were treated as in (A).Following treatment, cells were harvested and apoptotic cell death determined via Annexin V staining. Experiments were conducted 3 times and data are presented as the mean ± SEM *P < 0.05, **P < 0.005, and ***P < 0.001.
During the course of our studies that were presented above, it came to light that numerous vendors had unknowingly sold to the research community (including us) an incorrectly synthesized isomer of bosutinib (Bos-I), rather than the “authentic” bosutinib.20 The 2 compounds differed only in the arrangement of the same R groups around the aniline ring. Authentic bosutinib is designated 2, 4 dichloro, 5-methoxy, while bosutinib isomer is 3, 5 dichloro, 4-methyoxy (Fig. S1C).20 This was somewhat problematic since in our screen (MTS, clonogenic and apoptosis assays, as shown above) we had used the isomer of bosutinib rather than the authentic drug. However, subsequent studies with authentic bosutinib showed it too had chemosensitization activity (see below). Given the novelty of Bos-I and because it provided the greatest chemosensitizing activity of the clinically relevant inhibitors tested, we focused our study on this inhibitor.
Chemosensitization occurs through off-target activities
To investigate the mechanism of chemosensitization by Bos-I, we queried a database (www.reactionbiology.com/webapps/largedata/) containing the inhibitory activities of 178 kinase inhibitors (including Bos-I) against a panel of 300 recombinant human kinases.13 From this database, we found that Bos-I inhibited 84/300 kinases by >50%. We obtained the kinase target list of another Src/Abl inhibitor, dasatinib, that did not exhibit chemosensitization activity (Fig. 1A). Dasatinib inhibited 50/300 kinases by >50% and comparison of the Bos-I and dasatinib targets showed an overlapping set of Src and related kinases (Table 2). This common set of kinases were unlikely to be targets responsible for Bos-I sensitization. Indeed, dasatinib failed to sensitize cells to gemcitabine when used up to 5 μM (Fig. S2A). However, it was still possible that Bos-I’s chemosensitization activity was due to its increased potency, relative to dasatinib, toward one or more of these common targets. Indeed, we found that EGFR, HGK, MINK1, RET1, and ERBB4 kinases were inhibited more potently by Bos-I compared with dasatinib (>2-fold), that could explain the difference in sensitization between these 2 compounds (Table 2). This possibility was eliminated because querying the database showed that there were other kinase inhibitors which did not chemosensitize but inhibited at least one of these kinases to a similar degree as with Bos-I (Table 3). This then suggested that Bos-I’s sensitization activity was mediated through kinase(s) that were not inhibited by dasatinib. Since Wee1 and Chk1 were uniquely inhibited by Bos-I and, given their roles in maintaining genomic integrity and DNA damage checkpoint activation,21 they became the potential targets of chemosensitization. We next tested whether Chk1 and Wee1 were targets of Bos-I in a cellular context. Treatment of PANC1 cells with gemcitabine induced the DNA damage checkpoint that was reflected in the activation of Chk1 (measured via autophosphorylation of Chk1S296) and Wee1 (measured via phosphorylation of cdc2Y15). Importantly, the addition of Bos-I abolished gemcitabine-induced p-Chk1S296 and p-cdc2Y15. By contrast, gemcitabine-induced Chk1S296 or cdc2Y15 phosphorylation were unaffected by dasatinib (Fig. 2A). Taken together, these studies provide evidence that Bos-I can inhibit Chk1 and Wee1 both in vitro and in cells.
Table 2. A list of kinases that are inhibited (>50%) by bosutinib and dasatinib as determined by in vitro profiling.
| Kinases inhibited >50% by Bos-I | Kinases inhibited >50% by Dasatinib | ||||
|---|---|---|---|---|---|
| Kinase | % Residual activity | Kinase | % Residual activity | ||
| ABL1 | 1.8 | ABL1 | 2.77 | ||
| ABL2/ARG | 2.19 | ABL2/ARG | 2.74 | ||
| ACK1 | 3.54 | ACK1 | 2.62 | ||
| ARK5/NUAK1 | 11.43 | ALK1/ACVRL1 | 15.93 | ||
| AXL | 37.63 | ARAF | 33.76 | ||
| BLK | 20.73 | BLK | 3.04 | ||
| BMX/ETK | 14.48 | BMX/ETK | 2.53 | ||
| BTK | 5.69 | BRK | 4.59 | ||
| CAMK1d | 16.57 | BTK | 2.09 | ||
| CHK1 | 38.9 | c-Kit | 3.27 | ||
| CHK2 | 21.7 | CSK | 7.11 | ||
| CK1d | 10.86 | c-SRC | 3.52 | ||
| CK1epsilon | 11.44 | DDR2 | 0.24 | ||
| c-MER | 28.92 | EGFR | 21.08 | ||
| CSK | 12.33 | EPHA1 | 2.72 | ||
| c-SRC | 6.05 | EPHA2 | 6.82 | ||
| EGFR | 3.84 | EPHA3 | 3.46 | ||
| EPHA2 | 9.58 | EPHA4 | 1.63 | ||
| EPHA3 | 31.66 | EPHA5 | 0.24 | ||
| EPHA4 | 27.01 | EPHA8 | 4.13 | ||
| EPHA5 | 37.14 | EPHB1 | 4.8 | ||
| EPHA6 | 4.38 | EPHB2 | 0 | ||
| EPHA8 | 10.52 | EPHB3 | 0.99 | ||
| EPHB1 | 11.8 | EPHB4 | 1.17 | ||
| EPHB2 | 17.43 | ERBB4/HER4 | 4.03 | ||
| EPHB3 | 49.42 | FGR | 1.7 | ||
| EPHB4 | 12.48 | FMS | 0.77 | ||
| ERBB2/HER2 | 32.95 | FRK/PTK5 | 0 | ||
| ERBB4/HER4 | 1.53 | FYN | 1.96 | ||
| FES/FPS | 35.71 | HCK | 0.79 | ||
| FGR | 11.49 | HGK MAP4K4 | 49.2 | ||
| FLT1/VEGFR1 | 44.54 | KHS MAP4K5 | 3.07 | ||
| FLT3 | 30.26 | LCK | 0.31 | ||
| FLT4/VEGFR3 | 44.1 | LIMK1 | 16.71 | ||
| FMS | 43.09 | LYN | 0.43 | ||
| FRK/PTK5 | 29.2 | LYN B | 1.6 | ||
| FYN | 29.42 | MINK/MINK1 | 46.54 | ||
| GCK MAP4K2 | 3.16 | NEK11 | 21.66 | ||
| HCK | 11.88 | NLK | 27.49 | ||
| HGK MAP4K4 | 1.03 | P38a/MAPK14 | 36.53 | ||
| IRR/INSRR | 36.9 | PDGFRa | 1.82 | ||
| KHS MAP4K5 | 1.72 | PDGFRb | 2.48 | ||
| LCK | 1.88 | RAF1 | 39.66 | ||
| LOK/STK10 | 12.61 | RET | 45.99 | ||
| LRRK2 | 30.12 | RIPK2 | 25.2 | ||
| LYN | 2.13 | SIK2 | 1.91 | ||
| LYN B | 6 | SRMS | 16.98 | ||
| MEK2 | 8.43 | TEC | 11.28 | ||
| MEKK2 | 23.47 | TXK | 1.37 | ||
| MEKK3 | 11.8 | YES/YES1 | 1.94 | ||
| MELK | 25.8 | ||||
| MINK/MINK1 | 1.21 | ||||
| MLCK2/MYLK2 | 32.05 | ||||
| MLK1/MAP3K9 | 16.48 | ||||
| MLK2/MAP3K10 | 22.88 | ||||
| MLK3/MAP3K11 | 13.47 | ||||
| MST1/STK4 | 7.4 | ||||
| MST2/STK3 | 13.86 | ||||
| MST3/STK24 | 11.02 | ||||
| MST4 | 5.9 | ||||
| MYO3b | 40.28 | ||||
| NEK1 | 15.17 | ||||
| NEK2 | 8.56 | ||||
| NEK4 | 32.6 | ||||
| PAK1 | 11.68 | ||||
| PAK3 | 9.9 | ||||
| PKCa | 49.46 | ||||
| PKCd | 40.27 | ||||
| PKCmu/PRKD1 | 43.71 | ||||
| PKCnu/PRKD3 | 42.25 | ||||
| PKD2/PRKD2 | 35.54 | ||||
| PYK2 | 32.56 | ||||
| RET | 18.94 | ||||
| RSK3 | 44.2 | ||||
| SIK2 | 1.93 | ||||
| SLK/STK2 | 21.59 | ||||
| STK25/YSK1 | 10.1 | ||||
| SYK | 21.1 | ||||
| TAK1 | 28.29 | ||||
| TRKC | 15.25 | ||||
| TXK | 27.53 | ||||
| TYRO3 SKY | 47.75 | ||||
| WEE1 | 40.47 | ||||
| YES/YES1 | 7.29 | ||||
| Bos-I | Dasatinib | ||||
| Kinase inhibited% of overlap | 84 | 35 | |||
| 41.7 | |||||
| Remaining Kinase activity (%) | |||||
| Kinases inhibited by: | Bos-I | Dasatinib | Inhibitor potency (Dasatinib / Bos-I) | ||
| ABL1 | 1.8 | 2.77 | 1.5 | ||
| ABL2/ARG | 2.19 | 2.74 | 1.3 | ||
| ACK1 | 3.54 | 2.62 | 0.7 | ||
| ARK5/NUAK1 | 11.43 | 15.93 | 1.4 | ||
| BLK | 20.73 | 3.04 | 0.1 | ||
| BMX/ETK | 14.48 | 2.53 | 0.2 | ||
| BTK | 5.69 | 2.09 | 0.4 | ||
| CSK | 12.33 | 7.11 | 0.6 | ||
| c-SRC | 6.05 | 3.52 | 0.6 | ||
| EGFR | 3.84 | 21.08 | 5.5 | ||
| EPHA2 | 9.58 | 6.82 | 0.7 | ||
| EPHA3 | 31.66 | 3.46 | 0.1 | ||
| EPHA4 | 27.01 | 1.63 | 0.1 | ||
| EPHA5 | 37.14 | 0.24 | 0.0 | ||
| EPHA8 | 10.52 | 4.13 | 0.4 | ||
| EPHB1 | 11.8 | 4.8 | 0.4 | ||
| EPHB2 | 17.43 | 0 | 0.0 | ||
| EPHB3 | 49.42 | 0.99 | 0.0 | ||
| EPHB4 | 12.48 | 1.17 | 0.1 | ||
| ERBB4/HER4 | 1.53 | 4.03 | 2.6 | ||
| FGR | 11.49 | 1.7 | 0.1 | ||
| FMS | 43.09 | 0.77 | 0.0 | ||
| FRK/PTK5 | 29.2 | 0 | 0.0 | ||
| FYN | 29.42 | 1.96 | 0.1 | ||
| HCK | 11.88 | 0.79 | 0.1 | ||
| HGK MAP4K4 | 1.03 | 49.2 | 47.8 | ||
| KHS MAP4K5 | 1.72 | 3.07 | 1.8 | ||
| LCK | 1.88 | 0.31 | 0.2 | ||
| LYN | 2.13 | 0.43 | 0.2 | ||
| LYN B | 6 | 1.6 | 0.3 | ||
| MINK/MINK1 | 1.21 | 46.54 | 38.5 | ||
| RET | 18.94 | 45.99 | 2.4 | ||
| SIK2 | 1.93 | 1.91 | 1.0 | ||
| TXK | 27.53 | 1.37 | 0.0 | ||
| YES/YES1 | 7.29 | 1.94 | 0.3 | ||
The % residual activity is shown. A comparison of inhibitor potency of dasatinib / Bos-I is shown (right) and values shown in red denote kinases inhibited more potently (>2-fold) inhibited by Bos-I compared with dasatinib.
Table 3. Table of screened compounds which also inhibit the kinases that are more potently inhibited (>2-fold) by Bos-I compared with Dasatinib. (as identified in Table 2).
| Remaining Kinase activity (%) | |||||
|---|---|---|---|---|---|
| Bos-I | Erlotinib | Gefitinib | Lapatinib | Sunitinib | |
| EGFR | 3.84 | 4.21 | 2.97 | 8.02 | 89.17 |
| HGK MAP4K4 | 1.03 | 76.48 | 97.78 | 111.21 | 12.44 |
| MINK/MINK1 | 1.21 | 85.68 | 102.32 | 110.74 | 10.46 |
| RET | 18.94 | 48.57 | 76.94 | 99.77 | 3.43 |
| ERBB4/HER4 | 1.53 | 61.41 | 24.15 | 11.19 | 94.81 |
Kinases inhibited by >50% are highlighted in red.
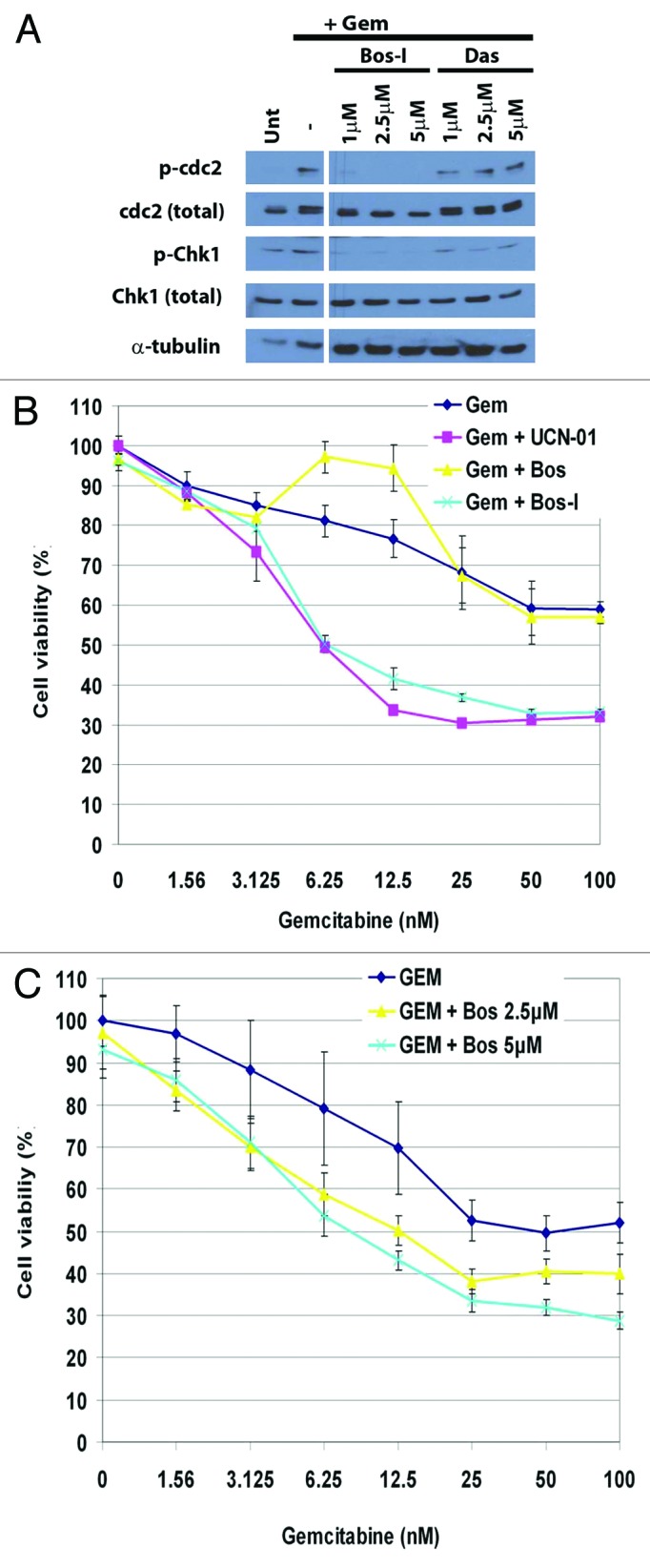
Figure 2. Chemosensitization occurs through off-target activities. (A) PANC1 cells were treated with gemcitabine (100 nM) for 24 h followed treatment with the indicated kinase inhibitors for an additional 6 h. Lysates were probed for p-Cdc2 (Y15), Cdc2, pChk1 (S296), Chk1, and α-tubulin. (B) PANC1 cells were treated with increasing concentration of gemcitabine (24 h) followed by addition of Bos-I or bosutinib (both 1 μM) for a further 48 h. Cell viability was assessed using MTS assay. Assays were performed in triplicate a minimum of 3 times and data are presented as the mean ± SD (C) Cells were treated as in (B), except bosutinib was used at 2.5 and 5 μM. Assays were performed in triplicate 3 times and data are presented as the mean ± SD.
Given the structural similarity of the 2 bosutinibs, we next compared the activities of the 2 drugs in a variety of assays. Using >50% inhibitory activity as a cut-off, we generated kinase inhibitory profiles of the authentic bosutinib (Tocris Bioscience) and the isomer (LC Labs) against a panel of 300 recombinant kinases. We found that both compounds inhibited a common set of 72 kinases, which include the Src family kinases. In addition, bosutinib more potently inhibited 16 kinases while Bos-I more potently inhibited 31 kinases, including Chk1 and Wee1 (Table 4). We next compared the inhibitory effects of the 2 compounds by determining their IC50 values for recombinant Src, Abl, Chk1 and Wee1 kinases. We found that authentic bosutinib was a more potent inhibitor of Src and Abl compared with Bos-I (IC50 for Src: bosutinib 40.5 ± 19.5 pM and Bos-I 4130 ± 900 pM; IC50 for Abl: bosutinib 32.4 ± 24 pM and Bos-I 566 ± 69.1 pM). However, Bos-I was more potent toward Chk1 and Wee1 compared with bosutinib (IC50 for Chk1: Bos-I 221 ± 45.4 nM and bosutinib 785 ± 136.6 nM; IC50 for Wee1: Bos-I 54.8 ± 12 nM and bosutinib 644 ± 195 nM). The ability of bosutinib to inhibit Chk1 and Wee, as well as its greater activity toward Wee1 compared with Chk1, is consistent with recent large scale kinase profiling studies.21 We next compared the chemosensitization properties of bosutinib vs. Bos-I. When used at 1 μM, Bos-I but not bosutinib synergized with gemcitabine, as determined by MTS assay (Fig. 2B). However, consistent with the authentic bosutinib’s relatively weaker in vitro inhibitory activities against Chk1 and Wee1, increasing its concentration to 2.5 or 5 μM sensitized PANC1 cells to gemcitabine (Fig. 2C). These findings support the idea that Wee1 and Chk1 are the targets responsible for chemosensitization and more potently inhibited by Bos-I compared with bosutinib.
Table 4. A list of kinases that preferentially inhibited (>50%) by bosutinib or Bos-I or those kinases equally inhibited (>50%) by both compounds.
| Bosutinib selective (>50% inhibitory activity) | Bos-I selective (>50% inhibitory activity) | Kinases inhibited in common (>50% inhibitory activity) |
|---|---|---|
| ALK | CAMK1g | ABL1 |
| CAMK2a | CHK1 | ABL2/ARG |
| CLK1 | FGFR1 | ACK1 |
| CLK3 | FGFR2 | ARK5/NUAK1 |
| DDR2 | FLT1/VEGFR1 | AXL |
| EPHB3 | FLT3 | BLK |
| IKKe/IKBKE | FLT4/VEGFR3 | BMX/ETK |
| MLCK/MYLK | IR | BTK |
| PHKg1 | IRR/INSRR | c-MER |
| STK33 | KDR/VEGFR2 | c-SRC |
| TBK1 | MELK | CAMK1d |
| TEC | MLCK2/MYLK2 | CHK2 |
| TRKA | MLK1/MAP3K9 | CK1d |
| TRKB | MLK2/MAP3K10 | CK1epsilon |
| ULK1 | NEK1 | CSK |
| ZAK/MLTK | NEK2 | DDR1 |
| PAK1 | EGFR | |
| PAK2 | EPHA2 | |
| PAK3 | EPHA3 | |
| PKCepsilon | EPHA4 | |
| PKCmu/PRKD1 | EPHA5 | |
| PKCnu/PRKD2 | EPHA6 | |
| RET | EPHA8 | |
| RSK4 | EPHB1 | |
| STK32B/YANK2 | EPHB2 | |
| TAK1 | EPHB4 | |
| TAOK1 | ERBB2/HER2 | |
| TTBK1 | ERBB4/HER4 | |
| TTBK2 | FES/FPS | |
| WEE1 | FGR | |
| FMS | ||
| FRK/PTK5 | ||
| FYN | ||
| GCK MAP4K2 | ||
| HCK | ||
| HGK/MAP4K4 | ||
| HPK1/MAP4K1 | ||
| KHS/MAP4K5 | ||
| LCK | ||
| LOK/STK10 | ||
| LRRK2 | ||
| LYN | ||
| LYN B | ||
| MARK4 | ||
| MEK1 | ||
| MEK2 | ||
| MEKK2 | ||
| MEKK3 | ||
| MINK/MINK1 | ||
| MLK3/MAP3K11 | ||
| MST1/STK4 | ||
| MST2/STK3 | ||
| MST3/STK24 | ||
| MST4 | ||
| MYO3b | ||
| NEK4 | ||
| PAK3 | ||
| PKCd | ||
| PYK2 | ||
| RSK3 | ||
| SIK1 | ||
| SIK2 | ||
| SLK/STK2 | ||
| STK25/YSK1 | ||
| SYK | ||
| TNIK | ||
| TRKC | ||
| TXK | ||
| TYK1/LTK | ||
| TYK2 | ||
| TYRO3/SKY | ||
| YES/YES1 |
Phenotypic and biochemical studies demonstrate Chk1 and Wee1 are targets of Bos-I
We next sought cellular and biochemical evidence that Chk1 and Wee1 are targets of Bos-I. First, timelapse microscopy was used to directly assess the effects of Bos-I (and other kinase inhibitors; Fig. S3A) on gemcitabine-treated PANC1 cells. Gemcitabine inhibits DNA replication, causing an S phase checkpoint arrest that block entry into mitosis. However, addition of Bos-I or UCN-01 to gemcitabine-arrested cells resulted in an increase in the number of mitotic cells over the duration of filming (24 h) (Fig. 3A), consistent with these compounds causing checkpoint override. This data was corroborated by flow cytometry which also showed an increase in mitotic cells (p-MPM2 positive) after addition of Bos-I to gemcitabine-treated cells (Fig. S3B). Moreover, videos of PANC1 cells expressing GFP-Histone H2B (GFP-H2B) treated with gemcitabine followed by Bos-I showed that they entered an aberrant mitosis and either died in mitosis or exited with highly multinucleated cells, indicative of mitotic catastrophe (Fig. 3B). Second, the mitotic figures of these cells displayed a signature defect that is exhibited by cells forced to enter mitosis with incompletely replicated genomes.22 Early EM studies showed that cells forced into mitosis with unreplicated genomes (MUGs) exhibited highly fragmented chromosomes that included separation of the centromere/kinetochore complex.23 Using light and electron microscopy, we previously reported observing similar features for cells forced into mitosis after gemcitabine and UCN-01 treatment.22 We therefore compared the mitotic figures of gemcitabine-arrested cells treated with Bos-I and UCN-01. Both treatments caused highly fragmented chromosomes and detachment of centromeres from the bulk of the chromosomes (Fig. 3C). The combined results clearly establish that Bos-I forces gemcitabine-arrested cells to enter a catastrophic mitosis, consistent with it targeting kinases involved in the DNA damage checkpoint pathway.

Figure 3. Phenotypic and biochemical studies demonstrate Chk1 and Wee1 are targets of Bos-I. (A) PANC1 cells were treated with gemcitabine (100nM) for 24h before the addition of UCN-01 (100 nM) or Bos-I (1 µM). Cell fates were tracked by video-microscopy for 24 h, with images collected every 5 min. Representative montages of cells that were untreated, treated with gemcitabine or gemcitabine followed by Bos-I or UCN-01 and quantification of the percentage of cells that entered mitosis during filming is shown. A minimum of 100 cells were scored. (B) PANC1 cells expressing GFP-H2B were used for live cell videomicroscopy. Following treatments, distinct cell fates were observed and montages of such examples are shown. Quantification of the cell fates following treatments from >100 cells is shown. (C) PANC1 cells treated as indicated were fixed and stained for CENP-F (kinetochores), α-tubulin (mitotic spindle) and counter-stained with DAPI (DNA). 100% of the mitotic cells examined exhibit the same defects. (D) PANC1 cells were treated with gemcitabine (10 nM) for 24 h followed by Bos-I (1 µM) for an additional 3 h. Cells were fixed and immunostained with γH2AX and RAD51 antibodies. The percentage of RAD51 positive cells is shown from >100 cells per treatment. *P < 0.001 compared with gemcitabine alone. Quantitative analysis of the average γH2AX intensity per cell. ***P < 0.001, **P < 0.005 vs. gem 10 nM; *P < 0.001 vs. gem 2 µM. (E) Mitotic figures generated after indicated treatments were immunostained with γH2AX antibody. 100% of the aberrant mitotic figures were positive for γH2AX. Representative images are shown.
We performed 2 additional assays to demonstrate that Bos-I can inhibit Chk1 and Wee1 in cells. In addition to their roles in checkpoint control, Chk1 and Wee1 are essential for maintaining the stability of stalled replication forks to facilitate fork restart. Inhibition of either kinase results in the collapse of stalled forks and generation of double strand breaks (DSBs) that are detectable by an increase in phospho-H2AX (γH2AX) foci.24,25 In this context, γH2AX serves as a pharmacodynamic marker of Chk1 or Wee1 inhibition in cells undergoing replication stress. When PANC1 cells were treated with gemcitabine at 10 nM (24 h) or 2 μM (2 h, then washed out), γH2AX foci increased as expected.24 Notably, addition of Bos-I to gemcitabine-treated cells further increased γH2AX foci formation by ~3-fold, as similarly seen for UCN-01 (Fig. 3D). As a confirmation, we depleted endogenous Wee1 with an siRNA that targeted its 3′UTR, and in parallel transfected a plasmid that co-expressed FLAG-tagged murine Wee1 (3xFLAG-mWee1) and GFP-H2B, allowing identification of transfected cells (Fig. S3C). Cells transfected with control siRNA and treated with gemcitabine (10 nM) showed in a modest increase in γH2AX. In cells that were depleted of Wee1 and treated with gemcitabine, γH2AX was increased 3-fold compared with gemcitabine-treated controls. Importantly, γH2AX was reduced ~2.5-fold in cells transfected with mWee1 (GFP-H2B positive) compared with neighboring untransfected cells (GFP-H2B negative) (see Fig. S3D; arrow). Moreover, Bos-I treatment failed to increase γH2AX staining in the mWee1 transfected cells (GFP-H2B positive) that were treated with gemcitabine. This suggested that excess Wee1 attenuated Bos-I promoted collapse of stalled forks (Fig. S3D) and confirmed the relationship between Wee1, Bos-I and stabilization of stalled forks. The second assay was to assess the effects of Bos-I on Rad51 foci formation, a critical component of error-free homologous recombination that is used to repair stalled and collapsed forks.26 Previous studies showed that RAD51 forms foci upon gemcitabine treatment and this localization pattern is dependent on Chk1.27 Consistent with those studies, gemcitabine treated cells exhibited increased Rad51 foci formation in approximately 65% of cells. However, addition of Bos-I reduced the percentage of RAD51 positive cells to approximately 15% (Fig. 3D).
The combined cell-based and biochemical assays strongly suggest that Bos-I inhibits the DNA damage checkpoint, by targeting Chk1 and Wee1. This causes irreparable DSBs that lead to chromatin and centromere fragmentation when gemcitabine treated cells are forced to prematurely enter mitosis. Moreover, these observations provide a mechanistic explanation for why cells undergoing premature mitosis have extensive γH2AX staining (Fig. 3E).22
Chemosensitization is observed in multiple cell lines and with different chemotherapeutic agents
Previous studies have shown that Chk1 inhibitors can synergize with a variety of DNA damaging agents.1,3,9 To explore whether Bos-I could similarly be used to enhance the effects of other DNA damaging agents, PANC1 cells were treated with the DNA cross-linking agent cisplatin and the topoisomerase II inhibitor doxorubicin. Both agents dose-dependently impaired cell cycle progression, as expected (Fig. S4). Consistent with the observation for gemcitabine, Bos-I enhanced the cytotoxic effects of both cisplatin- and doxorubicin (Fig. 4A). Mia PaCa-2 cellular sensitivity to gemcitabine and doxorubicin was also found to be enhanced after treatment with Bos-I (Fig. 4B). However, in normal RPE1 cells treated with gemcitabine or doxorubicin treatments (Fig. 4C), the addition of Bos-I did not further reduce proliferation. Finally, to address whether the chemosensitization effect of checkpoint override inhibitors was dependent on DNA damaging agents, PANC1 cells were treated with increasing concentrations of paclitaxel (taxol), which impairs microtubule dynamics and arrests cells in mitosis (Fig. S4). We found that in cells treated with taxol none of the inhibitors further reduced proliferation (Fig. 4D).

Figure 4. Chemosensitization is observed in multiple cell lines and with different chemotherapeutic agents (A) PANC1, (B) MiPaCa-2 (MP-2), (C) RPE1-hTERT cells were treated with increasing concentrations of increasing concentrations of indicated drugs (24 h) before the addition of Bos-I. Cell viability was assessed 48 h later using MTS assay. (D) PANC1 cells were tested for taxol sensitization by Bos-I and UCN-01. All experiment (A–D) was done in triplicate and experiments were performed at least 3 independent times. Results are presented as the mean ± SD.
Bosutinib isomer is a potent sensitizer of gemcitabine in pancreatic cancer xenografts
To explore the clinical relevance of Bos-I, we tested its effectiveness as a chemosensitizer in cells that were generated from a patient’s pancreatic adenocarcinoma (PDAC).22 We confirmed by immunofluorescence that EGF-1 cells treated with gemcitabine resulted in a reduced mitotic index but that the checkpoint arrest was overridden by Bos-I as judged by an increase in the mitotic index (Fig. 5A). Consistent with checkpoint override, EGF-1 cellular sensitivity to gemcitabine was enhanced by Bos-I (Fig. 5B). Based on these data, we used the EGF-1 cells to perform xenograft studies to evaluate the in vivo efficacy of Bos-I alone and in combination with gemcitabine. Animals were treated on a weekly schedule consisting of gemcitabine (50 mg/kg) on day 1 followed by Bos-I (100 mg/kg) on days 2 and 3—the rationale being that Bos-I would be maximally effective after cells were arrested in the cell cycle by gemcitabine. As shown in Figure 5C, tumors in vehicle treated animals grew linearly while Bos-I alone significantly reduced tumor growth, demonstrating significant efficacy as a single agent up until day 17 (P = 0.013). However, after 17 d, tumors resumed growth in animals treated with either gemcitabine or Bos-I as single agents (P = 0.006 and P < 0.001, respectively) suggesting the development of drug resistance. In marked contrast, the combination of gemcitabine and Bos-I, significantly attenuated tumor growth when compared with either single agent treatment alone (P < 0.02). To explore whether the mechanism of action for sensitization in vivo was similar to that in vitro, we used the phamacodynamic assay as employed above to measure γH2AX. In animals treated with gemcitabine alone there was an increase in γH2AX in the tumor cells compared with tumor cells from vehicle treated animals (P < 0.01). Moreover, γH2AX levels were highest in the tumor cells from animals treated with the combination of gemcitabine and Bos-I (P < 0.0001) (Fig. 5D). We also observed that the mitotic index was reduced in tumors from animals treated with gemcitabine as compared with vehicle treated animals (vehicle 2.2 ± 0.7% of mitotic cells vs. gem-treated 0.4 ± 0.4% of mitotic cells; P = 0.03). However, in tumors from animals treated with the combination of gemcitabine and Bos-I, the mitotic index was increased compared with animals treated with gemcitabine alone (gem-treated 0.4 ± 0.4% of mitotic cells vs. gem+Bos-I treated 2.0 ± 0.6% of mitotic cells; P = 0.05), consistent with the in vitro data. Furthermore, the extent of DNA damage as assessed by γ-H2AX was approximately 2-fold higher in mitotic tumor cells in animals treated with gemcitabine and Bos-I compared with animals treated with vehicle alone (Fig. 5E). These findings show that Bos-I can enhance inhibition of tumor growth by gemcitabine in vivo.

Figure 5. Bosutinib isomer is a potent sensitizer of gemcitabine in pancreatic cancer xenografts. (A) A patient derived pancreatic cell line, EGF-1, was treated with gemcitabine (100 nM) for 24h before the addition of UCN-01 (100 nM) or Bos-I (1 μM) for a further 9 h. Cells were then fixed and immunostained with α-tubulin (red) and DAPI (blue). Quantification of the mitotic index from a minimum of 100 cells is provided. (B) EGF-1 cell viability was assessed after treatment with increasing concentrations of gemcitabine for 24 h followed by Bos-I (1 μM) for a further 48 h before addition of MTS reagent. (C) Xenograft studies using EGF-1 cells to test combinatorial treatment were performed. Tumors were allowed to grow to 100mm3 before being randomized and treated with either vehicle, gemcitabine alone (50 mg/kg), Bos-I alone (100 mg/ml) or a combination of the two. The tumor growth index was used to determine efficacy of drug treatments. *P = 0.013 compared with vehicle treated animals on day 17. **P = 0.006 compared with gemcitabine-treated animals at day 17. ***P < 0.001 compared with Bos-I-treated animals at day 17. †P < 0.001 compared with either Bos-I or gemcitabine treated animals. Data are presented as the mean ± SEM (D) The average γH2AX intensity per tumor cell was determined via IHC staining after 1 round of treatments. A minimum of 100 cells were quantified and the data are presented as average ± SEM *P = 0.01, **P = 0.0001 compared with vehicle treated animals. (E) As for (D), except the average γH2AX intensity per mitotic tumor cell is shown ± SEM. A minimum of 25 mitotic cells were quantified. *P = 0.0015.
The gatekeeper residue in Wee1 contributes to inhibitor specificity
To understand the molecular basis for the differences in the specificity of bosutinib and Bos-I for Chk1 and Wee1, molecular models were constructed to visualize the interactions between the inhibitors and the ATP binding pocket. Structurally, the 2 compounds only differ in the positioning of the Cl and methoxy atoms on the aniline ring (Fig. S1), suggesting that the spatial position of these groups dictate their interaction within the ATP binding pocket. It is well established that the gatekeeper residue of kinases can dictate the ability for inhibitors to bind.28 Our molecular models showed that the methoxy group of authentic bosutinib (position 5) may sterically clash with gatekeeper residue in Chk1 (Leucine, L84) and Wee1 (Asparagine, N376) and possibly reduce its efficacy. By contrast, the methoxy group in Bos-I (position 3) is positioned away from the gatekeeper (Fig. 6A). This suggests that the spatial relationship between the gatekeeper residue and the methoxy group in bosutinib and Bos-I may dictate the ability of the inhibitors to bind and thereby inhibit. These observations were supported by relative binding energy calculations derived from our models of bosutinib and Bos-I bound to Chk1 and Wee1. Consistent with cellular and biochemical data presented above, the calculations indicated that Bos-I had a stronger preference for both Wee1 and Chk1 compared with bosutinib (Table 5).

Figure 6. The gatekeeper residue in Wee1 contributes to inhibitor specificity. (A) Ribbon diagrams of published structures of Chk1 (PDB code 2HY0) and Wee1 (PDB code 3BI6) bound with the inhibitors bosutinib or Bos-I which are superimposed (main). Insets show enlarged view of the pocket bound to their inhibitors (gray backbone). The gatekeeper residues are shown in pink (black arrow) for both kinases. (B) In vitro kinase assays were used to assess the ability of bosutinib and Bos-I (0.5–10 µM) to inhibit recombinant Wee1 WT and Wee1 gatekeeper mutants. Phosphorylation of purified cdc2 (p-Cdc2 Y15) was used as a read-out for Wee1 activity. Kinase reactions were performed at least twice. A representative western blot is shown. (C) Ribbon diagram of the Wee1 (PDB code 3BI6) gatekeeper mutant N376F (shown in pink, highlighted with arrow). Insets show enlarged view of bosutinib and Bos-I and the positioning of the gatekeeper residue.
Table 5. Binding Energy of 2 inhibitors bound to 3 kinases.
| Kinase | PDB | Kinase with Bosutinib | Kinase with Bos-Isomer |
|---|---|---|---|
| Wee1 | 3BI6 | 390 | 448 |
| Chk1 | 2HY0 | 449 | 574 |
| Abl | 3UE4 | 536 | 526* |
| Binding energy of inhibitors (kcal/mol) | |||
The binding energy as assessed by the program Yasara36 using the AMBER 2003 force field without long range electrostatics enabled and BindEnergy command. The BindEnergy command calculates the binding energy of the inhibitor with respect to the rest of the protein according to the current force field.37 The binding energy is obtained by calculating the energy at infinite distance (between the inhibitor and solvent, i.e., the unbound state) and subtracting the bound state. The more positive the binding energy, the more favorable the interaction in the context of the chosen force field. The starting coordinates for the 3 proteins were obtained from the listed PDB codes.
To explore the importance of the gatekeeper residue in determining the sensitivity to bosutinib or Bos-I binding, we used the aforementioned kinase inhibitor database to identify kinases that are preferentially inhibited (>50%) by Bos-I or bosutinib and then compared the gatekeeper residues. For Bos-I, the gatekeeper residues were predominantly methionine, or valine, while similar analysis of bosutinib sensitive kinases identified predominantly methionine, threonine or phenylalanine as gatekeeper residues (Fig. S5). This analysis clearly demonstrated that the gatekeeper residue of bosutinib-sensitive kinases were enriched for threonine or phenylalanine and provided us with an opportunity to test whether these gatekeeper residues might dictate selectivity for bosutinib over Bos-I. We performed in vitro kinase assays using recombinant Wee1 wild-type (WT) as well as gatekeeper mutants N376F and N376T and tested the inhibitory activity of bosutinib and Bos-I using phosphorylation of Cdc2, its physiological substrate, as a read out. We observed that phosphorylation of Cdc2Y15 by Wee1 WT was potently inhibited by Bos-I, while approximately 4-fold more bosutinib was needed to achieve similar inhibition (IC50 values: Bosutinib 6.9 µM vs. Bos-I 1.8 µM). Importantly, both the N376T and the N376F mutant exhibited increased sensitivity to bosutinib as its IC50 was reduced to 1.9 µM and 0.9 µM, respectively. The sensitivity of the N376T mutant to Bos-I was unchanged (Bos-I Wee1 WT 1.8 µM vs. N376T 1.6 µM), while the N376F mutant was now less sensitive to Bos-I (IC50: WT 1.8 µM vs. N376F 8.3 µM). These findings demonstrate that while the N376T mutant improves sensitivity to bosutinib, the sensitivity to the N376F mutant toward the inhibitors is flipped, compared with the Wee1 WT. We next generated molecular models of both inhibitors bound to the mutants. For the N376F mutant, the orientation of the benzyl group of the phenylalanine differed depending on which inhibitor was bound. When bosutinib was bound, the benzyl group was rotated away from the 5-methoxy group leading to reduced steric hindrance. In contrast, when Bos-I was bound, the benzyl group of N376F was orientated such that it was proximal to the 4-methoxy group. This orientation would generate a steric clash between the benzyl group and the 4-methoxy group that could explain the resultant decrease in ability to inhibit Wee1. In performing similar analysis of the 376T mutant (Fig. S6), we were unable to identify structural differences that may account for why Bos-I is slightly more effective compared with the WT Wee1, nor for the dramatic increase in effectiveness of bosutinib as compared with WT Wee1. Using a combination of in vitro kinase reactions and modeling studies, we provide evidence that both inhibitors can bind Wee1, albeit to slightly different abilities. Furthermore, the gatekeeper residue appears to be an important, but unlikely to be the sole determinant, for sensitivity,
Discussion
We took advantage of the promiscuity of kinase inhibitors13 to identify compounds that would enhance cellular sensitivity to gemcitabine. Our screen revealed BEZ-235, dovitinib, and Bos-I as compounds that were found to enhance killing of PANC1 cells to a sub-lethal concentration of gemcitabine. BEZ-235 is a PI3K/mTOR inhibitor that was reported to have off target effects against the related ATR and ATM/DNA PKcs kinases that result in radio-sensitization.18,19 Thus, the sensitization activity of BEZ-235 to gemcitabine observed in our screen was likely due to the off-target activities toward ATM and ATR. Dovitinib and Bos-I inhibit RTKs and Src/Abl kinases, respectively, and are not previously known to impinge upon the DNA damage checkpoint pathway. Tellingly, dasatinib, a potent inhibitor of Src/Abl, did not exhibit sensitization activity. This argues against Src/Abl as the targets for sensitization. Further arguing against Abl/Src is the fact that Bos-I was a more potent chemosensitizer (2.5–5-fold) than bosutinib, but was a weaker inhibitor of Src (100-fold) and Abl (17-fold) in vitro.
Bos-I was recently reported to have been unknowingly synthesized and sold as bosutinib, a drug approved by the FDA for the treatment of CML. The existence of the isomer was uncovered by crystallographic studies of bosutinib bound to Abl kinase. The anomalous electron density map led to the discovery that the drug used for the studies was in fact the isomer.20 Bos-I is estimated to have been unknowingly circulated among the research community for approximately 6 y and many companies have since been identified to have unknowingly sold the isomer instead of the authentic drug (http://www.pkcpharma.com/TwoOrMoreBosutinibs.html).
Cell-based assays confirmed that Bos-I was could cause drug-arrested cells to override the DNA damage checkpoint. To identify candidate targets responsible for checkpoint override activity, we queried a database13 and found that dovitinib and Bos-I inhibited Chk1 by >50%. Interestingly, Bos-I was one of only 2 compounds in the database (178 compounds total) found to inhibit Wee1 kinase. The other compound was SB218078, a particularly promiscuous inhibitor which inhibited 174/300 of the kinases tested but has not been tested in clinical trials. This observation strongly suggests that the inhibitory activity of bosutinib and Bos-I toward Wee1 is specific, given the very selective profile of Wee1.
When testing the authentic bosutinib for sensitization activity, we found that a 2.5-fold higher concentration was required to achieve a similar degree of sensitization as observed with Bos-I. The weaker sensitization activity of bosutinib is consistent with the ~4 and ~11-fold weaker inhibitory activity against Chk1 and Wee1 kinases, respectively, as compared with Bos-I. Nonetheless, both these compounds have off-target activity toward Chk1 and Wee1. This off target activity toward Chk1 and Wee1 has previously been reported but only in the context of comprehensive kinase profiling screens where 72 inhibitors were tested against a panel of >400 kinases. These studies used a competitive binding assay to analyze kinase inhibitor selectivity and identified that bosutinib binds Wee129 (www.discoverx.com) and Chk1,29 albeit less potently compared with its main targets of Abl and Src. Moreover, the affinity of bosutinib was reported to be greater for Wee1 vs. Chk1 (Kd Wee1 = 510 nM, Kd Chk1 3000 nM),29 paralleling our in vitro kinase inhibitory data. Taken together, the findings from these large scale screens are fully consistent with our data that showed bosutinib and Bos-I can inhibit Chk1 and Wee1. This finding thus provides a mechanistic basis for checkpoint override and chemosensitization that we detail. However, is has been shown that inhibition of the upstream kinases ATR/ATM can also cause drug-arrested cells to prematurely enter mitosis23 and enhance IR-induced cell death through this process.18,19 From our studies, we cannot rule out bosutinib or Bos-I having additional inhibitory activity toward ATR or ATM as these kinases were not present in the kinase profiling screen performed.
The findings presented here shows that p53-deficient cell lines forced to enter a defective mitosis is an effective way to induce cell death. This is in accordance with previous studies using drug treatments to induce DNA damage (via doxorubicin), followed by checkpoint override (via UCN-01) followed by paclitaxel (mitotic arrest), representing a rational approach to selectively kill p53-deficient tumors.30 These approaches confirm that exploitation of a defective mitosis represents an effective approach to kill p53 deficient cells.
We sought to understand the structural basis as to why Wee1 is more potently inhibited by Bos-I as compared with bosutinib. Given the role of the gatekeeper residue in regulating inhibitor accessibility to the ATP pocket,28 we studied its involvement further. By detailing the gatekeeper residues of kinases more favorably inhibited by Bos-I over bosutinib, and vice versa, we discovered that bosutinib-favorable gatekeeper residues were phenylalanine, threonine or methionine. In our study of Wee1, we hypothesized that since the gatekeeper is asparagine, mutating it to more bosutinib-favorable gatekeeper residues may enhance its sensitivity to bosutinib. Indeed, the N376F and N376T mutants both showed increased sensitivity (~7.5-fold and ~3.5-fold, respectively) to bosutinib compared with Wee1WT. We attempted to use the Wee1 mutants to test whether we could recapitulate in cells the differential sensitivity we observed in vitro. This approach was confounded by the fact that overexpression of wild-type Wee1 rendered cells insensitive to bosutinib or Bos-I even up to concentrations of 10 µM, as judged by γ-H2AX staining (data not shown). Nonetheless, our structural and in vitro data are consistent with a study that showed the importance of the gatekeeper residue in dictating specificity of bosutinib toward the Src kinase.31 This structural study showed that the threonine gatekeeper in Src was critical for its sensitivity toward bosutinib, and mutating it to asparagine (which is coincidently the gatekeeper residue in Wee1) significantly reduced its potency. The model put forth by the authors propose that water molecules within the ATP binding pocket can form hydrogen bond interactions with side chains of inhibitors, in this case with the nitrile group of bosutinib and Bos-I. Alteration of the gatekeeper residue, thus altering the pocket size, can disrupt the bosutinib-water interaction and weaken binding, and less inhibitory activity. However, the same gatekeeper substitutions that disrupted binding of bosutinib did not affect binding of Bos-I.31 Thus, although the gatekeeper is known to be a crucial factor in dictating kinase inhibitor sensitivity, it is clear other factors are involved.
Bosutinib and Bos-I are not as potent as inhibitors specifically designed to target Chk16,10 and Wee1.32 Accordingly, higher concentrations of these inhibitors are required to achieve sensitization relative to bona fide Wee1 and Chk1 inhibitors. Thus, the checkpoint override and chemosensitization properties of the 2 bosutinibs may be due to the dual inhibition of Chk1 and Wee1, despite both kinases acting in the same pathway. Consistent with this notion, chemosensitization by MK1775, a Wee1 inhibitor that is currently in Ph2 trials, was further enhanced with Chk1 inhibitors.8,32
Given that our goal was to improve treatment options for pancreatic cancer patients, we used a patient derived pancreatic tumor xenograft (PDX) to test the ability of Bos-I alone or in combination with gemcitabine to suppress tumor growth. The results show that Bos-I, when used alone, suppressed tumor growth for ~17 d at which point the tumors grew at rates comparable to vehicle treated animals and subsequent treatments failed to blunt tumor growth. A previous study using bosutinib (retrospectively thought to be the isomer based on where it was purchased) showed it had significant activity as a single agent against a spontaneous metastatic thyroid cancer mouse model in which Src is constitutively active.33 The treatment regimen that inhibited tumor growth and increased survival was 150 mg/kg, 5 times a week. Although inhibition of Src was proposed to be the mechanism of growth inhibition, it is possible that the off-target activity toward Chk1 and Wee1 may also have been a contributory factor. The more conservative regimen employed in our study (100 mg/kg, twice a week) may explain why we did not observe a more durable response with Bos-I as single agent. Nonetheless, taken together these findings substantiate the notion that Bos-I can have significant activity as a single agent. These findings are consistent with in vitro and in vivo studies demonstrating that pharmacologic inhibition of Chk1 alone is sufficient to kill tumor cells exhibiting replication stress.33 Furthermore, Chk1 inhibitor efficacy was found to correlate with Myc levels.34 It is notable that the EGF-1 cells were found to have an amplification of the Myc locus, suggesting a mechanistic basis for why Bos-I demonstrated single agent activity. Of pertinence, we found that Bos-I given in combination with gemcitabine significantly suppressed tumor growth for the duration of the 30 d experiment. Our findings suggest that the dose-scheduling may be an important parameter for combinatorial efficacy. Based on our cellular studies, we specifically dosed animals with Bos-I 24h after gemcitabine to try and achieve 2 aims. First, since Bos-I should only be effective after DNA damage-mediated checkpoint activation, when Chk1 and Wee1 are active, maximal kinase inhibitory activity should be observed. Consistent with this notion, a recent study demonstrated that the Chk1 inhibitor GNE-900 potentiated the effects of gemcitabine in vitro and in vivo and that the optimal dose-scheduling required a lag period (24 or 36 h) between gemcitabine administration followed by Chk1 inhibition.35 Second, by limiting Bos-I treatment to twice a week, this scheduling may help to reduce toxicities as observed with prolonged treatment of Chk1 inhibitors in clinical trials. This study did not address whether the combinatorial treatment induced senescence and/or indirectly affected tumor growth, for example through disrupting angiogenesis. Clearly these are important factors that can dictate tumor growth and affecting these processes could contribute to the efficacy observed.36 Our studies used gemcitabine because it is a major modality of treatment for pancreatic cancer patients. However, we believe that the choice of which chemotherapy to be used along with bosutinib or Bos-I should be explored further. Indeed, not all chemotherapeutics can be potentiated with the Chk1 inhibitor GNE-900 and that even with agents that induce DNA damage (antimetabolite, alkylating, topoisomerase inhibitors), there is a variation in chemosensitizing ability.37 Optimizing bosutinib and Bos-I with other DNA damaging agents may identify combinatorial therapies which yield greater efficacy. Finally, we propose that although Bos-I has not yet been tested in humans, it is possible that the current FDA approved bosutinib might improve response of cancer patients treated with genotoxic agents.
Materials and Methods
Reagents
The following reagents were obtained from Sigma: doxorubicin, cisplatin, UCN-01, and Tween-80, propidium iodide. Gemcitabine was obtained from the FCCC pharmacy. The inhibitors dovitinib, erlotinib, gefitinib, lapatinib, sunitinib, dasatinib, BEZ-235, and bosutinib isomer-1 (Bos-I) was purchased from LC Labs while bosutinib (authentic) and paclitaxel from Tocris.
Cell culture
Cell lines were obtained from ATCC and banked at Fox Chase Cancer Center (FCCC) until use. Mycoplasma testing was conducted at FCCC prior to studies. PANC1 (p53 mutant), Mia PaCa-2 (p53 mutant), RPE1-hTERT (p53 WT) cells were grown in DMEM supplemented with 10% FBS, 2 mM glutamine and 1% penicillin, streptomycin and kanamycin. EGF-1 cells were derived and used as previously described.22
In vitro kinase assays
Kinase inhibitory profiles of bosutinib and Bos-I were conducted by Reaction Biology Corp using the ‘HotSpot” assay platform as previously described.13 IC50 values and curve fits were obtained by performing a 10 dose titration of each drug (30 µM – 1.52 nM) (n = 4 independent experiments) and data analyzed by Prism (GraphPad Software). Data available from Reaction Biology Corp (www.reactionbiology.com/webapps/kir/).
In vitro kinase reactions were performed with baculovirus expressed GST-murine Wee1 (the pENTR plasmid was kindly provided by G Enders, FCCC38) and cdc2 as substrate. Site-directed mutagenesis was used to generate N376F and N376T mutants. The mutants were generated, purified and used in in vitro assays as for Wee WT. Inhibitors at the indicated concentrations were pre-incubated with GST-Wee1 for 20 min on ice before addition of cdc2 and 50 μM ATP and 3× reaction mix (MgCl2 30 mM, ATP 150 μM, DTT 3 mM) Reactions were then transferred to 37 °C for 30 min before termination by the addition of 3× SDS buffer. Samples were subjected to SDS-PAGE and immunoblotted for Cdc2 phospho-Y15 (New England Biolabs), total Cdc2 (Santa Cruz), Wee1, and GST (Cell Signaling Technology). In vitro assays were performed at least twice for each mutant. Densitometry was performed using Image J (http://www.NIH.gov) on the p-cdc2 signal relative to total cdc2, allowing calculation of IC50 values of each inhibitor.
Wee1 rescue studies
pENTR murine Wee1 (a kind gift from Dr G Enders38) was used to generate a 3xFLAG-mWee1 co-expressing GFP-H2B construct which was transfected into PANC1 cells using Fugene 6 (Roche). Control siRNA #4 (Dhamacon) or siRNA targeting the 3′UTR of human Wee138 was introduced into cells using HiPerfect transfection reagent (Qiagen). Twenty-four hours (24 h) later, cells were reseeded onto glass coverslips. Cells were treated with or without gemcitabine (10 nM) for 24 h before being harvested. Where indicated, Bos-I (1 μM) was added for the last 6 h of treatment. Validation of transfected cells was performed by immunofluorescence to demonstrate coincident GFP-H2B and FLAG (Sigma) positive cells.
Molecular modeling to assess inhibitor binding
Structure of Chk1 (PDB code 2HY0) and Wee1 (PDB code 3BI6) were superimposed on Abl protein with bound bosutinib (PDB code 3UE4). The exact position of bosutinib was copied into the Chk1 and Wee1 structures. Second the structure of Bos-I was copied from Stk10 kinase with Bos-I bound (PDB code 4BC6) and this inhibitor was superimposed onto the bosutinib position in the active sites of both proteins. Each of the 2 proteins with either inhibitor were subjected to energy minimization following removal of water molecules, using the AMBER 2003 force field with long-range electrostatics disabled in the program Yasara39 and BindEnergy command. The BindEnergy command calculates the binding energy of the inhibitor with respect to the rest of the protein according to the current force field.40 The binding energy is obtained by calculating the energy at infinite distance (between the inhibitor and solvent, i.e., the unbound state) and subtracting the bound state. Binding energy was assessed with the same force field and reported in units of kcal/mole in Table 5. The more positive the binding energy, the more favorable the interaction in the context of the chosen force field. Gatekeeper residues in Wee1 were substituted in Yasara and energy minimizations performed with the same methodology as above, in the presence of the corresponding inhibitor. The resulting side-chain rotamer best satisfied the steric and energy considerations. Identification of gatekeeper residues in Bos-sensitive and Bos-I-sensitive kinases was obtained from Huang et al.41
Cell viability assays
For screening of kinase inhibitors we used the MTS assay (Promega) as previously employed.5 Briefly, cells seeded into 96 well plates were treated with vehicle or gemcitabine (10 nM) for 24 h before the addition of kinase inhibitors (all at 1 μM except for UCN-01 which was 100 nM) for a further 48 h. Cell viability was then determined by measuring the absorbance at 490 nm. All data points were normalized to untreated cells, allowing us to ascertain that the effects of the inhibitors alone was negligible (<10%). All treatments were performed in triplicate and performed a minimum of 3 times.
For clonogenic assays, 1000 cells per well were seeded into a 6 well plate. Cells were treated with gemcitabine (10 nM) for 24 h. Kinase inhibitors (all at 1 μM except for UCN-01 which was 100 nM) were added for a further 3 h before replacing with drug-free medium. Cells were allowed to grow for 10 d before being fixed and stained with crystal violet for quantitation as previously described.5 To quantify apoptosis, the Guava Nexin assay (Millipore) was employed to detect the percentage of cells positive for Annexin V. Experiments were conducted 3 independent times.
Microscopy
For time-lapse studies, PANC1 cells stably expressing GFP-histone H2B were seeded into 12 well plates and thymidine (2 mM) added for 18 h to block cells at the G1/S boundary. Thymidine was washed out and 3 h later various genotoxic drugs were added to arrest cells the cell cycle. Twenty-four hours (24 h) later, kinase inhibitors were added before the time-lapse commenced. The multiwall plate was placed into a heated chamber and brightfield and fluorescent images were taken every 5 min for up to 48 h, using a Nikon TE2000S microscope (Nikon) controlled by Metamorph (Molecular Devices). To quantify the fates of cells for each condition, movies were examined frame by frame and a minimum of 100 cells were counted, and a minimum of 3 independent movies were conducted. Selected frames were chosen for montages to highlight distinct cell fates.
For immunofluorescence, cells were seeded onto coverslips 24 h prior to drug treatment. Cells were synchronized with thymidine prior to drug treatments as described above. 6–9 h after addition of checkpoint inhibitors, cells were fixed (4% paraformaldehyde/PBS) and stained as previously described.5 Antibodies to α-tubulin (Sigma–Aldrich), and CENP-F were used.42 Alexa Fluor-conjugated secondary antibodies (488, 555, 647 nm) were used at a final concentration of 1 μg/ml (Invitrogen) and cells were counterstained with DAPI. Images were captured using a 40× or 100× objective mounted on an inverted microscope (Eclipse TE2000S; Nikon) with a charge-coupled device camera (Photometrics Cascade 512F; Roper Scientific) using Nikon Elements 2.0 (Nikon).
Flow cytometry
Cells treated with indicated drugs were harvested and processed for DNA content analysis as previously performed.5 For dual-DNA content analysis, cells were fixed in 70% ethanol. After washing with PBS and PBS + 0.5% Tween20, cells were blocked with PBS + 0.5% BSA. Cells were then incubated with FITC conjugated phospho-MPM2 (Upstate Biotechnology, now Millipore) for 1 h on ice. Cells were washed once with PBS and the protocol for propidium iodide staining was followed.5 DNA content was acquired from 10 000 events using Becton Dickinson single laser three-fluorescence FACScan flow analyzer and Cell Quest software (Becton Dickinson) and FlowJo software was used to process the data.
Western blotting
PANC1 cells were treated with gemcitabine (100 nM) for 24 h followed by the addition of Bos-I or dasatinib at indicated concentrations for 6 h. Cell lysates were generated and 40 µg of protein was used for SDS-PAGE. Following transfer onto PVDF membrane, the following antibodies were used for immunoblotting: Cdc2 (Santa Cruz), phospho-cdc2 Y15 (New England Bioloabs), phospho-Chk1 S296 (Cell Signaling), Chk1 (Abcam), and α tubulin (Sigma).
Pharmacodynamic analysis of γH2AX and RAD51
Cells were plated onto coverslips and treated with gemcitabine (10 nM) for 24 h. Cells were then treated with or without kinase inhibitor (1 µM) for a further 6 h before coverslips were harvested and fixed as described above. Coverslips were subjected to immunostaining with γH2AX (Upstate Biotechnology, now Millipore) and RAD51 (Genetex) antibodies. Alexa Fluor-conjugated secondary antibodies were used at a final concentration of 1 μg/ml (Invitrogen) and cells were counterstained with DAPI. For quantitative assessment of γH2AX, images were taken using NIS-Elements AR software (Nikon). Using DAPI to define the nuclei of an individual cell as a “region of interest”, the sum intensity of γH2AX for each ROI/cell was determined. For each condition, >100 were scored and the sum intensity averaged. For tumors samples were paraffin-embedded, and samples were generated and processed as previously described43,44 using the γH2AX antibody according to the manufacturer’s recommendation. For quantitative analysis of interphase and mitotic cells, a 100× objective was used and a z-series was taken for each field of view (0.33 µm increments) and the maximum projection image was then subjected to γH2AX quantification as detailed for cells.
Animal studies
Male CB.17/scid mice aged 6–8 wk were housed in the Fox Chase Cancer Center breeding colony. All experiments were performed according to protocols approved by the FCCC institutional animal use committee (protocol #IRB 12-822). After mice were transplanted s.c. in each flank with fragments of cryopreserved EGF-1 F2 tumors and grew to a size of ~100 mm3, mice were randomized to receive vehicle or different drugs (n = 6 per treatment group). Gemcitabine (50 mg/kg i.p.) was administered once a week on day 1, followed by Bos-I administered by oral gavage (100 mg/kg in 0.4% Tween-80) on day 2 and 3 of a 7-d cycle. Mice were subjected to a total of 4 cycles of therapy. Treatment-to-control ratio of the tumor volumes and % growth inhibition (V = 0.5 × [greatest diameter] × [shortest diameter]2) were used to determine and compare efficacy of therapies against tumor grafts. To estimate difference in tumor growth between vehicle and active agents, we used generalized linear models estimated by generalized estimating equations as described. Mice were sacrificed early if tumors reached >2000mm3 or if they exhibited signs of distress.
Statistics
For MTS and clonogenic assays, a paired t test was performed to determine statistical significance, where P < 0.05 was considered significant. For animal studies, we used growth curve analysis. In order to make the distribution of the data more normal, we first transformed the growth data by taking the log. Next, we fit a generalized linear model of growth using the log-transformed data. We assumed a Gaussian family and identity link. In order to account for correlated observations over time, we estimated the model using Generalized Estimating Equations assuming a Markov working correlation matrix.45
Supplementary Material
Disclosure of Potential Conflicts of Interest
There are no competing interests for any of the authors. A patent application (US application # PCT/US2013/031344) was filed by Brian Cocca (Stradley Ronon Stevens and Young, LLP) and Inna Khartchenko (FCCC) on behalf of the applicants Neil Beeharry and Timothy Yen.
Acknowledgments
We appreciate the comments and critical reading from Drs Enders and Dunbrack. Dr Enders also provided the mouseWee1 cDNA and human Wee 1 siRNA. We wish to acknowledge the technical assistance of Drs Fink, Duong-Ly, and Yang. We also acknowledge the Core supported Imaging, Molecular Modeling, High-throughput Screening, Cell Sorting, Histopathology, Animal and Tissue Culture facilities at Fox Chase Cancer Center for equipment and technical support. This work is dedicated to the memory of Elena Gitelson MD PhD.
Financial Disclosure
This work was supported in part by NIH CA169706, core grant CA06927, DoD OC100172, an Appropriation from Common Wealth of PA, the PA CURE (T.J.Y.); the Plain and Fancy Board of Associates Fellowship, FCCC (N.B.); the Pew Charitable Fund, by a generous gift from Mrs Concetta Greenberg to Fox Chase Cancer Center, by Tobacco Settlement funding from the State of Pennsylvania, the Bucks County Board of Associates, NIH K22 CA160725, R21 CA164205, and a career development award from Genentech (I.A.) and R01 GM083025 (J.R.P.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007;67:339–45. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 2.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tse AN, Schwartz GK. Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–44. doi: 10.1158/0008-5472.CAN-04-0841. [DOI] [PubMed] [Google Scholar]
- 4.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65:6835–42. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 5.Beeharry N, Rattner JB, Bellacosa A, Smith MR, Yen TJ. Dose dependent effects on cell cycle checkpoints and DNA repair by bendamustine. PLoS One. 2012;7:e40342. doi: 10.1371/journal.pone.0040342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, Grant S, Ninkovic S, Chen P, Nichols T, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 8.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–39. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 9.Montano R, Chung I, Garner KM, Parry D, Eastman A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol Cancer Ther. 2012;11:427–38. doi: 10.1158/1535-7163.MCT-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrassa L, Damia G. Unleashing Chk1 in cancer therapy. Cell Cycle. 2011;10:2121–8. doi: 10.4161/cc.10.13.16398. [DOI] [PubMed] [Google Scholar]
- 11.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–81. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 13.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–45. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isham CR, Bossou AR, Negron V, Fisher KE, Kumar R, Marlow L, Lingle WL, Smallridge RC, Sherman EJ, Suman VJ, et al. Pazopanib enhances paclitaxel-induced mitotic catastrophe in anaplastic thyroid cancer. Sci Transl Med. 2013;5:ra3. doi: 10.1126/scitranslmed.3004358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blay JY. A decade of tyrosine kinase inhibitor therapy: Historical and current perspectives on targeted therapy for GIST. Cancer Treat Rev. 2011;37:373–84. doi: 10.1016/j.ctrv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Blagosklonny MV. A new science-business paradigm in anticancer drug development. Trends Biotechnol. 2003;21:103–6. doi: 10.1016/S0167-7799(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 17.Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001;61:1065–72. [PubMed] [Google Scholar]
- 18.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–7. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM- and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–43. doi: 10.1593/neo.111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7:e29828. doi: 10.1371/journal.pone.0029828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guertin AD, Martin MM, Roberts B, Hurd M, Qu X, Miselis NR, Liu Y, Li J, Feldman I, Benita Y, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. 2012;12:45. doi: 10.1186/1475-2867-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beeharry N, Rattner JB, Caviston JP, Yen T. Centromere fragmentation is a common mitotic defect of S and G2 checkpoint override. Cell Cycle. 2013;12:1588–97. doi: 10.4161/cc.24740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinkley BR, Zinkowski RP, Mollon WL, Davis FM, Pisegna MA, Pershouse M, Rao PN. Movement and segregation of kinetochores experimentally detached from mammalian chromosomes. Nature. 1988;336:251–4. doi: 10.1038/336251a0. [DOI] [PubMed] [Google Scholar]
- 24.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, Schwartz G, Tse A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 25.Domínguez-Kelly R, Martín Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsels LA, Morgan MA, Tanska DM, Parsels JD, Palmer BD, Booth RJ, Denny WA, Canman CE, Kraker AJ, Lawrence TS, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009;8:45–54. doi: 10.1158/1535-7163.MCT-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horváth Z, Kéri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 29.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 30.Blagosklonny MV. Sequential activation and inactivation of G2 checkpoints for selective killing of p53-deficient cells by microtubule-active drugs. Oncogene. 2002;21:6249–54. doi: 10.1038/sj.onc.1205793. [DOI] [PubMed] [Google Scholar]
- 31.Levinson NM, Boxer SG. A conserved water-mediated hydrogen bond network defines bosutinib’s kinase selectivity. Nat Chem Biol. 2014;10:127–32. doi: 10.1038/nchembio.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, Cole KA. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–84. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim WG, Guigon CJ, Fozzatti L, Park JW, Lu C, Willingham MC, Cheng SY. SKI-606, an Src inhibitor, reduces tumor growth, invasion, and distant metastasis in a mouse model of thyroid cancer. Clin Cancer Res. 2012;18:1281–90. doi: 10.1158/1078-0432.CCR-11-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108:3336–41. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackwood E, Epler J, Yen I, Flagella M, O’Brien T, Evangelista M, Schmidt S, Xiao Y, Choi J, Kowanetz K, et al. Combination drug scheduling defines a “window of opportunity” for chemopotentiation of gemcitabine by an orally bioavailable, selective ChK1 inhibitor, GNE-900. Mol Cancer Ther. 2013;12:1968–80. doi: 10.1158/1535-7163.MCT-12-1218. [DOI] [PubMed] [Google Scholar]
- 36.Xiao Y, Ramiscal J, Kowanetz K, Del Nagro C, Malek S, Evangelista M, Blackwood E, Jackson PK, O’Brien T. Identification of preferred chemotherapeutics for combining with a CHK1 inhibitor. Mol Cancer Ther. 2013;12:2285–95. doi: 10.1158/1535-7163.MCT-13-0404. [DOI] [PubMed] [Google Scholar]
- 37.Li C, Andrake M, Dunbrack R, Enders GH. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol. 2010;30:116–30. doi: 10.1128/MCB.01876-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blagosklonny MV. The power of chemotherapeutic engineering: arresting cell cycle and suppressing senescence to protect from mitotic inhibitors. Cell Cycle. 2011;10:2295–8. doi: 10.4161/cc.10.14.16819. [DOI] [PubMed] [Google Scholar]
- 39.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 40.Meagher KL, Redman LT, Carlson HA. Development of polyphosphate parameters for use with the AMBER force field. J Comput Chem. 2003;24:1016–25. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 41.Huang D, Zhou T, Lafleur K, Nevado C, Caflisch A. Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics. 2010;26:198–204. doi: 10.1093/bioinformatics/btp650. [DOI] [PubMed] [Google Scholar]
- 42.Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ. CENP-F is a. ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil Cytoskeleton. 1993;26:214–26. doi: 10.1002/cm.970260305. [DOI] [PubMed] [Google Scholar]
- 43.Yang DH, McKee KK, Chen ZL, Mernaugh G, Strickland S, Zent R, Yurchenco PD. Renal collecting system growth and function depend upon embryonic γ1 laminin expression. Development. 2011;138:4535–44. doi: 10.1242/dev.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang DH, Smith ER, Roland IH, Sheng Z, He J, Martin WD, Hamilton TC, Lambeth JD, Xu XX. Disabled-2 is essential for endodermal cell positioning and structure formation during mouse embryogenesis. Dev Biol. 2002;251:27–44. doi: 10.1006/dbio.2002.0810. [DOI] [PubMed] [Google Scholar]
- 45.Shults A, Ratcliffe SJ, Leonard M. Improved generalized estimating equation analysis via xtqls for implementation of quasi-least squares in Stata. Stata J. 2007;7:147–66. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.