

Abstract
Mutations in the Cockayne syndrome A (CSA) protein account for 20% of Cockayne syndrome (CS) cases, a childhood disorder of premature aging and early death. Hitherto, CSA has exclusively been described as DNA repair factor of the transcription-coupled branch of nucleotide excision repair. Here we show a novel function of CSA as transcription factor of RNA polymerase I in the nucleolus. Knockdown of CSA reduces pre-rRNA synthesis by RNA polymerase I. CSA associates with RNA polymerase I and the active fraction of the rDNA and stimulates re-initiation of rDNA transcription by recruiting the Cockayne syndrome proteins TFIIH and CSB. Moreover, compared with CSA deficient parental CS cells, CSA transfected CS cells reveal significantly more rRNA with induced growth and enhanced global translation. A previously unknown global dysregulation of ribosomal biogenesis most likely contributes to the reduced growth and premature aging of CS patients.
Keywords: Cockayne syndrome, RNA polymerase I transcription, DNA repair, premature aging, growth, ribosomopathy
Introduction
The devastating childhood disorder Cockayne syndrome (CS) is characterized by a failure to thrive, high UV sensitivity, and neurological degeneration with deafness and mental retardation, eventually leading to cachexia and childhood death.1,2 CS is a recessive polygenic disorder; functionally relevant mutations of 5 different genes cause the disease. These genes are crucial for the repair of UV-induced DNA lesions by transcription-coupled repair (TCR), a branch of nucleotide-excision repair (NER) and, if mutated, are responsible for the elevated UV sensitivity of CS patients. However, the failure to grow and the prevailing manifestation of severe neurological degeneration cannot be conclusively explained by non-repaired UV-induced DNA damage in CS patients.
Hypersensitivity to UV light represents a key feature in CS patients with cells undergoing apoptosis at a relatively low dose of UV irradiation compared with cells from healthy donors. Additionally, CS cells do not recover from a block of transcription after UV light, a feature often explained by a “road block” for the elongating RNA polymerases caused by UV-lesions in the transcribed DNA strand. Nonetheless, even undamaged DNA is not transcribed in UV-irradiated CS cells,3 indicating that not only a failure of DNA repair, but a failure of transcription regulation per se might account for this cellular pathology. In addition, the assumption that these cellular pathologies contribute to the neuronal degeneration and premature aging symptoms of Cockayne syndrome patients was challenged by the observation that patients with the mild cutaneous disease UV-sensitive syndrome still reveal hypersensitivity to UV light and block of transcription after UV light.4,5 These results argue that the hitherto described cellular abnormalities of CS cells are not involved in the pathophysiology of premature aging and cachexia in this disease.
Accordingly, it was previously postulated that the CS proteins, besides their role in TCR, may play a role as transcription factors, as the CS proteins XPB and XPD are components of the basal transcription factor TFIIH. There is increasing evidence that all CS proteins except CSA execute transcriptional functions in RNA polymerase II transcription;6,7 however, a gene with all CS proteins participating in transcription has not been identified yet. Moreover, microinjection of CSA antibodies did not impair RNA polymerase II transcription, indicating that CSA might not participate in gene expression of protein-coding genes.8 It is currently unresolved whether CSA together with other CS proteins regulates RNA polymerase I transcription, the rate-limiting step in ribosomal biogenesis. Four CS proteins have been identified to influence transcription by RNA polymerase I, which comprises up to 60% of total transcription in the cell.9-11 Three CS proteins, the TFIIH subunits XPB and XPD and CSB, have been shown to participate in transcription elongation of RNA polymerase I, whereas the fourth, XPG, exerts an epigenetic role in demethylation and activation of the rDNA promoter.11-13 Here, we describe a previously unreported role of CSA on rDNA transcription by RNA polymerase I. This observation was supported by our findings that CSA was identified in nucleoli, the subcellular location of ribosomal biogenesis, and that cells from CSA patients showed a reduced RNA polymerase I transcription. Moreover, knockdown of CSA by shRNA significantly reduced rDNA transcription. ChIP experiments revealed that CSA binds to promoter and gene-internal regions of the active fraction of the rDNA gene. Subsequently, CSA recruits the elongation factors CSB and TFIIH to the rDNA promoter and stimulates re-initiation of RNA polymerase I transcription. Stable transfection of CSA in CS cells is followed by an increase in ribosome numbers, translation activity, and cellular growth. Collectively, these data suggest a role of CSA in ribosomal biogenesis, and that non-functional CSA leads to reduced ribosomal biogenesis and most likely promotes premature aging in CS.
Results
CSA localizes to the nucleolus and influences RNA polymerase I transcription
To investigate a possible involvement of CSA in RNA polymerase I transcription, we first studied whether CSA localizes to the nucleolus. The CSA interaction partners CSB and TFIIH are part of the transcription apparatus of RNA polymerase I in the nucleolus.9,10 HA staining of the stable HA–CSA transfected Cockayne syndrome cell line CS3BE revealed a uniform nuclear staining for CSA without omission of the nucleoli. Moreover, counterstaining with nucleolin depicted specific nucleolar staining for HA-CSA (Fig. 1A). The nucleolar staining of CSA was enriched by proteasomal inhibition with MG132, indicating that nucleolar CSA might be a target of ubiquitinylation and degradation. Moreover, a CSA antibody reported not to stain the nucleolus10 displayed a co-staining with nucleolin after proteosomal inhibition (Fig. 1A). Quantitative confocal analysis determined an overlap coefficient of 0.66 for nucleolin and nucleolar CSA (Supplemental Material). Thus, CSA localizes to the nucleolus.
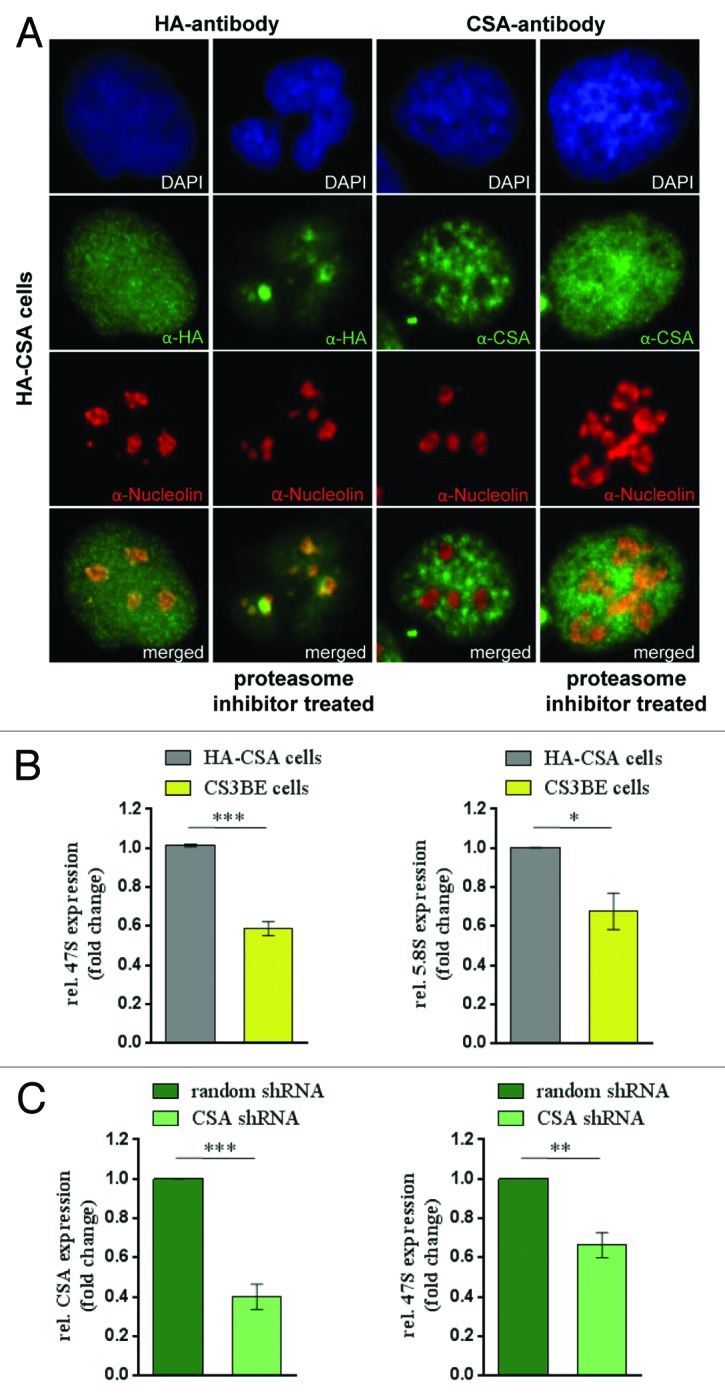
Figure 1. CSA localizes to nucleoli and CSA mutation impairs rDNA transcription. (A) Immunofluorescence stainings with anti-HA and CSA antibody showing CSA in green and nucleolin in red in HA-CSA reconstituted CS3BE cells before and after proteasomal inhibition by MG-132. (B) QPCR analysis of the 47S precursor rRNA and the gene internal 5.8S/ internal transcribed spacer expression in CS3BE cells and CS3BE cells reconstituted with HA-tagged CSA. (C) Inhibition of CSA expression reduces rRNA transcription. QPCR detection of CSA expression and the 47S precursor rRNA expression after anti-CSA shRNA transfection and antibiotic selection in secondary fibroblasts. Values are normalized against RPL13 expression and the respective IgG controls. Values are mean ± s.e.m. of 3 independent experiments. (*P > 0.05; **P > 0.01; ***P > 0.001)
TFIIH and CSB contribute as elongation factors to RNA polymerase I transcription.12,13 To assess whether CSA is involved in rDNA transcription, we determined the rate of rRNA synthesis in CSA-deficient (CS3BE) and reconstituted (HA-CSA) cells by qPCR against different regions of the 47S rRNA precursor. The precursor is post-transcriptionally processed and indicates ongoing RNA polymerase I transcription at the time of harvest. The 47S and 5.8S/internal transcribed spacer (ITS) analysis revealed that cells lacking CSA have markedly reduced rRNA synthesis (Fig. 1B). Moreover, the transcriptional reduction occurs both for the first ITS (47S) and for the late ITS (5.8S), suggesting a failure in the initiation and elongation of RNA polymerase I transcription. To further evaluate a possible role of CSA in RNA polymerase I transcription, the expression of endogenous CSA was silenced by shRNA in secondary fibroblasts. After antibiotic selection for stably transfected cells CSA expression and synthesis of the 47S rRNA precursor were monitored by qPCR. As shown in Figure 1C impaired CSA expression results in a distinctly reduced rRNA transcription, indicating a function for CSA in RNA polymerase I transcription. Collectively, our results show that CSA is localized in nucleoli and stimulates RNA polymerase I transcription.
CSA binds the rDNA and associates with RNA polymerase I
TFIIH and CSB bind to the promoter and gene-internal regions of the rDNA as revealed by ChIP analysis.12,13 To further determine whether CSA also binds to the rDNA, ChIP experiments with chromatin of CSA-reconstituted CS3BE (HA-CSA) and the parental cells (CS3BE) were performed. QPCR analysis of the precipitated chromatin identified CSA to bind to the rDNA promoter and gene-internal sequences of the rDNA but not the intergenic spacer (Fig. 2B). There is a clear enrichment of rDNA promoter and coding regions in the precipitate of CSA reconstituted cells. These results are novel, as no gene-specific binding of CSA has been described thus far.

Figure 2. CSA associates with the active rDNA promoter and RNA polymerase I. (A) Schematic representation of a rDNA gene and the primers used in this study (IGS, intergenic spacer). (B) QPCR analysis of a representative ChIP experiment precipitated with the RNA polymerase I or HA-tag antibodies from chromatin of reconstituted (HA-CSA) or parental CS3BE cells. Values are normalized against input and IgG controls. (C) Methylation sensitive restriction analysis of ChIPed DNA reveals that CSA associates with the active, HpaII-digestable rDNA fraction like RNA polymerase I. (D) ChIP-re-ChIP experiment showing that CSA occupies the same molecules of rDNA as RNA polymerase I. Values are normalized against input and IgG controls. (E) ChIP–western experiment with the above indicated antibodies demonstrate that RNA polymerase I and CSA occupy the same rDNA molecules. (F) Co-immunoprecipitation with the above indicated antibodies and subsequent western blot analysis of 2 experiments with RNA polymerase I- and HA–CSA-specific antibodies. Pictures are representative of at least 3 independent experiments. Values are mean ± s.d. (*P > 0.05; **P > 0.01; ***P > 0.001; ****P > 0.0001)
Approximately half of the rDNA copies in the cell are silenced by promoter methylation14 and can be distinguished by different sensitivity to digestion by the isoschizomeric restriction enzymes HpaII and MspI. The RNA polymerase I and CSA-precipitated chromatin were digested by these enzymes followed by the amplification of the rDNA promoter. Whereas 60% of the input rDNA promoter was digested by the methylation-sensitive enzyme HpaII representing the unmethylated active fraction of the rDNA, HpaII nearly completely digested the promoter sequences precipitated by the RNA polymerase I or CSA antibody (Fig. 2C). This indicates that CSA binds the active (unmethylated) fraction of the rDNA promoter and suggests that CSA might play a role in transcription by RNA polymerase I.
To further address the question whether CSA binds the same rDNA molecules as RNA polymerase I, ChIP-re-ChIP experiments were performed. After the first round of precipitation, the antibody-bound chromatin was released by incubation with DTT and subjected to a second round of precipitation by the respective other antibody. As depicted in Figure 2D RNA polymerase I and CSA isolated rDNA promoter fragments can be re-ChIPed by the respective other antibody. Thus, RNA polymerase I and CSA occupy the same molecules of the rDNA promoter. These results were further corroborated by ChIP–western experiments, as shown in Figure 2E. RNA polymerase I precipitated chromatin was decrosslinked and analyzed for the presence of CSA using western blot analysis. Notably, CSA was detected in RNA polymerase I-precipitated chromatin.
Furthermore, CSA associates with RNA polymerase I in solution in the absence of DNA as shown by co-immunoprecipitation of RNA polymerase I and CSA from nuclear extracts (Fig. 2F).
We identified a previously unreported facet of the Cockayne syndrome protein CSA, which, in the absence of DNA damage, binds to the active fraction of the rDNA promoter and, in fact, co-localizes with RNA polymerase I on the same rDNA molecule.
CSA stimulates re-initation of RNA polymerase I
To gain further insight into the molecular mechanism of CSA function as a transcription factor, we used the RNA polymerase I in vitro transcription assay. Flag-tagged CSA protein was overexpressed in HEK cells and purified to near homogeneity by isolation with flag-M2-beads under high-salt conditions and was found to migrate as a single 50-kDa sized band when subjected to SDS-PAGE and silver staining (Fig. 3A). This protein was analyzed by western blot analysis to determine the purity of our preparation (Fig. 3B). The high-salt washed protein was not associated with DDB1 or CSB, RNA polymerase I, and TFIIH, indicating that our purification procedure either removed association partners, or that a fraction of overexpressed CSA might not associate with other proteins. These CSA preparations were used to reconstitute RNA polymerase I in vitro transcription with nuclear extracts of CSA-deficient cells. CSA at a concentration of 3 ng already stimulated in vitro transcription of CS3BE nuclear extracts 4-fold, while preincubation with a CSA antibody (Santa Cruz) completely abolished this stimulation (Fig. 3C). To further specify this stimulation we renatured CSA-overexpressed in Sf9 cells after excision of a 50-kDa region of a SDS-PAGE. This CSA preparation stimulated RNA polymerase I in vitro transcription as shown in Figure 3D. These results clearly show that CSA activates RNA polymerase I transcription at the rDNA promoter.
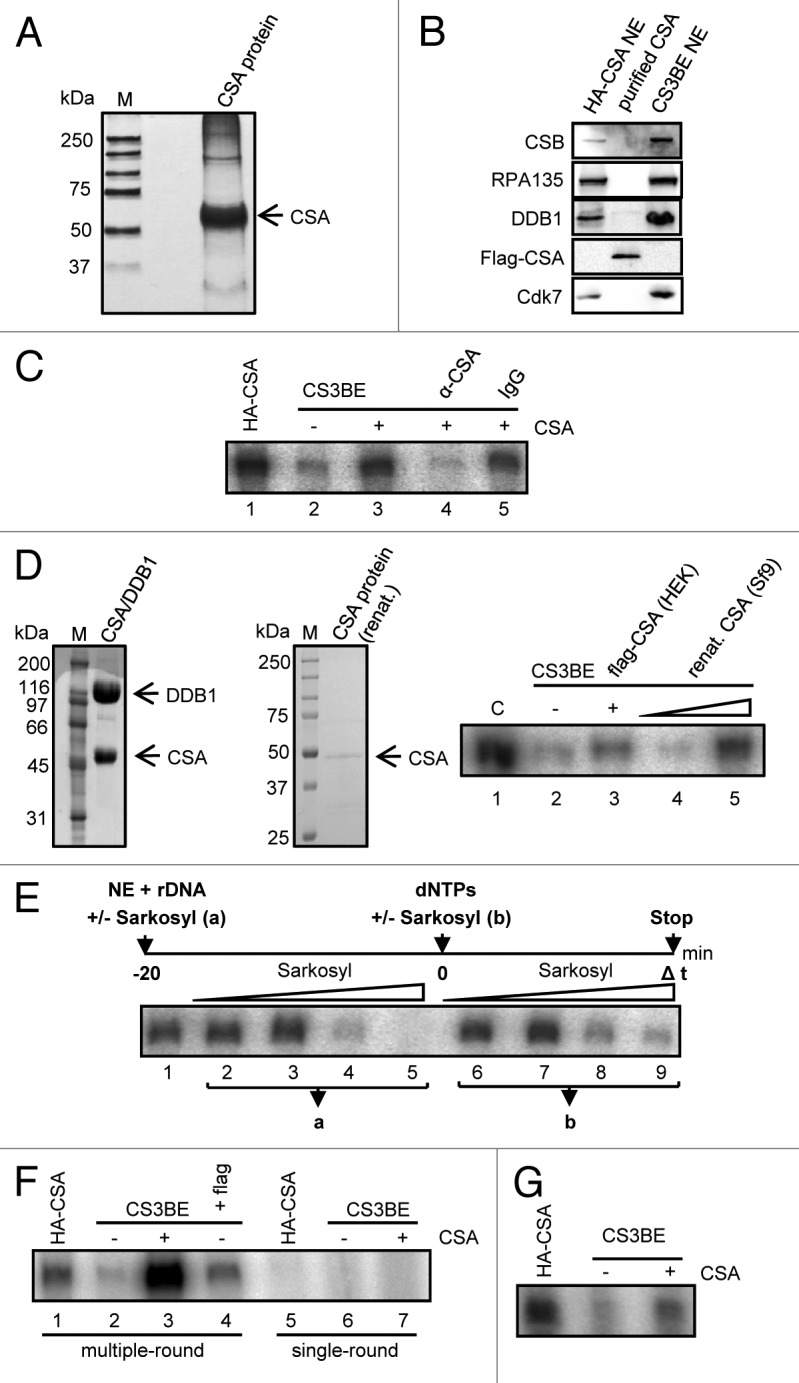
Figure 3. CSA stimulates re-initiation of RNA polymerase I transcription. (A) Silver staining of purified CSA protein after SDS-PAGE. (B) Western blot analysis of purified CSA demonstrating that high-salt washed CSA is free of CSB, RNA polymerase I, DDB1, and TFIIH (cdk7). (C) In vitro RNA polymerase I transcription with the indicated nuclear extracts and addition of purified CSA and CSA-specific antibodies that specifically block stimulation. (D) Sf9-expressed CSA stimulates RNA polymerase I transcription. Coomassie staining of the Sf9 expressed DDB1/CSA complex and the renatured CSA. In vitro transcription reaction with renatured CSA protein. (E) Sarkosyl titration before (a) and after (b) initiation complex formation reveals the critical sarkosyl concentration for single-round transcription (compare lane 5 to 9). (F) Single-round transcription is not stimulated by CSA. Multiple but not single-round transcription is enhanced by addition of CSA. (G) Transcription with immobilized template showing that CSA enhances transcription after initiation complex formation. Bead-bound template was incubated with nuclear extract in the absence of CSA. After removal of the nuclear extract and washing of the preformed initiation complexes CSA was added and in vitro transcription performed. Pictures are representative of at least 3 independent experiments.
Next we asked whether stimulation of RNA polymerase I transcription by CSA might be due to an increase in initiation complex formation and performed single-round transcription by adding sarkosyl to 0.025% after preincubation of nuclear extracts with the template. Sarkosyl at this concentration precludes initiation and exclusively allows one round of transcription of formed initiation complexes (Fig. 3E; compare lane 5 and 9). The strength of the resulting signal is proportional to the amount of newly formed initiation complexes. As depicted in Figure 3F, CSA stimulated multiple-round transcription (lane 3) but failed to enhance single-round transcription (lane 7). This is in line with the observation that HA–CSA nuclear extract harboring CSA did not build up more initiation complexes than CS3BE extract lacking full-length CSA (compare lane 5 with 6). Thus, CSA does not stimulate initiation complex formation at the rDNA promoter. This result was further corroborated by in vitro transcriptions with immobilized complexes. Nuclear extracts with or without CSA were incubated with rDNA template immobilized on magnetic beads. The beads were collected by magnetic attraction, the nuclear extracts removed, and the immobilized complexes were washed with low-salt buffer. CSA was added with the nucleotides at transcription start. Interestingly, CSA was able to stimulate in vitro transcription from preformed initiation complexes when nuclear extracts were already removed (Fig. 3G). No additional initiation complexes can be formed under these conditions, while the preformed complexes can be forced to perform better in multiple-round transcription. This demonstrates that CSA stimulates the re-initiation step of RNA polymerase I transcription.
CSA recruits the elongation factors CSB and TFIIH and stimulates ribosomal biogenesis and growth
Template immunoprecipitation assays13 were used to further analyze CSA at the rDNA promoter. The combination of in vitro transcription and chromatin immunoprecipitation allows further study of the binding dynamics of a given protein at a defined gene. Kinetics of CSA binding to the rDNA promoter after transcription start revealed that CSA stayed bound to the promoter when the polymerase starts to migrate from the promoter in this single-round transcription (Fig. 4A). Moreover, when CSA was titrated to CS3BE extract lacking full-length CSA, the CSB protein shows an increased binding to the rDNA. This was also the case for TFIIH (Fig. 4B). Consistent with a CSA-dependent re-initiation of RNA polymerase I transcription, CSA might stimulate the recruitment of the elongation factors CSB and TFIIH to the rDNA promoter.
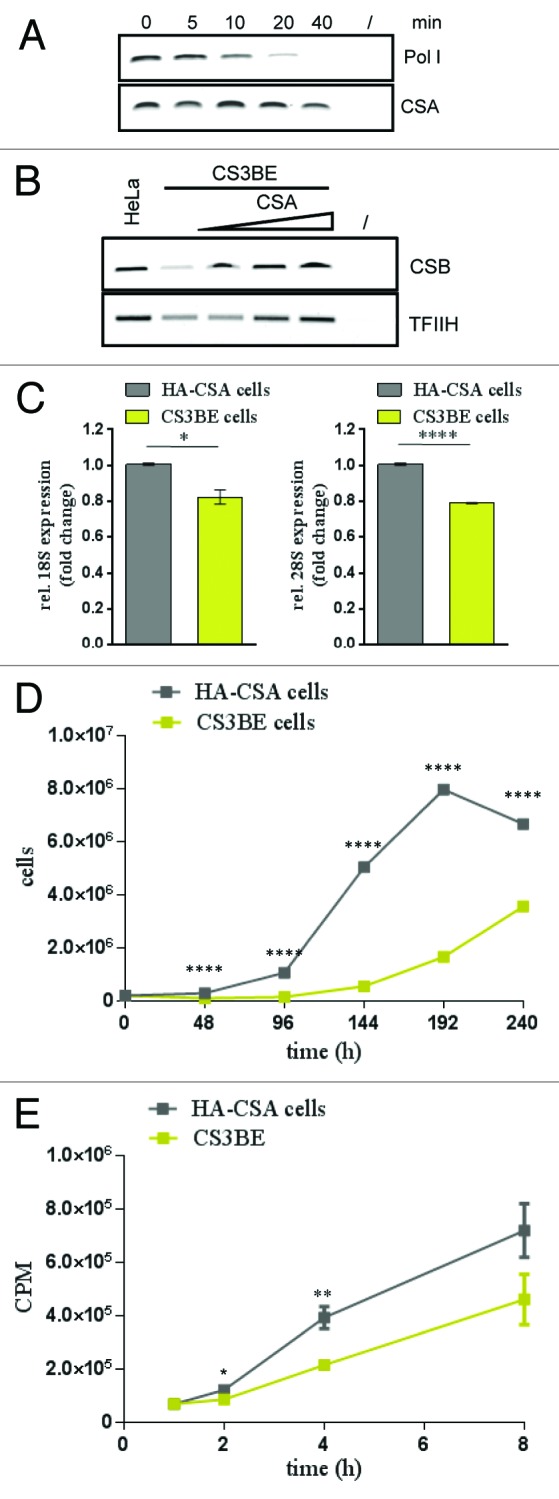
Figure 4. CSA recruits CSB and TFIIH to the rDNA promoter and loss of CSA retards ribosomal biogenesis and growth. (A) Template-immunoprecipitation experiment (13) showing the kinetics of CSA at the rDNA promoter after start of transcription. (B) Template immunoprecipitation experiments demonstrating CSB and TFIIH binding to the rDNA promoter after addition of CSA. (C) QPCR analysis of 18S and 28S rRNA amounts in cells lacking CSA (CS3BE) and CSA reconstituted cells. Values are normalized against RPL13 expression and the respective IgG controls and then normalized to HA-CSA cells. (D) Growth kinetics of CS3BE cells with or without CSA expression. (E) Metabolic labeling and translation kinetics of the indicated cells show that CSA stimulates protein biosynthesis. Pictures are representative of at least 3 independent experiments (A, B, D, and E) and values are mean ± s.e.m. of 3 independent experiments (C). (*P > 0.05; **P > 0.01; ***P > 0.001; ****P > 0.0001)
To investigate the consequences of a reduced 47S pre-rRNA transcription on the steady state of ribosomes in CSA-deficient vs. CSA-competent cells, we used qPCR to quantify the most abundant RNA species, the 18S and 28S rRNA. As demonstrated in Figure 4C, the loss of CSA is accompanied by a significant reduction in mature 18S and 28S rRNA. As a reduced ribosomal biogenesis is postulated to reduce growth of Cockayne syndrome cells, we compared the growth properties of CSA-deficient with CSA-competent cells. Growth kinetics of CSA-deficient CS3BE cells in comparison to HA–CSA cells clearly revealed a reduced growth capability of CS3BE cells when CSA competent cells entered the exponential growth phase (Fig. 4D). To investigate if this growth deficit could be caused by a reduced translation capacity of the CS cells we performed metabolic labeling experiments. After withdrawal of the amino acids methionine and cysteine for 20 min, 35S-labeled methionine and cysteine were added and incorporated radioactivity measured after the indicated timepoints (Fig. 4E). The data demonstrate the significantly reduced protein synthesis in parental CSA-deficient cells compared with CSA-competent cells. This indicates that the translation capacity and thus protein synthesis is significantly reduced by loss of CSA.
These results show a previously unreported function of CSA in RNA polymerase I transcription and ribosomal biogenesis. Most interestingly, Cockayne syndrome mutations in CSA substantially impair ribosomal biogenesis and cellular growth.
Discussion
In this study the DNA repair factor CSA was identified to serve a previously unreported function as a transcription factor of RNA polymerase I essentially required for normal ribosomal biogenesis and cellular growth. This important finding predicts that CSA mutations causal for Cockayne syndrome, a premature aging condition, lead to key features of severe cachexia and growth retardation, and this is exactly the case for CS patients. Our finding that CSA is critical for RNA polymerase I transcription and ribosomal biogenesis occurring in the nucleolus is supported by several lines of evidence. First, as shown by immunostaining, CSA locates to the nucleolus. This result, while confirmed by Saijo et al.,15 is in contrast to a former report which did not detect any nucleolar staining for CSA.10 This apparent discrepancy is most likely due to differences in the sensitivity of the antibodies used. To further support this view and to exclude that the observed nucleolar CSA staining might be an artifact of overexpressed CSA, we stained HA–CSA cells overexpressing CSA with the CSA antibody previously used in the study by Bradsher et al.10 In contrast to a clear nucleolar CSA staining with the herein used HA antibody, the CSA antibody used by Bradsher and coworkers did not stain CSA in the nucleoli of comparable cells. These data support the notion that different sensitivities of the reported antibodies explain the controversial results. Interestingly, proteasomal inhibition with MG-132 for 15 h led to positive nucleolar CSA staining by the “Bradsher” CSA antibody. These data may indicate that this antibody recognizes exclusively CSA accumulated in the nucleoli following inhibition of its proteosomal degradation. The even nuclear and nucleolar staining of CSA was not resistant to transcription inhibition by actinomycin D, indicating that the localization depends on ongoing transcription by RNA polymerase I. Pre-treatment of cells with triton X100 resulted in a loss of CSA staining. As repair-engaged CSA shows triton resistance,15 the nucleolar CSA might not be in a repair mode. Moreover, co-staining with TFIIH or CSB revealed nucleolar co-localization (Supplemental Material).
Second, our data show that increased CSA expression significantly enhanced the level of RNA polymerase I transcription. A former study investigated the effect of CSA antisera on transcription after microinjection and did not observe any inhibitory effect.8 A reduction of transcription via antibodies might be dependent on the epitope specifically recognized by the antibody. Therefore, this study does not necessarily contradict our finding that CSA induces RNA polymerase I transcription. Our results indicate that CSA is an important factor for enhanced RNA polymerase I transcription required under conditions of growth, while in the absence of wtCSA there is still enough rRNA synthesis to ensure at least viability.
Third, using ChIP experiments, we show that CSA is endowed with the capacity to bind to specific genes like the rDNA. As CSA occupies the complete coding region, is associated with the active fraction of the rDNA-like RNA polymerase I, and is able to form a complex with the Pol I enzyme, it is possible that CSA is a part of the initiating and elongating enzyme complex. The kinetic data shown in the template immunoprecipitation assays, however, argue against this hypothesis, as CSA under single-round transcription conditions does not leave the template but stays associated with the rDNA throughout transcription. The finding that CSA only covers active rDNA suggests that CSA has an affinity to open chromatin structures as represented by active rDNA copies.16 Alternatively, CSA might be recruited to the rDNA by the elongation and chromatin remodeling factor CSB.12,17 While binding of CSA to the initiating form of RNA polymerase II was previously reported,18 the question whether CSA, in a CSB-dependent fashion, also binds to RNA polymerase II and III genes in the absence of DNA damage, so far has not been addressed. Fourth, following CSA overexpression and purification under high-salt conditions or renaturation from SDS-PAGE after expression in Sf9 cells, we purified CSA free of known interaction partners, which stimulate RNA polymerase I in vitro transcription. Western blot analysis demonstrates that the CSA preparation contains neither DDB1 or the RNA polymerase I elongation factors CSB and TFIIH nor RNA polymerase I itself. ChIP analysis with a DDB1 antibody did not reveal any rDNA sequences (see Supplemental Materials). Together, these data support the view that CSA does not bind as part of the DDB1–CSA complex to the rDNA. The findings that essentially pure CSA and Sf9-expressed CSA markedly stimulated RNA polymerase I in vitro transcription with extracts from CSA-mutant cells and that CSA antibodies abrogated this stimulation further confirm that CSA plays a crucial, so-far-unknown role in ribosomal biogenesis. Based on the observation that CSA did not influence single-round transcription, but enhanced transcription after initiation complex formation, we conclude that re-initiation, possibly by contact with TFIIH and CSB does account for the stimulation of RNA polymerase I transcription. In fact, TFIIH migrates with the polymerase from the promoter,13 while CSA remains bound throughout transcription, suggesting that CSA enhances transcription by recruiting TFIIH and CSB-bound polymerase to the intiation site.
Fifth, CSA influences the steady state of mature rRNA, translation capacity, and growth of fibroblasts. Cockayne syndrome cells mutant in CSA show a reduced content of 28S/18S rRNA accompanied by markedly reduced overall protein biosynthesis and reduced growth. As growth retardation is a hallmark of Cockayne syndrome patients,1,2 impaired ribosomal biogenesis and reduced protein synthesis might be responsible for this key symptom in CS patients. Our data may even suggest that Cockayne syndrome, at least in part, can be interpreted as a consequence of a ribosomopathy. Other ribosomopathies like the Treacher–Collins syndrome and Diamond–Blackfan anemia that exhibit congenital cranial malformation and bone marrow failure, respectively, are not classified as premature aging diseases. Treacher–Collins syndrome (TCS) can be caused by mutations in a RNA polymerase I/III subunit19 or in the RNA polymerase I transcription/processing factor treacle.20 Severe clinical features may include hearing loss21 and mental retardation, symptoms that are also found in Cockayne syndrome. Treacle plays a tissue-specific essential role in ribosome biogenesis as demonstrated in vivo.22 Mice haploinsufficient for Tcof1 (treacle) exhibited diminished levels of 28S rRNA in neural ectoderm and neural crest, leading to apoptosis and craniofacial abnormalities. This phenotype can be rescued by blocking neuroepithelial apoptosis through inhibition of p53.23 This indicates that a tissue-specific inhibition of RNA polymerase I transcription is followed by craniofacial malformation that can be overcome by inhibition of p53. As p53 might also play a role in the pathogenesis of Cockayne syndrome and aging in general,24,25 elevated p53 levels caused by systemically impaired ribosomal biogenesis might, in fact, contribute to degeneration and premature aging in Cockayne syndrome. As CSA and CSB also directly regulate p53 turnover by physical interaction,24,26 and the CSB protein is critically involved in the hypoxic response modulating p53,27 Cockayne syndrome cells seem to be in a constant state of p53 hyperactivation and stress.25 CSB on the other side is also involved in regulating stability of triplet repeats.28 However, inactivating mutations in CSA affect re-initiation of RNA polymerase I transcription and might be sensed by the cell as nucleolar stress that is followed by an imbalance between rRNA and ribosomal proteins. An excess of ribosomal proteins stabilize p53 and mediates cell cycle arrest and apoptosis.29 Reduced ribosomal biogenesis, nucleolar stress, and consecutive apoptosis instead of growth and proliferation might contribute to retarded growth and premature aging in Cockayne syndrome.
Materials and Methods
Cell lines and extracts
CS3BESV cells and CS3BESV cells stably transfected with HA-tagged CSA as well as HeLa, HEK cells, and secondary fibroblasts 1306 were maintained in DMEM, supplemented with 10% FCS, penicillin, and streptomycin. HA–CSA cells were cultivated with 400 µg/ml G418. Nuclear extracts preparation followed the protocol of Dignam.30
Antibodies
Anti-HA high-affinity (Roche), anti-CSA, anti-CSB, anti-flag, and anti-cdk7 from Santa Cruz, nucleolin antibody from Abcam, anti-DDB1 from Cell Signaling, anti-CSA antibody from Genetex, and anti-CSA antibody from F. Coin were used. Production of the RPA-135 is described in Assfalg et al.13
Immunofluorescence
Cells seeded on slides were fixed with 4% PFA for 15 min at 4 °C and subsequently permeabilized with 0.5% Triton X100 for 7 min at RT. After blocking with 5% BSA, the first antibody was incubated over night at 4 °C. After subsequent washing, the secondary antibody was added for 1 h at room temperature.
RNA preparation, primer
RNA from growing cells was prepared by using RNeasy kit (Qiagen, Hilden). Primer used were: 47S: h47S F:TGTCAGGCGT TCTCGTCTC
R:AGCACGACGT CACCACATC; 5.8S F: TCGTGCGTCG ATGAAGAACG CAG
5.8S R:ATTGATCGGC AAGCGACGCT CAG; CSA-F: CTAGAGGACC CAAAGTACAA
CSA-R: TGACTTTTTC CCATTATGTT. The other primers were published by O`Sullivan et al.31
shRNA
shRNA was from Sigma (TRCN0000342530) with the following sequence: CCGGGCGCTA ATGCTTGAAC TCTTTCTCGA GAAAGAGTTC AAGCATTAGC GCTTTTTG
ChIP
Chromatin immunoprecipitations were performed as described by Assfalg et al.13
For ChIP-re-ChIP, samples were released after the first round of precipitation and washed by incubation in 10 mM DTT. Then the samples were diluted and incubated with the respective other antibody. For ChIP–western analysis, the precipitated chromatin was heated in Laemmli buffer for 45 min at 99 °C prior to SDS-PAGE.
Immunoprecipitation
Co-immunoprecipitation experiments were performed with the indicated antibodies by incubation of nuclear extracts for 2 h at 4 °C and subsequent addition of protein-A/G agarose (Roche) and additional overhead rotation for 60 min. Beads were harvested by centrifugation, transferred to columns, and washed with buffer AM100. Bound proteins were released by addition of Laemmli buffer and analyzed by SDS-PAGE and western blotting.
In vitro transcription, template immunoprecipitation
In vitro analysis followed the protocol in reference 13 and references therein.
CSA purification
CSA protein was expressed after transient transfection of HEK cells with plasmid p3xFlag-CSA. Cells were harvested in buffer AM600 and sonicated 3 × 15 s. NP40 was added to 0.5%, and lysate was clarified by centrifugation. Flag-M2 beads (Sigma) were used to isolate 3xflag CSA by incubation overnight. The resin was sequentially washed with buffers AM containing 1000/400 mM KCl and 0.5%NP40 and flag-CSA was eluted in buffer AM300 0.1% NP40 with 0.2 µg/µl flag-peptide. Sf9-expressed CSA protein was excised from a SDS gel, minced and eluted 3 h in renaturation buffer (20 mM Tris, pH8, 1 mM EDTA, 100 mM NaCl, 5 mg/ml BSA, 1 µM DTT, 1% Triton X100, protease inhibitor mix [Roche]) at 37 °C. After centrifugation glycerol was added to 10% and the protein snap frozen. For SDS-gel analysis, BSA was omitted from the renaturation buffer.
Metabolic labeling
Cells were harvested by trypsination and counted. 200 000 cells per sample were starved for 20 min by incubation with DMEM/dialyzed FCS devoid of methionine/cysteine and subsequently labeled by the addition of 35S methionine/cysteine (10 µCi/ml). At the indicated time points the reaction was stopped by washing with ice-cold PBS and subsequent TCA precipitation for 90 min. Precipitates were spotted on glass filters, washed, and incorporated radioactivity was quantified in a szintillation counter.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Meinhard Wlaschek for stimulating discussion and critical reading of the manuscript and Frederic Coin for the CSA antibody. The flag-CSA plasmid was a kind gift of Luca Proietti-de-Santis. Sf9-expressed DDB1/CSA protein was a kind donation of Nicholas Thomä. Angelika Rück and Carmen Hauser from ILM Ulm helped by confocal microscopy. This work was supported by the German Research Foundation DFG, IB 83/2-2, and IB 83/3-1. S.I. was supported by the “Perspektivförderung” of Baden-Wuerttemberg.
Glossary
- Abbrevations
CSA, Cockayne syndrome protein A
- CS
Cockayne syndrome
- CSB
Cockayne syndrome protein B
- TFIIH
transcription factor IIH
- TCR
transcription coupled repair
- NER
nucleotide excision repair
- UV
ultraviolet
- XPB/XPD/XPG
xeroderma pigmentosum complementation group B/D/G
- ChIP
chromatin immunoprecipitation
- rDNA
ribosomal DNA
- QPCR
quantitative polymerase chain reaction
- ITS
internal transcribed spacer
- DDB1
DNA damage binding protein 1
References
- 1.Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 2.Laugel V, Dalloz C, Durand M, Sauvanaud F, Kristensen U, Vincent MC, Pasquier L, Odent S, Cormier-Daire V, Gener B, et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat. 2010;31:113–26. doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- 3.Proietti-De-Santis L, Drané P, Egly JM. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006;25:1915–23. doi: 10.1038/sj.emboj.7601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itoh T, Ono T, Yamaizumi M. A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat Res. 1994;314:233–48. doi: 10.1016/0921-8777(94)90068-X. [DOI] [PubMed] [Google Scholar]
- 5.Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15410–5. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le May N, Mota-Fernandes D, Vélez-Cruz R, Iltis I, Biard D, Egly JM. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Le May N, Fradin D, Iltis I, Bougnères P, Egly JM. XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol Cell. 2012;47:622–32. doi: 10.1016/j.molcel.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 8.van Gool AJ, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, Egly JM, Bootsma D, Hoeijmakers JH. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997;16:5955–65. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iben S, Tschochner H, Bier M, Hoogstraten D, Hozák P, Egly JM, Grummt I. TFIIH plays an essential role in RNA polymerase I transcription. Cell. 2002;109:297–306. doi: 10.1016/S0092-8674(02)00729-8. [DOI] [PubMed] [Google Scholar]
- 10.Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, Egly JM. CSB is a component of RNA pol I transcription. Mol Cell. 2002;10:819–29. doi: 10.1016/S1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- 11.Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schäfer A, Grummt I, Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33:344–53. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Lebedev A, Scharffetter-Kochanek K, Iben S. Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J Mol Biol. 2008;382:266–74. doi: 10.1016/j.jmb.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Assfalg R, Lebedev A, Gonzalez OG, Schelling A, Koch S, Iben S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012;40:650–9. doi: 10.1093/nar/gkr746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santoro R, Grummt I. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol Cell. 2001;8:719–25. doi: 10.1016/S1097-2765(01)00317-3. [DOI] [PubMed] [Google Scholar]
- 15.Saijo M, Hirai T, Ogawa A, Kobayashi A, Kamiuchi S, Tanaka K. Functional TFIIH is required for UV-induced translocation of CSA to the nuclear matrix. Mol Cell Biol. 2007;27:2538–47. doi: 10.1128/MCB.01288-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dammann R, Lucchini R, Koller T, Sogo JM. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:2331–8. doi: 10.1093/nar/21.10.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan X, Feng W, Imhof A, Grummt I, Zhou Y. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol Cell. 2007;27:585–95. doi: 10.1016/j.molcel.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 18.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–67. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 19.Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer-Haas C, Maiwald R, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43:20–2. doi: 10.1038/ng.724. [DOI] [PubMed] [Google Scholar]
- 20.The Treacher Collins Syndrome Collaborative Group Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996;12:130–6. doi: 10.1038/ng0296-130. [DOI] [PubMed] [Google Scholar]
- 21.Phelps PD, Poswillo D, Lloyd GA. The ear deformities in mandibulofacial dysostosis (Treacher Collins syndrome) Clin Otolaryngol Allied Sci. 1981;6:15–28. doi: 10.1111/j.1365-2273.1981.tb01782.x. [DOI] [PubMed] [Google Scholar]
- 22.Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103:13403–8. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125–33. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latini P, Frontini M, Caputo M, Gregan J, Cipak L, Filippi S, Kumar V, Vélez-Cruz R, Stefanini M, Proietti-De-Santis L. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle. 2011;10:3719–30. doi: 10.4161/cc.10.21.17905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frontini M, Proietti-De-Santis L. Interaction between the Cockayne syndrome B and p53 proteins: implications for aging. Aging (Albany NY) 2012;4:89–97. doi: 10.18632/aging.100439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berquist BR, Bohr VA. Cockayne syndrome, underlying molecular defects and p53. Cell Cycle. 2011;10:3997–8. doi: 10.4161/cc.10.23.18352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frontini M, Proietti-De-Santis L. Cockayne syndrome B protein (CSB): linking p53, HIF-1 and p300 to robustness, lifespan, cancer and cell fate decisions. Cell Cycle. 2009;8:693–6. doi: 10.4161/cc.8.5.7754. [DOI] [PubMed] [Google Scholar]
- 28.Kovtun IV, Johnson KO, McMurray CT. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY) 2011;3:509–14. doi: 10.18632/aging.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donati G, Bertoni S, Brighenti E, Vici M, Treré D, Volarevic S, Montanaro L, Derenzini M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene. 2011;30:3274–88. doi: 10.1038/onc.2011.48. [DOI] [PubMed] [Google Scholar]
- 30.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Sullivan AC, Sullivan GJ, McStay B. UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol Cell Biol. 2002;22:657–68. doi: 10.1128/MCB.22.2.657-668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.