

Abstract
Allosteric targeting of protein kinases via displacement of the structural αC helix with type III allosteric inhibitors is currently gaining a foothold in drug discovery. Recently, the first crystal structure of CDK2 with an open allosteric pocket adjacent to the αC helix has been described, prospecting new opportunities to design more selective inhibitors, but the structure has not yet been exploited for the structure-based design of type III allosteric inhibitors. In this work we report the results of a virtual screening campaign that resulted in the discovery of the first-in-class type III allosteric ligands of CDK2. Using a combination of docking and post-docking analyses made with our tool BEAR, 7 allosteric ligands (hit rate of 20%) with micromolar affinity for CDK2 were identified, some of them inhibiting the growth of breast cancer cell lines in the micromolar range. Competition experiments performed in the presence of the ATP-competitive inhibitor staurosporine confirmed that the 7 ligands are truly allosteric, in agreement with their design. Of these, compound 2 bound CDK2 with an EC50 value of 3 μM and inhibited the proliferation of MDA-MB231 and ZR-75–1 breast cancer cells with IC50 values of approximately 20 μM, while compound 4 had an EC50 value of 71 μM and IC50 values around 4 μM. Remarkably, the most potent compound 4 was able to selectively inhibit CDK2-mediated Retinoblastoma phosphorylation, confirming that its mechanism of action is fully compatible with a selective inhibition of CDK2 phosphorylation in cells. Finally, hit expansion through analog search of the most potent inhibitor 4 revealed an additional ligand 4g with similar in vitro potency on breast cancer cells.
Keywords: Cyclin-dependent kinase 2, protein kinase, allosteric inhibitors, virtual screening, structure-based drug design, BEAR
Introduction
Owing to their crucial role in the modulation of cell pathways, protein kinases are important targets for anticancer drug discovery.1 The classic approach of targeting the ATP binding site has recently come up against selectivity issues, which can be considerably reduced by following an allosteric modulation approach. As a matter of fact, novel approaches that target “truly” allosteric sites (i.e., distinct from the ATP site) are currently gaining a foothold in protein kinase drug discovery. Allosteric modulators bind to sites that are less conserved across the kinome and only accessible upon conformational changes; therefore, they are thought to provide higher selectivity and extended drug target residence times.2 Of these, type III inhibitors bind exclusively to allosteric pockets located in the proximity of the αC helix, which is a central mediator of allosteric activation/inactivation.3 This helix is located in the N-lobe beside the active site and is usually swung outwards when the kinase is in an inactive conformation.4 Being closely related to protein kinase inactivation, allosteric targeting via displacement of the conserved structural αC helix allows a direct and specific modulation mechanism. Allosteric modulation mechanisms involving this helix have been structurally elucidated in a variety of protein kinases, such as MEKs, PDK1, PKCζ, AKT1, and more recently CDK2.3,5,6
CDKs have been intensively investigated as anticancer targets, but none of the ATP-competitive CDKs inhibitors have yet been approved for clinical use, mainly because of selectivity issues.7 In fact, the majority of the inhibitors tested so far were able to inhibit several CDKs with comparable IC50, but due to undesired toxicity, their use at effective doses was limited. Nevertheless, the relatively more selective (albeit not as selective as an allosteric inhibitor would be) CDK4/CDK6 inhibitor PD-0332991 was recently reported to triple the progression-free survival in combination with an aromatase inhibitor in patients with estrogen receptor-positive breast cancer, further supporting the notion that higher selectivity in CDK inhibitors may lead to clinically relevant candidates.8 Moreover, it was reported that CDK isoform- and substrate-selective inhibition can be achieved through the use of cyclin groove inhibitors, E2F peptides, and other molecules that block recruitment of phosphorylated retinoblastoma and/or E2F.9-12
CDK2 is an important member of the CDK family, which plays an important role in controlling the G1-to-S-phase checkpoint and in DNA replication13 and, therefore, represents an important pharmacological target for arresting or recovering control of the cell cycle in dividing cells.7,14 In CDK2, the αC helix is involved in Cyclin recruiting to form an active CDK2/Cyclin complex. By inducing or stabilizing an outward orientation of the αC helix with an allosteric ligand, the CDK–Cyclin interface would be disrupted and kinase activation would be impaired.3 Recently, crystal structures of CDK2 bound with the extrinsic fluorophore 8-anilino-1-naphtalene sulfonate (ANS) have been reported.15,16 In the crystals, 2 molecules of ANS bind a cavity formed by αC and the nearby strands β4 and β5 and induce a remarkable outward displacement of the αC helix, ultimately resulting in disruption of the recognition and binding site of Cyclin A. Ternary crystal structures of CDK2 in complex with ANS and type I inhibitors (JWS648, SU9516, and staurosporine) confirmed the truly allosteric nature of this fluorophore and resulted in CDK2 conformations nearly identical to that of the binary CDK2–ANS complex.15,16 This is the first time that a completely allosteric ligand proved to be able to displace the αC helix and to inactivate a CDK kinase via an allosteric mechanism, prospecting a new strategy to design inhibitors with potentially improved selectivity. However, ANS is readily displaced from CDK2 upon Cyclin binding, because its affinity for CDK2 (Kd = 37 μM) is significantly lower than that of Cyclin A.15 Therefore, the design of compounds able to bind this newly discovered allosteric pocket with higher affinity and with better drug-like properties is highly desirable.
In this work we have performed a virtual screening of commercially available compounds (Asinex collection, ~600 000 compounds) in the allosteric pocket of the CDK2–ANS binary complex. The screening was conducted by using a combination of docking (AutoDock 4)17 and our in-house post-docking tool BEAR (Binding Estimation After Refinement).18,19
Of the 35 best-ranking compounds tested experimentally, 7 were able to bind CDK2 in the micromolar range and with the expected allosteric mechanism. In fact, these molecules bind to the allosteric site with a dose-dependent response, and competition experiments performed in the presence of the type I inhibitor staurosporine excluded the possibility that the identified compounds could be ATP-competitive inhibitors, thus confirming their allosteric nature. Some of them proved to be able to inhibit the growth of breast cancer cell lines in the micromolar range, and the most potent compound of our set (compound 4) was able to selectively inhibit CDK2-mediated Rb phosphorylation in cells, thus supporting its mechanism of action. Finally, hit expansion of compound 4 provided an additional active candidate with similar in vitro activity.
Results and Discussion
Using the crystal structure of CDK2 in complex with the allosteric probe ANS15 (PDB code 1PXF), a virtual screening of commercially available compounds (the Asinex collection, >600 000 compounds) in this newly discovered allosteric pocket was performed using a combination of docking (AutoDock 4) and post-docking (BEAR)18,19 methods. BEAR has been extensively validated and used in prospective and retrospective drug discovery applications. By integrating the structural refinement of the docked poses with molecular dynamics and a more accurate ranking of potential ligands with MM-PBSA, BEAR proved to be able to significantly enrich ranked lists with biologically active compounds18–20 and to discover active hits in previous virtual screening campaigns.21
Before database screening, the docking protocol was tested for its ability to reproduce the crystallographic orientation of ANS. Re-docking of ANS retrieved a binding mode that was almost identical to that of the co-crystallized ligand (RMSD of 0.529 Å). Of the 2 ANS molecules bound to the crystal, AutoDock placed the ligand in the inner (residue 305.A in 3PXF, named ANS1) and conceivably more favorable binding site, with a score of −7.4 kcal/mol. According to BEAR, the predicted binding free energies (calculated without the inclusion of the entropic term) of ANS in the inner (ANS1) and outer (ANS2) pockets were −37.7 and −24.6 kcal/mol, respectively, confirming that the inner pocket provides higher binding, at least with this ligand.
The selection of compounds for biological evaluation was made from the hundred top-scoring compounds ranked according to the MM-PBSA scoring function implemented in BEAR. In the top hundred compounds, MM-PBSA scores ranged from −59.1 to −47.1 kcal/mol. A common feature to all top-scoring compounds was a strong ionic interaction with Lys33, i.e., the catalytic lysine of CDK2 that lines one side of the inner ANS1 pocket. Moreover, all compounds occupied the ANS1 pocket with a combination of complementary electrostatic (Lys33) and hydrophobic (Leu55, Phe146, Tyr15, Phe80, Leu78, Leu148, Leu66) interactions, many of them extending, in all or in part, to the outer ANS2 pocket, thus creating additional favorable contacts. In the outer pocket, a number of top-scoring compounds formed additional electrostatic interactions with Lys56 (αC helix) and His71 (β4 sheet), and hydrophobic interactions with Ile52, Leu76, Leu78, Leu37, Val69, and Ile35. To promote the selection of structurally diverse compounds, potential hits were grouped into chemical classes on the basis of Tanimoto similarity indexes and clustering analyses. Then, compound selection was performed by: (1) selecting at least one representative of each cluster of compounds; (2) the visual inspection of the binding modes; (3) the analysis of chemical groups interacting with Lys33. A total of 35 compounds were finally purchased and submitted to biological assays.
Compounds were initially screened for their ability to bind CDK2 and displace ANS from the allosteric pocket. Intrinsically fluorescent compounds were not considered for further characterization. Competition experiments showed that 7 compounds (compounds 1–7; Chart 1) had a concentration-dependent ability to displace ANS from CDK2, with EC50 values in the micromolar range. Selected computational and biological properties of these compounds are reported in Table 1. Four of the 7 hits have significantly diverse structures (having average pairwise Tanimoto similarity coefficients of around 0.2) and have been classified as representatives of carbonyl, nitro, and carboxylic acid classes of compounds depending on the chemical group interacting with Lys33. ADMET predictions performed with QikProp (Schrödinger Suite 2011) showed that almost all the predicted properties of these compounds fall within the 95% range of similar values for known drugs. Therefore, the identified compounds may constitute valuable starting points for further hit-to-lead optimization.

Chart 1. Chemical structures of the allosteric inhibitors 1–7 and those resulting from the hit expansion of compound 4.
Table 1. Predicted free energies of binding, VS rankings, chemical classification, occupation of the ANS1 and ANS2 pockets, selected physico–chemical properties, and biological properties of the 7 allosteric inhibitors discovered in the primary virtual screening (compounds 1-7) and of the nine analogs of compound 4 selected from hit expansion (compounds 4a-4i).
| Cpd # | Asinex # | ΔG’binda | Rank #b | Classc | ANS1d | ANS2d | MWe | PSAf | logPg oct/w | EC50 (μM)h | IC50 (μM)i MDA-MB231 | IC50 (μM)i ZR-75–1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BAS01060589 | −55.9 | 2 | carb. acid | x | x | 492.0 | 132.9 | 4.8 | 7 ± 3 | >50 | >50 |
| 2 | BAS00631909 | −51.6 | 14 | nitro | x | x | 515.4 | 102.8 | 5.6 | 3 ± 1 | 20.5 ± 4.4 | 16.7 ± 2.2 |
| 3 | BAS00590245 | −51.4 | 16 | carbonyl | x | 514.3 | 64.1 | 4.7 | 45 ± 16 | 18.6 ± 2.5 | 25.9 ± 1.0 | |
| 4 | BAS00380830 | −50.9 | 21 | nitro | x | 438.5 | 108.9 | 5.0 | 71 ± 28 | 4.0 ± 0.1 | 4.5 ± 0.2 | |
| 5 | BAS00434932 | −49.5 | 29 | nitro | x | 385.5 | 61.5 | 4.2 | 27 ± 6 | >50 | >50 | |
| 6 | BAS01060577 | −49.2 | 33 | nitro | x | x | 449.9 | 134.2 | 3.7 | 32 ± 7 | 49.2 ± 1.2 | >50 |
| 7 | BAS01123443 | −54.2 | 5 | carb. acid | x | x | 463.9 | 134.8 | 3.9 | 48 ± 18 | >50 | >50 |
| Hit expansion of compound 4 | ||||||||||||
| 4a | BAS02102259 | −50.8 | 2l | nitro | x | 506.6 | 127.1 | 5.6 | >100 | 21.1 ± 1.5 | 43.3 ± 9.5 | |
| 4b | BAS00619651 | −50.7 | 3 | nitro | x | 486.6 | 112.6 | 6.0 | 88 ± 57 | >50 | >50 | |
| 4c | BAS01404025 | −49.8 | 6 | nitro | x | 502.6 | 121.6 | 5.7 | 49 ± 21 | >50 | >50 | |
| 4d | BAS02102292 | −49.4 | 7 | nitro | x | 520.6 | 126.8 | 5.7 | 23 ± 12 | >50 | >50 | |
| 4e | BAS02102245 | −49.4 | 8 | nitro | x | 520.6 | 124.8 | 5.7 | 46 ± 14 | >50 | >50 | |
| 4f | BAS01547732 | −48.9 | 13 | nitro | x | 492.5 | 130.1 | 5.1 | 76 ± 39 | 19.4 ± 1.9 | 21.1 ± 1.2 | |
| 4g | BAS00111586 | −48.8 | 15 | nitro | x | 446.5 | 111.2 | 5.0 | 60 ± 37 | 6.5 ± 1.0 | 5.6 ± 0.5 | |
| 4h | BAS00916022 | −48.0 | 20 | nitro | x | 508.6 | 114.0 | 6.4 | >100 | >50 | >50 | |
| 4i | BAS00381203 | −47.2 | 24 | nitro | x | 398.5 | 110.2 | 4.3 | >100 | 23.4 ± 5.5 | 21.4 ± 0.3 | |
a Predicted free energy of binding of the ligand (kcal/mol) according to BEAR (MM-PBSA). bRanking position according to BEAR (MM-PBSA). cClassification of the ligand depending on the chemical group interacting with the conserved catalytic Lys33. dOccupation of the inner (ANS1) and outer (ANS2) pocket. eMolecular weight (range 95% of drugs 130/725). fPolar surface area (range 95% of drugs 7/200 Ǻ2). gLog of the octanol/water partition coefficient (range 95% of drugs -2/6.5). hEC50 values (μM) determined from binding competition with ANS, with standard deviation. iIC50 values (μM) obtained in cell-based assays performed with MDA-MB231 and ZR-75–1 breast cancer cell lines, with standard deviation. lRanking position within the focused library of 2217 analogs of compound 4.
Figure 1A shows the dose-response curves obtained from 3 independent experiments. All the compounds showed a typical displacement curve. EC50 values obtained from these curves range from 3 μM for compound 2 to 71 μM for compound 4. Compounds 1 (EC50 of 7 μM) and 2 (EC50 of 3 μM) bound CDK2 with an affinity higher than ANS (Kd = 37 μM),15 while compounds 3 and 5–7 had similar affinity. To confirm the truly allosteric nature of these ligands, competition experiments between compounds 1–7 and ANS were repeated in the presence of a potent ATP-competitive type I inhibitor that blocks the ATP site, i.e., staurosporine (Fig. S1 reported as supplementary information). The presence of staurosporine did not significantly modify the ANS displacement curves of the 7 compounds, indicating that the latter compounds do not occupy the ATP site. Rather, they compete with ANS for the allosteric site previously described by crystallography of the CDK2–ANS complex.15
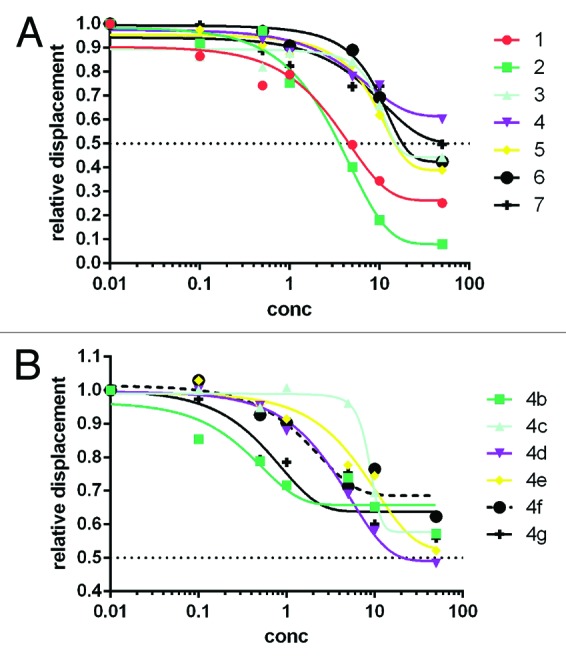
Figure 1. Concentration-dependent displacement of ANS from CDK2. Panel (A) reports the displacement activity of compounds 1–7, while (B) shows the activity of six (4b–4g) of the nine compounds derived from the hit expansion of compound 4. Compounds 4a, 4h, and 4i did not show appreciable displacement activity.
When tested for their ability to inhibit the CDK2/Cyclin A kinase activity, all the 7 hits did not show significant inhibitory activity (data not shown) at concentrations showing ANS displacement. Ligands were also tested at higher concentrations (up to 100 μM), depending on compound solubility. Moreover, pre-incubation of CDK2/Cyclin A with compounds 1–7 for 1 h did not show inhibition. Likewise, ANS did not inhibit CDK2/Cyclin A activity up to a concentration of 200 μM. In the same experimental conditions, the ATP-competitive inhibitor staurosporine showed a clear concentration-dependent ability to inhibit CDK2/Cyclin A kinase activity, with an IC50 of 10 nM. In the experimental conditions described above, the lack of direct inhibition of the catalytic activity shown by compounds 1–7 and ANS is not surprising, considering that this assay makes use of pre-formed CDK2/Cyclin A complexes in which the kinase is in the active conformation. In the CDK2/Cyclin A active conformation the ANS allosteric site is inevitably closed and therefore not accessible to ligands. This is demonstrated by the comparison of plenty of crystal structures of CDK2 in the active (CDK2/Cyclin A or E complexes) vs. inactive (CDK2) conformations available in the Protein Data Bank. As a matter of fact, detection of allosteric ligands using standard kinase activity assays is known to be challenging, as these methods preferentially detect compounds that bind to active kinases in an ATP-competitive manner.22,23 Nevertheless, a 30% inhibition of kinase activity was achieved when a 100 μM concentration of compound 4 was pre-incubated with CDK2 before the addition of Cyclin A and the substrate Rb, confirming the ability of this compound to bind and partially inhibit CDK2 activity when pre-formed CDK2/Cyclin A complexes are not used (data not shown). Considering the tight binding of Cyclin A for CDK2, it is conceivable that more potent analogs of compounds 1–7 may inhibit complex formation with cyclins more effectively.
The 7 compounds were then tested for their ability to inhibit the growth of human breast cancer cells in vitro, using 2 different human breast cancer cell lines, MDA-MB231 and ZR-75–1. As reported in Table 1, the most effective compound was 4, with a concentration able to inhibit the growth by 50% (IC50) of 4 and 4.5 μM for MDA-MB231 and ZR-75–1, respectively. Four of the 7 compounds (1, 5–7) did not show appreciable activity (IC50> 50 μM). Compounds 2 and 3 showed IC50 values around 20 μM. Compounds 1 and 2 were the most effective in displacing ANS from CDK2, but only 2 showed appreciable activity in cell-based assays. Moreover, compound 4 was the less efficient in binding CDK2 but the most active in the cell-based assays. These findings indicate the lack of a clear correlation between in vitro CDK2 binding characteristics of these compounds and their effect on cells, a commonly observed phenomenon generally arising from differential cell membrane permeability of compounds.24,25 Nevertheless, compound 4 was able to effectively inhibit CDK2-mediated phosphorylation of Retinoblastoma at a concentration of 4 μM (its IC50 in cells). In fact, Figure 2 shows that treatment of MDA-MB231 cells with the specific CDK2 inhibitor SU9516 (10 μM) or compound 4 resulted, after 24 h, in a clear reduction of T821-Rb phosphorylation, a CDK2 preferred site,26 without affecting the phosphorylation of a CDK4/CDK6 preferred site (S780-Rb). These data demonstrate that compound 4 inhibits CDK2 activity in cells at concentrations inhibiting cell growth and provide proof of principle that its mechanism of action is fully compatible with a selective inhibition of CDK2 phosphorylation.
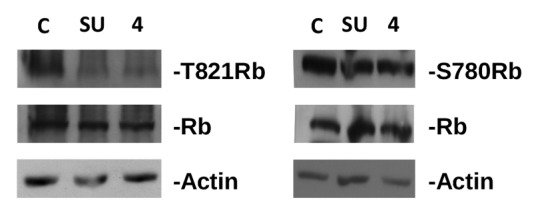
Figure 2. Inhibition of Rb phosphorylation by compound 4 and SU9516 in MDA-MB231 cells. Proteins were separated on SDS page, electroblotted to nitrocellulose, and hybridized with antibodies against phoshorylated Rb (T821, left panel and S780, right panel). Blots were subsequently hybridized with antibodies against total Rb and actin (used as loading control). C, control, untreated cells; SU, cells treated with SU9516 (10 μM); 4, cells treated with compound 4 (4 μM).
The predicted binding modes of the 7 active hits were confirmed by performing a more exhaustive sampling and clustering of docked orientations compared with the original conditions applied for virtual screening. The results are collectively reported in Tables S1 and S2, and the overall best-scoring solutions according to BEAR/MM-PBSA chosen as representative binding modes of each compound are graphically shown in Figure 3. Compounds 1 and 7 are predicted to bind the inner catalytic Lys33 with the 6-carboxylate group and the outer Lys56 group with the 2-nitro group of the 2-nitro-phenylsulfanyl moiety. The remaining interactions were mainly hydrophobic contacts with Tyr15, Ile35, Ile52, Leu55, Val69, Leu76, Leu78, and Phe80. Compounds 2 and 6 bind Lys33 with the nitro group of the 2-nitro-phenylsulfanyl moiety, and Lys56 and His71 with the carboxylate group. Being characterized by 2 polar heads separated by a hydrophobic core, head-to-tail orientations of compounds 1/7vs.2/6 having each polar group interacting once with Lys33 and once with Lys56 are reasonable. Other interactions are mainly hydrophobic. Compound 3 binds Lys33 with the pyrimidin-4-one carbonyl and interacts favorably with Tyr15, Ile35, Leu55, Leu66, Leu78, Phe80, Leu148, Val154, and Val156. The nitro group of compound 4 interacts with Lys33, and the carbonyl group of the cyclohex-2-enone ring hydrogen bonds with the Tyr15 hydroxyl. The nitrophenyl ring establishes stacking interactions with Tyr15, while the rest of the molecule is in contact with Ile35, Ile52, Leu55, Val64, Leu66, Leu78, Val154, and Val156. Compound 5 binds Lys33 with the nitro group, while the remainder of the molecule interacts with Tyr15, Ile35, Ile52, Leu55, Ile63, Val64, Leu66, Phe80, Phe146, Leu148, Val154, and Val156.

Figure 3. Predicted binding modes of compounds 1–7 in the allosteric pocket of CDK2, highlighting (left) the location of each compound in the overall fold, (right) the close up of the allosteric site with selected residues interacting with the ligands. For comparative purposes, the first box shows the binding mode of the 2 ANS molecules present in the crystal structure of the binary complex (PDB code 1PXF).
Hit expansion of compound 4
With the aim of potentially identifying additional biologically active hits and to provide a first exploration of their SAR, commercially available analogs of compound 4, i.e., the compound showing the most interesting activity in cancer cell-based assays, were searched for in the Asinex database. To this end, a focused library of commercial analogs of 4 was prepared by including compounds with a similarity Tanimoto coefficient (Tc) of at least 0.7 with this reference compound. This operation resulted in 2217 compounds, 1536 of which were not post-processed with BEAR in the primary virtual screening owing to the AutoDock energy score cut-off of −11 kcal/mol imposed when selecting molecules for post-docking refinement and rescoring. The entire focused library (2217 compounds) was then submitted to post-docking using the same procedure described for the primary screening. According to MM-PBSA, compound 4 ranked first in this focused library, but a number of analogs with interesting scores emerged, some of which were not processed before because their AutoDock scores were less favorable than the cut-off value. Nine analog compounds (compounds 4a–4i, included in Chart 1) with favorable MM-PBSA scores and binding modes consistent with that of the parent compound 4 were finally selected for biological evaluation. Their chemical and biological properties are included in Table1.
Three compounds (4a, 4h, 4i) did not show appreciable ANS displacement activity (Table 1). The remaining 6 analogs had EC50 values ranging from 23 to 88 μM. In some cases, e.g., compound 4d, the affinity for CDK2 was slightly improved with respect to that of the parent compound 4. The dose-response curves of the 6 active analogs obtained from 3 independent experiments are shown in Figure 1B. Moreover, the compounds showing appreciable ANS displacement activity were also tested in the presence of staurosporine to confirm that they bind in the allosteric ANS pocket. Again, the displacement ability of the 6 tested compounds did not change in the presence of staurosporine (Fig. S2), confirming their allosteric binding.
When tested in cell based assays on both MDA-MB231 and ZR-75–1 human breast cancer cell lines, compound 4g showed IC50 values similar to those of compound 4, while compounds 4a, 4f, and 4i had IC50 values around 20 μM. The remaining 5 compounds did not show appreciable antiproliferative activity (Table 1).
Conclusions
The recent availability of the first crystal structure of a member of the CDK family with an open allosteric pocket in proximity of αC helix allowed us to apply a virtual screening protocol that led to the successful identification of the first-in-class allosteric inhibitors of CDK2. Using a combination of high-throughput docking and refinement and rescoring performed with our post-docking tool BEAR, we rationally selected and tested 35 top-ranked compounds and finally identified 7 ligands able to bind CDK2 in the allosteric ANS pocket, corresponding to a hit rate of 20%.
The active ligands were able to displace ANS from CDK2 at micromolar concentrations. The ligands competed with ANS for the allosteric pocket previously demonstrated by crystallography of the CDK2–ANS complex,15 thus showing a mechanism fully compatible with the expected allosteric binding. Competition experiments repeated in the presence of the ATP-competitive inhibitor staurosporine confirmed the truly allosteric nature of these ligands. All tested compounds did not show appreciable direct inhibition of CDK2/Cyclin A kinase activity at the concentrations used in our experiments. In fact, when the assay makes use of pre-formed CDK2/Cyclin A complexes, the kinase is in the active conformation and the CDK2 allosteric site is inevitably closed and therefore not accessible to allosteric ligands. On the contrary, a 30% inhibition of kinase activity was observed when a 100 μM concentration of compound 4 was pre-incubated with CDK2 before the addition of Cyclin A and the substrate Rb, confirming the ability of this compound to bind and partially inhibit CDK2 activity when pre-formed active CDK2/Cyclin A complexes are not used. It is not excluded that stronger allosteric binders would achieve more effective competition with Cyclin A and consequently show up higher direct inhibition by shifting the equilibrium toward the inactive conformation. This possibility will be explored when tighter allosteric binders will be available. Therefore, future efforts will be directed toward hit optimization and generation of more potent analogs.
Importantly, some of these ligands inhibited the proliferation of MDA-MB231 and ZR-75-1 breast cancer cells with IC50 values in the low micromolar range. In particular, compounds 4 and 2 had IC50 values of 4 μM and 20 μM, respectively. Remarkably, treatment of MDA-MB231 cells with the most potent compound 4 resulted in a clear reduction of a CDK2 preferred phosphorylation site (T821-Rb) but not of a CDK4/6 preferred site (S780-Rb). This finding indicates that 4 is able to selectively inhibit CDK2-mediated Rb phosphorylation and confirms that its mechanism of action is fully compatible with a selective inhibition of CDK2 phosporylation in cells. In cells, the amount of CDK2 complexed with Cyclin A changes in response to varying Cyclin A levels, and CDK2 is not always complexed with Cyclin.27–29 Therefore, in cells, uncomplexed CDK2 in the inactive conformation can effectively bind allosteric inhibitors, and this, in turn, would reduce its binding with Cyclin A.
Finally, hit expansion of compound 4 provided an additional ligand 4g with similar in vitro potency. No clear correlation between in vitro CDK2 binding and antiproliferative activity in cells was observed, as commonly reported in the literature because of differential cell membrane permeability of compounds.24,25 Indeed, several factors can influence the antiproliferative activity of a compound including its stability, its intracellular accumulation, and metabolism. Therefore, further optimization of drug potency, cellular penetration, and SAR investigations on the newly identified hits will be required to further develop these compounds.
Materials and Methods
Compound database
The Asinex collection of commercially-available compounds was downloaded in Mol2 format from the ZINC database (“Usual” subset) (July 2011).30 This data set contained a total of 600 722 compounds including protonation variants and tautomers at pH values from 5.75 to 8.25, corresponding to 545 609 unique ZINC IDs.
Protein preparation
The crystallographic coordinates of CDK2 in complex with 2 molecules of 8-anilino-1-naphtalene sulfonate (ANS) used for docking were taken from the Protein Data Bank (PDB), PDB code 3PXF.15 The 2 ANS and all water molecules were removed from the structure. Hydrogen atoms were added to the protein structure using the Leap module of Amber10.31 All Asp and Glu were negatively charged and all Lys and Arg were positively charged. This structure was used as starting point in both docking and post-docking calculations.
Docking
Docking calculations were performed with AutoDock 4.2.3.17 The protein structure was converted into pdbqt file by merging non-polar hydrogen atoms to heavy atoms and assigning Gasteiger partial charges and atom types with MGLTools 1.5.4.32 For docking, each molecule in the Asinex database was assigned Gasteiger partial charges with MGLTools 1.5.3. On the contrary, the original CM2 charges present in ZINC were retained for the post-docking analyses made with BEAR (see below). AutoGrid was employed to build grid maps around the allosteric site of CDK2, with a spacing of 0.375 Å and centered on the center of mass of the 2 ANS molecules. Grid dimensions were set so as to include the 2 ANS molecules and a number of buried water molecules around them (W318, 327, 338, 374, 406, 473, 478, 496 in 3PXF) that could be displaced by the docked compounds. The number of resulting grid points were 62 × 46 × 44 (0.375 Å spacing). The Lamarckian genetic algorithm was used for the conformational sampling. For each compound, 10 runs were performed with a population size of 150, 250 000 energy evaluations, and 27 000 maximum generations. Clustering was performed with a cluster tolerance of 2 Å. Re-docking of ANS with these parameters retrieved a binding mode that was almost identical with that of the co-crystallized ligand (RMSD of 0.529 Å). Of the 2 ANS molecules bound to the crystal, AutoDock placed the ligand in the inner (residue 305.A in 3PXF) and conceivably more favorable binding site, with a score of −7.4 kcal/mol.
The Asinex database was ranked according to the lowest-energy scoring docking solution.
Post-docking with BEAR
The best scoring docking complexes obtained with AutoDock were post-processed with BEAR (Binding Estimation After Refinement), our in-house structural refinement and rescoring tool based on molecular dynamics and MM-PBSA.18–21 Post-docking was performed on the top fraction of the ranked database obtained by applying an AutoDock energy score cut-off of −11 kcal/mol (~27 000 compounds, corresponding to ~5% of the initially docked database). The best AutoDock score was −15.6 kcal/mol. Each complex was submitted to the BEAR refinement and rescoring procedure using the Amber force field ff0333 for the protein and the General Amber force field (gaff)34 for small molecules. CM2 partial atomic charges were used for small molecules. The protocol consists of 3 steps based on molecular mechanics (MM) and dynamics (MD) cycles. In particular, 2000 steps of MM energy minimization of the whole protein–ligand complex were performed, followed by 100 ps MD where the ligand was allowed to move, and a final re-minimization of the entire complex. A distance-dependent dielectric constant ε = 4r and a non-bonded cut-off of 12 Å were used. At the end, the binding free energy of each compound in the refined complex was computed by using the MM–PBSA method.35 The Poisson–Boltzmann algorithm was used to solve the polar term of the solvation free energy, while the LCPO method was applied to calculate the solvent-accessible surface area for the non-polar contribution. Dielectric constants of 80 and 1 were used for solvent and solute, respectively. BEAR has been extensively validated and used in VS. Other details can be found in references 18–21.
Post-docking results were ranked according to the MM-PBSA binding free energy scores, and top ranked compounds were visually inspected with Chimera 1.6.2.36 Before selection, the top 100 compounds were clustered into chemical classes using the Canvas similarity and clustering utility available in Maestro 9.2,37 using default settings. Thirty-five compounds were finally selected, purchased, and submitted to biological evaluations.
Binding mode predictions of the 7 active hits
The binding modes of the active compounds 1–7 discovered by virtual screening were confirmed by performing a more exhaustive sampling and clustering of docked orientations compared with the original conditions applied for virtual screening (100 runs of Lamarckian genetic algorithm instead of 10). Since the ZINC database did not contain all possible stereoisomers of compounds 1–7, complete sets of stereoisomers were re-generated using the LigPrep utility available in Maestro (Schrödinger 2011 suite). For each enantiomer, 100 docking runs were performed with AutoDock, and the resulting orientations were clustered using a default RMS tolerance of 2 Å. For each enantiomer, clusters of orientations were ranked according to (1) the lowest energy score predicted by AutoDock and (2) the population of each cluster, i.e., the number of orientations that populated each cluster. Then, representative orientations of the 5 best-scoring clusters and of the 5 most-populated clusters were post-processed with BEAR. CM2 atomic charges present in the ZINC database were used for post-docking refinement. The docking energy (AD4) and MM-PBSA (BEAR) scores obtained for each post-processed orientation are reported in Tables S1 and S2 as supplementary information. Finally, the overall best-scoring solutions according to MM-PBSA (BEAR) were chosen as representatives of the binding mode of each compound.
Hit expansion of compound 4
With the aims of exploring the SAR of the identified hits and potentially discovering other biologically active hits, commercially available analogs of compound 4, i.e., the compound showing the most interesting activity in cancer cell-based assays, were searched in the Asinex database and then processed with the same docking and post-docking procedure described above. To prepare the focused library, compound 4 was taken as a reference for similarity searching calculations. Molecular ACCess System (MACCS) fingerprints38 as implemented in OpenBabel 2.3.139 were computed for the reference compound 4 and for the complete Asinex database. Then, similarity searching calculations made with OpenBabel were ranked according to Tanimoto coefficient (Tc) values computed for each database molecule vs. the reference compound 4, and compounds with a Tc value of at least 0.7 were selected. This selection led to a focused library of 1108 unique compounds, which corresponded to 2217 database entries by including multiple tautomers and protonation states. Of these, 681 were already processed in the primary virtual screening, while the remaining 1536 had been excluded owing to the AutoDock energy score cut-off of −11 kcal/mol imposed when selecting molecules for post-docking refinement and rescoring. The 2217 compounds were then submitted to docking and post-docking calculations using the same procedure described for the primary screening.
ADME predictions
ADME properties of the tested compounds were evaluated by using QikProp v.3.4 (Schrödinger suite).40
Biological assays
Recombinant CDK2 was purified from E. coli using a GST-CDK2 fusion construct. E. coli transformed with a GST–CDK2 plasmid were grown overnight in LB medium in selection antibiotic. Bacterial culture was then diluted 1:100 and grown at 37 °C until reached an absorbance value of 0.6 at 600 nM. At this point temperature was set down to 30 °C and induction with 0.5 mM IPTG (isopropyl-b-D-thiogalactoside) was performed for additional 6 h. Bacterial culture was centrifuged at 4000 rpm for 20 min and pellet resuspended in PBS. Bacterial cells were sonicated 5 times for 20 s and incubated in the presence of 1% triton X100 for 30 min at 4 °C. After centrifugation at 12 000 rpm for 10 min at 4 °C, GST beads were added to the supernatant and incubated for further 60 min at 4 °C. Lysates were washed 3 times in PBS and GST-CDK2 eluted with reduced GSH (10 mM in 50 mM Tris pH 8). The purity of the protein was checked by SDS-PAGE electrophoresis and the amount determined using the Protean assay (BIORAD).
ANS displacement assay was performed as described by Martin et al.16 The assays were performed in 96-well plates in a total volume of 50 microliters of 40 mM Hepes pH 7.5 ANS (final concentration 50 μM), and different concentrations of the different compounds (ranging from 0.01 to 50 μM) were added and the fluorescence (excitation 360 nM, emission 460 nM) measured using an Infinite M200 Microplate Reader (Tecan) to determine the intrinsic fluorescence of the compounds. Recombinant CDK2 (final concentrations 1.5 μM) was then added and a second measurement taken. Using the formula reported in reference 16 the relative displacement for each compound was determined.
To discriminate between type I/II and type III ligands, competition assays in the presence of Staurosporine (2.5 μM) were performed as described in reference 16.
The inhibition of CDK2/Cyclin A kinase activity was determined using a commercially available luminescent-based method (ADP-Glo™ Kinase Assay, Promega) determining the ability of recombinant CDK2/Cycin A complex to phosphorylate one of its major substrate, Rb. The assay was performed following the manufacturer’s instructions. Staurosporine was used as positive control. Luminescence was determined with a plate-reading luminometer (Promega). In some experiments compound 4 at a 100 μM concentration was pre-incubated for 30 min with CDK2 alone, before the addition of cyclin A and the substrate Rb.
Cell growth-inhibitory activity was determined using the MTS test. MDA-MB231 and ZR75–1 breast cancer cells were used for these experiments. Cells were seeded in 96-well plates at 35 000 cells/ml/well. Forty-eight hours after cell seeding different concentrations of the compounds were added, and the plates incubated at 37 °C for additional 72 h. At the end of the treatment period, MTS reagent (Promega) was added to each well, and the plates were incubated at 37 °C for 4 h in 5% CO2. Cell proliferation was evaluated by measuring the absorbance at 490 nm using an infinite M200 Microplate Reader (Tecan). A nonlinear regression method was used to calculate IC50 values (concentrations required to produce 50% growth inhibition).
Phosphorylation of CDK2 substrate Rb in cells was determined by western blotting. Cells were treated with compound 4, and after 24 h lysed in ice-cold buffer containing 50 mM TRIS-HCl pH 7.4, 250 mM NaCl, 0.1% Nonidet NP40, 5 mM EDTA and NaF 50 mM with a protease inhibitor cocktail (Sigma). Lysates were cleared by centrifuging at 13 000 rpm for 5 min. To detect phosphorylated Rb, 30 µg of total protein extracts were loaded on 8% SDS-PAGE and then transferred to a nitrocellulose membrane. The polyclonal anti-phospho-Rb T821 was from Invitrogen, the polyclonal anti-phospho-Rb S780 was from Cell Signaling, while anti-total Rb and anti-actin were from Santa Cruz Biotechnology. Bands were visualized by chemioluminescence (ECL, Amersham).
The tested compounds were purchased from Asinex.41 Vendor codes of the 35 purchased compounds are: BAS04379720; BAS04379667; BAS03379043; BAS03302486; BAS01840948; BAS01355752; BAS01213474; BAS01118120; BAS01060589; BAS01060577; BAS01052182; BAS01052061; BAS00732157; BAS00631909; BAS00619651; BAS00608642; BAS00590245; BAS00448634; BAS00434932; BAS00410884; BAS00410708; BAS00398159; BAS00380830; BAS00319966; BAS00261429; ASN24804207; ASN10813896; ASN07272942; ASN06130191; ASN06123095; ASN05298123; ASN05258995; ASN05115618; ASN04503530; BAS01123443. The vendor codes of the nine analogs of compound 4 purchased for hit expansion are: BAS00111586; BAS00381203; BAS01404025; BAS01547732; BAS02102292; BAS00619651; BAS00916022; BAS02102245; BAS02102259.
The vendor had verified compound purity by liquid chromatography-mass spectrometry (LC-MS) or nuclear magnetic resonance (NMR) experiments. All compounds had at least 95% purity except 1 (92%) and 3 (93%). All chiral compounds were purchased and tested as racemic mixtures.
Supplementary Material
{kind=link}
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Financial support from the Nando Peretti Foundation (grant NPF 2013–25) is gratefully acknowledged. Computational resources were provided by the ISCRA (Italian Super Computing Resource Allocation) grant “BEAR” Class A # HP10A6E2CI. We wish to thank Fabiana Caporuscio for critical reading of the manuscript.
Glossary
Abbreviations:
- CDK2
cyclin-dependent kinase 2
- MEKs
mitogen-activated protein kinase kinases
- PDK1
phosphoinositide-dependent kinase
- PKCζ
protein kinase Cζ
- AKT1
RAC-alpha serine/threonine-protein kinase
- ANS
8-anilino-1-naphtalene sulfonate
- BEAR
binding estimation after refinement
- SAR
structure–activity relationships
- Rb
Retinoblastoma
References
- 1.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 2.Eglen R, Reisine T. Drug discovery and the human kinome: recent trends. Pharmacol Ther. 2011;130:144–56. doi: 10.1016/j.pharmthera.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Palmieri L, Rastelli G. αC Helix Displacement as a General Approach for Protein Kinases Allosteric Modulation. Drug Discov Today. 2013;18:407–14. doi: 10.1016/j.drudis.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Rabiller M, Getlik M, Klüter S, Richters A, Tückmantel S, Simard JR, Rauh D. Proteus in the world of proteins: conformational changes in protein kinases. Arch Pharm (Weinheim) 2010;343:193–206. doi: 10.1002/ardp.201000028. [DOI] [PubMed] [Google Scholar]
- 5.Lamba V, Ghosh I. New directions in targeting protein kinases: focusing upon true allosteric and bivalent inhibitors. Curr Pharm Des. 2012;18:2936–45. doi: 10.2174/138161212800672813. [DOI] [PubMed] [Google Scholar]
- 6.Fang Z, Grütter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem Biol. 2013;8:58–70. doi: 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- 7.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–66. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 8.CDK Inhibitor Triples PFS in Breast Cancer Cancer Discov. 2013;3:4. [Google Scholar]
- 9.Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–31. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 10.McInnes C, Andrews MJ, Zheleva DI, Lane DP, Fischer PM. Peptidomimetic design of CDK inhibitors targeting the recruitment site of the cyclin subunit. Curr Med Chem Anticancer Agents. 2003;3:57–69. doi: 10.2174/1568011033353506. [DOI] [PubMed] [Google Scholar]
- 11.Andrews MJ, McInnes C, Kontopidis G, Innes L, Cowan A, Plater A, Fischer PM. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Org Biomol Chem. 2004;2:2735–41. doi: 10.1039/b409157d. [DOI] [PubMed] [Google Scholar]
- 12.Corsino P, Horenstein N, Ostrov D, Rowe T, Law M, Barrett A, Aslanidi G, Cress WD, Law B. A novel class of cyclin-dependent kinase inhibitors identified by molecular docking act through a unique mechanism. J Biol Chem. 2009;284:29945–55. doi: 10.1074/jbc.M109.055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 14.Echalier A, Endicott JA, Noble ME. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim Biophys Acta. 2010;1804:511–9. doi: 10.1016/j.bbapap.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schönbrunn E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem Biol. 2011;6:492–501. doi: 10.1021/cb100410m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin MP, Alam R, Betzi S, Ingles DJ, Zhu J-Y, Schönbrunn E. A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem. 2012;13:2128–36. doi: 10.1002/cbic.201200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rastelli G, Del Rio A, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem. 2010;31:797–810. doi: 10.1002/jcc.21372. [DOI] [PubMed] [Google Scholar]
- 19.Degliesposti G, Portioli C, Parenti MD, Rastelli G. BEAR, a novel virtual screening methodology for drug discovery. J Biomol Screen. 2011;16:129–33. doi: 10.1177/1087057110388276. [DOI] [PubMed] [Google Scholar]
- 20.Sgobba M, Caporuscio F, Anighoro A, Portioli C, Rastelli G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur J Med Chem. 2012;58:431–40. doi: 10.1016/j.ejmech.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 21.Degliesposti G, Kasam V, Da Costa A, Kang HK, Kim N, Kim DW, Breton V, Kim D, Rastelli G. Design and discovery of plasmepsin II inhibitors using an automated workflow on large-scale grids. ChemMedChem. 2009;4:1164–73. doi: 10.1002/cmdc.200900111. [DOI] [PubMed] [Google Scholar]
- 22.Lebakken CS, Reichling LJ, Ellefson JM, Riddle SM. Detection of allosteric kinase inhibitors by displacement of active site probes. J Biomol Screen. 2012;17:813–21. doi: 10.1177/1087057112439889. [DOI] [PubMed] [Google Scholar]
- 23.Simard JR, Getlik M, Grütter C, Schneider R, Wulfert S, Rauh D. Fluorophore labeling of the glycine-rich loop as a method of identifying inhibitors that bind to active and inactive kinase conformations. J Am Chem Soc. 2010;132:4152–60. doi: 10.1021/ja908083e. [DOI] [PubMed] [Google Scholar]
- 24.Gleeson MP, Hersey A, Montanari D, Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rastelli G. Emerging topics in structure-based virtual screening. Pharm Res. 2013;30:1458–63. doi: 10.1007/s11095-013-1012-9. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz NM, Leibundgut K, Hirt A. CDK2 catalytic activity and loss of nuclear tethering of retinoblastoma protein in childhood acute lymphoblastic leukemia. Leukemia. 2005;19:1783–7. doi: 10.1038/sj.leu.2403900. [DOI] [PubMed] [Google Scholar]
- 27.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 28.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arellano M, Moreno S. Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem Cell Biol. 1997;29:559–73. doi: 10.1016/S1357-2725(96)00178-1. [DOI] [PubMed] [Google Scholar]
- 30.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–68. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Darden TE, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, et al. (2008), AMBER 10, University of California, San Francisco. [Google Scholar]
- 32.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 33.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–74. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 35.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–97. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Suite 2011: Maestro, version 9.2, Schrödinger, LLC, New York, NY, 2011 [Google Scholar]
- 38.RDKit: Cheminformatics and Machine Learning Software. http://www.rdkit.org/
- 39.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suite 2011: QikProp, version 3.4, Schrödinger, LLC, New York, NY, 2011 [Google Scholar]
- 41.Asinex; Asinex Ltd: Moscow; http://www.asinex.com
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.