

Abstract
The DNA double-strand break repair and homologous recombination protein Rad51 is overexpressed in the majority of human cancers. This correlates with therapy resistance and decreased patient survival. We previously showed that constructs containing Rad51 promoter fused to a reporter gene are, on average, 850-fold more active in cancer cells than in normal cells. It is not well understood what factors and sequences regulate the Rad51 promoter and cause its high activity in cancerous cells. Here we characterized regulatory regions and examined genetic requirements for oncogenic stimulation of the Rad51 promoter. We identified specific regions responsible for up- and downregulation of the Rad51 promoter in cancerous cells. Furthermore, we show that Rad51 expression is positively regulated by EGR1 transcription factor. We then modeled the malignant transformation process by expressing a set of oncoproteins in normal human fibroblasts. Expression of different combinations of SV40 large T antigen, oncogenic Ras and SV40 small T antigen resulted in step-wise increase in Rad51 promoter activity, with all the 3 oncoproteins together leading to a 47-fold increase in expression. Cumulatively, these results suggest that Rad51 promoter is regulated by multiple factors, and that its expression is gradually activated as cells progress toward malignancy.
Keywords: Rad51 promoter, cancer, EGR1, p53, DNA repair, therapy resistance
Introduction
The ability of cancer cells to become resistant to and evade death due to chemo- and radiation therapy is a major problem in the treatment of cancer patients. Therapy resistance is based on several cellular processes. These include perturbations and loss of function in programed apoptotic cell death pathways,1 increases in the metabolism and removal of chemotherapeutic drugs,2,3 as well as cancer cell’s ability to repair DNA lesions induced by therapy.4 Changes in these cellular processes allow cancer cells to continue through the cell cycle, potentially producing more and more cells with genomic aberrations leading to more rapid evolution of resistant clones. Understanding the mutations and/or altered expression of the genes needed to support resistance is of utmost importance for the development of new anti-cancer therapies.
The majority of cancer therapeutics, such as cisplatin, etoposide, and radiation therapy, induce DNA damage and stalled replication forks,5 rapid cell cycle in cancer cells,6 and their lack of cell cycle checkpoints;7 these therapy-induced DNA lesions and stalled forks result in double-strand breaks, chromosomal instability, mitotic catastrophe, and, eventually, cell death.5 Cancer cells cope with increased levels of double-stranded breaks and stalled replication forks by overexpressing the recombinase and DNA repair protein RAD51.8,9 Expression of Rad51 is tightly controlled in normal human cells to decrease aberrant chromosomal recombination.10,11 The opposite is seen in the majority of human tumors and cancer cell lines, where Rad51 mRNA and protein levels as well as the promoter activity are very high.10,12-15 Rad51 overexpression not only allows the cancer cells to survive treatment,13,16 but is linked to metastasis17 and aggressiveness18 of the disease and poorer patient prognosis.19-21
Although the role of Rad51 in cancer development, progression and pathogenesis has been well established, very few studies have examined the mechanisms behind the differential regulation of Rad51 between cancerous and non-cancerous cells.10,22-25 We previously cloned a segment of the Rad51 promoter region spanning −2931 bp upstream to +3601 bp downstream relative to the start of transcription and analyzed promoter activity in a variety of non-cancerous and cancerous human cell lines. Promoter activity was measured by placing the open reading frame (ORF) of firefly luciferase under the transcriptional control of this cloned Rad51 promoter.10 Cancerous cells displayed an average 850-fold increase in promoter activity compared with non-cancerous cell lines, with a maximal difference of 12 500-fold. Due to such a large differential in promoter activity/transgene expression, we further extended the use of our cloned Rad51 promoter for use in targeted anti-cancer gene therapy. By replacing the luciferase gene with that of diphtheria toxin-A (DTA) to form pRad51-DTA, a number of human cancer cell lines from breast cancer, fibrosarcoma, cervical cancer, and prostate cancer were killed, while non-cancerous cells here relatively unharmed. These constructs were then used in vivo to visualize human cervical carcinoma tumors in mice and then treat them with favorable outcomes. These included reduction in tumor burden and ascites volume while increasing survival.24 Thus, we established the Rad51 promoter as a potent cancer-specific driver with potential for clinical applications in treatment and visualization of tumors.
Questions still remain as to why the Rad51 promoter construct showed such a large differential between cancerous and non-cancerous cells. The tumor suppressor p53, mutated in the majority of human tumors,26 had previously been shown to interact with the Rad51 promoter and downregulate both Rad51 expression and promoter activity.22,23,25 Although in these studies the re-introduction or overexpression of p53 decreased the activity of the Rad51 promoter in cancerous cell lines, it did not repress it to near background levels that we observed in non-cancerous human cells. Here, we set out to investigate both cis acting elements/regions on the Rad51 that promote or repress expression in normal and cancerous human cells as well as the trans acting factors that stimulate promoter activity during cancerous transformation of normal cells into cancerous.
In the present study, a series of deletions including the putative p5322 and EGR125 binding sites were introduced into the Rad51 promoter in the pRad51–Luc construct. These constructs were then tested in hTERT immortalized normal human fibroblasts HCA2-T and in the human cancer cell lines HT1080 and HeLa. No deletions were able to stimulate Rad51 promoter activity in HCA2-T cells, while several deletions including the EGR1 site deletion significantly decreased promoter activity in the cancer cell lines, although these activities were still substantially greater than detected in HCA2-T cells. As the EGR1 site-specific deletions lead to universal reductions in promoter activity for all 3 cell lines, and EGR1 transcription factor activity has been associated with prostate cancer growth,27 we further looked into EGR1 regulation of Rad51 expression. We established that the overexpression of EGR1 stimulates Rad51 promoter activity up to 4-fold in HCA2-T cells, and this induction is dependent on the presence of these EGR1 binding sequences. Due to the inability of any of the mutations to robustly stimulate Rad51 promoter activity in HCA2-T cells to levels seen in cancer cells, we predicted that trans acting factors may be causing the increased Rad51 promoter activity in cancer. To test this, we took HCA2-T cells through a stepwise transformation using SV40 T antigens to inhibit p53, pRb, and PP2A as well as pro-growth stimulation with oncogenic RasV12.28-30 Inhibition of p53 and pRb only slightly induced Rad51 promoter activity, while oncogenic Ras had no effect on its own. The combination of SV40 large T and small T antigens and oncogenic Ras resulted in a 47-fold increase in promoter activity. Taken together, trans acting factors are needed to stimulate Rad51 promoter activity during tumorigenesis, and this stimulation is dependent of cis-regulatory elements both in the 5′ and 3′ ends of the promoter region.
Results
Identification of activating and repressing regions in Rad51 promoter
We previously cloned Rad51 promoter (−2931 bp upstream to +3601 bp downstream from the start of transcription) in the pRad51-Luc plasmid and showed that it is highly active in human cancer cells and is repressed in non-cancerous cells.10,24 In order to identify the regions responsible for this differential regulation we constructed a series of deletion constructs using available restriction enzyme sites within the promoter region: −2931 to −23, −2010 to −207, −656 to -138, +173 to +265, and +1675 to +3313 bp both upstream and downstream from the start of transcription (Fig. 1A). In addition, specific sites hypothesized to interact with the transcription factors p53; −160 to −11522 and EGR1; −217 to -208 and +170 to +17725 were deleted by site-directed mutagenesis PCR (Fig. 1A).

Figure 1. Wild-type and deletion Rad51 promoter constructs and their activities in cancerous and normal human cells. (A) Rad51 gene promoter spanning −2931 to +3601 nucleotides relative to the start of transcription was cloned from normal human skin fibroblasts. This promoter fragment was placed upstream of the firefly luciferase gene (F. Luc) in the promoter-less pGL3 Basic plasmid forming pRad51-Luc. To characterize positive and negative regulatory cis-elements within the Rad51 promoter, various deletions were made by restriction digest followed by re-ligation (∆−2931 to −23, ∆ −2010 to −207, ∆ −656 to −138, ∆ +173 to +265, and ∆ +1675 to +3313) or by PCR site directed mutagenesis (∆ p53, ∆ EGR1). Black arrow inducates the start of transcription, 5′ UTR stands for the 5′ untranslated region that is exon 1. (B) The activity of the promoter constructs described above was tested in hTERT-immortalized normal human fibroblast cell line HCA2-T. (C) The activity of the promoter constructs in fibrosarcoma cell line HT1080. (D) The activity of the promoter constructs in HeLa cells. All experiments were repeated at least 4 times, and error bars show s.d. *Asterisk indicates P < 0.05.
Consistent with our earlier findings,10,24 the promoter activity was dramatically higher in the cancer cell lines than in the normal human fibroblasts cell line HCA2-T (Fig. 1B–D). No mutation or deletion had stimulating effect on promoter activity in HCA2-T (Fig. 1B), likely due to multiple repressive mechanisms controlling Rad51 in normal cells. Although not a robust change, deletion of the region −2010 to −207 resulted in a significant (P < 0.05) 50% reduction in promoter activity. The deletion of p53 and EGR1 sites from this non-cancerous cell line also resulted slight but significantly (P < 0.05) reduction in promoter activity by approximately 40% and 60%, respectively. The result for the p53 site deletion is surprising due to previous work showing that p53 downregulates Rad51 promoter activity at this site, albeit in a cancer cell line.22 This suggests p53 is not solely responsible for repression of Rad51 expression, and that p53’s role in downregulating Rad51 promoter activity may be cell type-specific or act indirectly to repress Rad51 promoter activity, as recently shown by Fong, et al.23 These results indicate that these regions are acting to promote Rad51 expression, and deletions of these regions result in repression of Rad51 activity in normal cells.
In the human cancer cells lines HT1080 and HeLa (Fig. 1C and D), large deletions at −2931 to −23 and −2010 to −207 led to an average decrease in promoter activity of 4-fold and 190-fold, respectively (P < 0.05). Site-directed deletion of the EGR1 sites, similar to what was seen in HCA2-T cells, led to significant decreases in Rad51 promoter activity of 50% in HT1080 and 70% in HeLa (Fig. 1C and D). One region deleted downstream from the start of transcription that had a dramatic effect on Rad51 promoter activity in both cancer cells lines was between +173 to +265. Deletion of this region resulted in a 15-fold decrease in promoter activity. This deletion removes the first exon/first intron border, as the first exon 5′ UTR ends at + 229 (Fig. 1A). This results in a new 3296 bp 5′ UTR compared with the 229 bp wild-type 5′ UTR and may influence the efficiency of translation and/or RNA stability.
Deletions at −656 to −138, −160 to −115 (p53 site) and +1675 to +3313 resulted in slight increase in Rad51 promoter activity in the 2 cancer cell lines (Fig. 1C and D). Taken together, the deletion data indicates the regions stimulating Rad51 promoter activity and allowing for overexpression in cancer cells are located in the region between −2931 to −656 and at the putative EGR1 biding sites.
EGR1 stimulates Rad51 promoter activity
Deletion of the 2 putative EGR1 biding sites reduced promoter activity in both normal and cancer cells (Fig. 1). Because overexpression of EGR1 has been linked to several cancers,27,31 we decided to closer examine EGR1’s role in Rad51 promoter activity in the HCA2-T cells. As shown in Figure 2, deleting each of the EGR1 sites separately reduced Rad51 promoter activity, but the effect did not reach statistical significance. The combination of the 2 EGR1 site deletions was synergistic and resulted in a significant reduction (Fig. 2A). We next examined if overexpression of EGR1 could stimulate Rad51 promoter activity and whether the stimulation is dependent on these EGR1 sites. Co-transfection of 2 µg of CMV-EGR1 overexpression plasmid along with wild-type pRad51-Luc resulted in a 4-fold increase in promoter activity (Fig. 2B) (P < 0.05). This increase in promoter activity by EGR1 overexpression was abolished when the 2 EGR1 sites were deleted on the Rad51 promoter in conjuncture with EGR1 overexpression, resulting in 67% decrease in promoter activity (P < 0.05) (Fig. 2B). These results indicate that EGR1 promotes Rad51 expression, and this stimulation of Rad51 promoter activity is requires nucleotides −217 to −208 and +170 to +177.

Figure 2. Putative EGR1 binding sites are necessary for maximum Rad51 promoter activity and for the stimulation by EGR1 overexpression. (A) Putative EGR1 binding sites contribute to Rad51 promoter activity. Promoter constructs containing deletions of the first site (−217 to −208) and the second site (+170 to +177) individually and in combination were tested in HCA2-T cells by luciferase assay. (B) EGR1 overexpression in HCA2-T cells stimulates Rad51 promoter activity, and this stimulation is dependent on the presence of the putative EGR1 biding sites. The wild-type pRad51-Luc plasmid was co-transfected with either 2.0 µg pCMV-EGFP (control) or 2.0 µg CMV-EGR1 plasmid into HCA2-T cells. Luciferase assays were performed 72 h post-transfection. All experiments were repeated 4 times, and error bars show s.d. *Asterisk indicates P < 0.05.
Tumorigenic transformation of HCA2-T cells activates the Rad51 promoter
Malignant transformation of human dermal fibroblasts can be achieved by ectopic overexpression of telomerase, oncogenic Ras, and the knockdown of the tumor suppressors p53 and pRb with SV40 Large T antigen and knockdown of PP2A and possibly constitutive mTOR activation with Small T antigen.28-30,32,33 Due to no mutations in the Rad51 promoter resulting in robust stimulation of promoter activity in the non-cancerous cells and EGR1 overexpression only giving a 4-fold increase, we examined whether the transfection of these oncogenic factors could activate Rad51 promoter activity (Fig. 3).
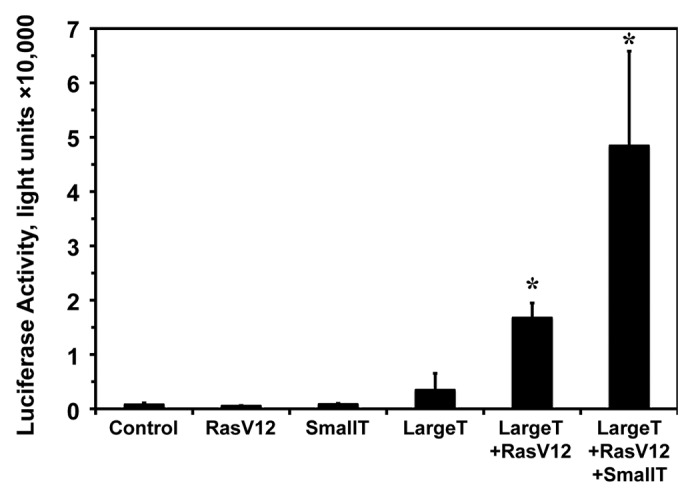
Figure 3. Step-wise oncogenic transformation of normal human fibroblasts stimulates Rad51 promoter activity. HCA2-T cells were transfected with wild-type pRad51-Luc and various combinations of plasmids encoding oncoproteins: SV40 Large T antigen (Large T), SV40 Small T antigen (Small T), oncogenic RasV12, or GFP (control) and Rad51 promoter activity was measured by luciferase assay. Luciferase assays were performed 72 h post-transfection. The experiments were repeated at least 3 times, and error bars show s.d. *Asterisk indicates P < 0.05.
Co-transfection of pRad51-Luc with oncogenic Ras (Ras G12V), a potent initiator of malignant transformation,34 did not change Rad51 promoter activity (Fig. 3). Similarly, co-transfection of pRad51-Luc with Small T antigen; resulted in non-significant increase in Rad51 promoter activity (Fig. 3). However, transfection of Large T antigen; which knocks down the activities of the tumor suppressors p53 and pRb, resulted in slight enhancement (2–4-fold) in Rad51 promoter activity (Fig. 3). This suggests that the large differential between non-cancerous and cancerous cells is not due entirely to repression by these 2 powerful tumor suppressors consistent with the deletion of the proposed p53 binding region being unable to enhance Rad51 promoter activity in the non-cancerous cell line (Fig. 1B). It was not until we combined Ras with Large T antigen, the minimum combination known to transform some mammalian cells, that we observed a dramatic; 16-fold, increase in Rad51 promoter activity (P < 0.05) (Fig. 3). Addition of the Small T antigen to Ras and Large T antigen regimen resulted in a 47-fold increase in Rad51 promoter activity over pRad51-Luc alone (P < 0.05) (Fig. 3). In summary, these results indicate that Rad51 promoter activity increases gradually as cells progress toward malignancy, with a dramatic increase observed in malignant cells.
Discussion
Rad51 promoter shows a dramatic, up to 850-fold difference in activity between normal and cancerous cells.10,24 Mechanisms responsible for this differential are poorly understood. In the present study we identified activating and repressing regions within Rad51 promoter in normal and cancerous cells. Interestingly, none of the promoter deletions were able to increase the activity in normal cells. In the cancerous cells, deletion of the regions −656 to −217, −208 to −115, and +1675 to +3313 resulted in increased promoter activity, suggesting that these regions downregulate the promoter. While deletion of the regions located between −2931 to −656, −217 to −208, and −115 to +1675 represses the promoter activity, suggesting that these regions positively regulate the promoter. Regions that downregulate expression are located between −656 to −217, −208 to −115, and +1675 to +3313 (Fig. 4). Interestingly, deletion of the p53 binding site did not stimulate the promoter in normal cells but showed a trend toward increase in promoter activity in the cancerous cells.
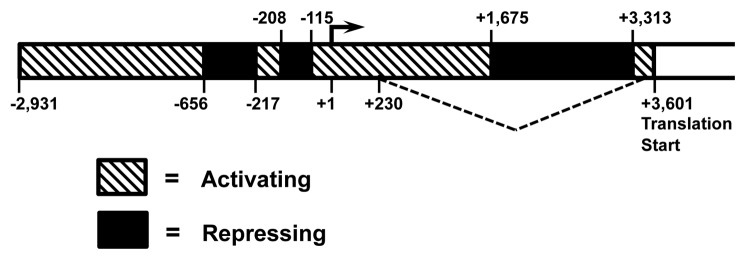
Figure 4. Diagram summarizing activating and repressing regions in Rad51 promoter in cancerous cells. The diagram is a compilation of data shown in Figure 1C and D, where the activity of Rad51 promoter constructs containing various deletions was tested in HT1080 and HeLa cells. Regions that activate expression are between −2931 to −656, −217 to −208, and −115 to +1675. Regions that downregulate expression are between −656 to −217, −208 to −115, and +1675 to +3313.
Previously, Rad51 overexpression in cancer cells was likened to transcriptional de-repression at the promoter12 by loss of functional p53 repressing at a proposed core promoter site of −160 to −11522,23 and loss of E2F4/p130 repression35 at base pairs −31 to −2125 due to hyperphosphorylation of p130 during cell cycle.36 This same sequence between −31 to −21 could then activate Rad51 expression via the oncogenic BCR/ABL fusion tyrosine kinase constitutively activating STAT5 transcription.25,37,38 Additionally, possible changes in CpG island methylation states between base pairs −116 to −92 as well as translational/posttranslational regulation were suggested to affect Rad51 expression and lead to the differential between cancerous and non-cancerous cells.39
While our data does not contradict these findings, our results and those of Fong, et al.,23 performed in different cell types using a longer Rad51 promoter region, suggest that the regulation of Rad51 expression is far more complex and cannot be explained by one or 2 factors changed during tumorigenesis affecting the level of transcription. This is evident in our experiment, which modeled the carcinogenesis process in HCA2-T cells by introducing SV40 Large T, Small T, and oncogenic Ras. Even though we obtained a robust (47-fold) increase in Rad51, this is still overshadowed by an average 850-fold increase in Rad51 promoter activity seen in cancer cells.10,24 Likewise, we showed that EGR1 upregulates the Rad51 promoter at the specific sites −217 to −208 and +170 to +177. However, this upregulation was only on the order of 4-fold (Fig. 2). These results suggest that other factors involved in tumorigenesis and cancer cell survival are perturbed during cancer progression to achieve this high level of Rad51 promoter activity.
The additional increase in Rad51 promoter activity conferred by expression of the Small T antigen could be due to both PP2A inhibition and inhibition of the 4E-BP1 translation factor. The latter leading to increased translation via eIF4E and mTOR activation.33,40 Interestingly, inhibition of eIF4E in cancer cells, but not in normal cells, sensitizes them to ionizing radiation by downregulating the translation of Rad51 mRNA in to protein.41
None of the deletions made to the Rad51 promoter completely shut it down in the 2 cancerous cell lines to levels detected in non-transformed HCA2-T cells (Fig. 1). Since these deletions encompass various areas of the Rad51 promoter region, including the entire upstream region, portions of the 5′ UTR and the 1st intron, this again suggests that regulation of the Rad51 expression is complex and involves cis and trans factors and elements operating at the level of transcription, RNA processing, stability, translational initiation, elongation, and posttranslational modifications.
Targeting expression of diagnostic reporter genes and cytotoxic/therapeutic genes specifically to cancerous cells would provide a powerful tool for the diagnosis and treatment of cancer. For this approach to be safe and effective, the expression must be driven by cancer-specific promoter/regulatory elements.42,43 This approach has been slow to transition into the clinic, mainly due to safety and efficacy issues. These promoter constructs must be highly specific to cancer cells as well as strong enough to express the transgene in high levels. Furthermore, safe and efficient gene delivery tools are needed. We previously validated Rad51 promoter constructs as potent drivers of cancer-specific gene expression.10,24 However, the Rad51 promoter region used in our earlier studies was too long for efficient viral-mediated delivery. The results of the present study will aid in developing next generation Rad51 promoter-based constructs by deleting segments that repress the promoter in cancer cells without increasing the activity in normal cells. Maintaining the regions that stimulate expression and removing those that represses the Rad51 promoter in normal cells could improve both the safety and efficacy of Rad51 promoter driven anti-cancer diagnostics and therapies and thus foster their translation into the clinic.
Materials and Methods
Cell culture
All cell lines were grown in monolayer on treated polystyrene cell culture dishes (Corning) at 37 °C in 3% O2, 5% CO2, and 97% relative humidity in HERA Cell 240 incubators. The human normal fibroblast line HCA2-T was immortalized by expression of hTERT from integrated pBABE-Puro retrovirus. HCA2-T cells were maintained in MEM (ATCC) supplemented with 15% FBS; FBS, (Gibco), and 1x Pen/Strep (Gibco). Human fibrosarcoma cell line HT1080 (ATCC) and human cervical carcinoma line HeLa (ATCC) were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco), 1× Pen/Strep (Gibco), and 1× nonessential amino acids (Gibco).
Cloning of the human Rad51 promoter region and construction of the wild-type pRad51-Luc plasmid
The 6532-bp wild-type Rad51 promoter, spanning nucleotides −2931 upstream to +3601 downstream relative to the start of transcription was cloned from human dermal fibroblast cell line HCA2 as previously described.10 This promoter region was engineered to control the expression of the reporter gene firefly luciferase by its incorporation into the pGL3 basic vector (Promega) by first introducting the restriction enzyme sites AgeI and AseI into the pGL3-Basic polylinker by site-directed mutagenesis (Stratagene) with the following primers: 5′-CCGGAAGCTT ACCGGTCGCC ACCATGGAAG ACGCC-3′ and 5′-GCCAAGCTTA ATTAATTCGC AGATCTCGAG CC-3′. This results in the pGL3-Basic(Age/Ase) vector. The full-length Rad51 promoter region was then subcloned by cutting its terminal 5′ and 3′ ends with the restriction enzymes AseI and AgeI and ligated into the same sites in pGL3-Basic to create pRad51-Luc, with the translational start of the firefly luciferase gene in frame with the first 12 amino acids of the Rad51 coding region and under the Rad51 promoter. Plasmid production of pRad51-Luc and subsequent mutant promoters was performed by transforming the plasmids into MAX Efficiency Stbl2 Chemically Competent Cells (Invitrogen) followed by isolation and purification with the EndoFree Plasmid Maxi Kit (Qiagen).
Construction of mutant Rad51 promoters
pRad51-Luc plasmids (in pGL3 Basic) were subjected to restriction enzyme digestion (New England Biolabs), gel purification using the QIAquick Gel Extration Kit (Qiagen) and subsequent creation of blunted ends with Klenow Enzyme (Roche) and rapid ligation using the Rapid DNA Ligation Kit (Roche) and plasmids produced using the MAX Efficiency Stbl2 chemically competent cells (Invitrogen) followed by isolation and purification with the EndoFree Plasmid Maxi Kit (Qiagen). Sites deleted and the enzymes used are as follows: −2931 to −23 (EcoR1 and AseI), −2010 to −207 (BglII and MluI), −656 to −138 (XhoI), +173 to +265 (SacII), and +1675 to +3313 (NdeI). Site-specific deletions of proposed p53 (−160 to −115) and EGR1 (−217 to −208 and +170 to +117) were accomplished using the manufacturer’s recommendations for the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). Sequences to be deleted were determined from previously published works identifying the p53 site22 and the EGR1 sites.25
Transfections and luciferase analysis comparing wild-type and mutant pRad51-Luc constructs
The pRad51-Luc and mutant plasmids were transfected into HCA2, HT1080, or HeLa cells by electroporation. Briefly, the equivalent of 2 µg of either wt or the mutant forms of pRad51-Luc (so all transfections contained the same number of plasmids) were transfected into 1 × 106 growing cells of each of the 3 cell lines by Amaxa Nucleofector II electroporation. The following nucleofector programs and transfection (Lonza/Amaxa) solutions were used for each cell line: HCA2, program U-20 and solution NHDF; HT1080, program L-005 and solution V; HeLa, program I-013 and solution V. After transfection, cells were incubated for 72 h as described above, then harvested, counted using a Z2 Coulter Counter (Beckman Coulter), and lysed using equal passive lysis buffer (Promega) at a ratio of 200 μL/1 × 106 cells. Twenty microliters (20 μL) of this extract was used in each individual luciferase activity assay (Promega) using a GloMax20/20 Luminometer (Promega). Each transfection was performed 2–3 times, and each individual sample was assayed in duplicate.
EGR1 stimulation of Rad51 promoter activity
EGR1 transcriptional regulation of Rad51 promoter activity was tested by transfecting 2 µg pRad51-Luc or EGR1 mutant pRad51-Luc plasmids into the 3 cell lines via electroporation and then assaying for luciferase activity. Additionally, EGR1 was overexpressed in HCA2-T and analyzed for its stimulation of Rad51 promoter activity by co-transfecting 2 µg of wild-type or mutant pRad51-Luc along with 2 µg of the EGR1 overexpression plasmid CMV-EGR1 (Origene SC128132) and analyzed for luciferase activity.
Tumorigenic transformation of HCA2-T cells and its effect on Rad51 promoter activity
Step-wise tumorigenic transformation and its effect on Rad51 promoter activity in HCA2-T cells was accomplished by co-transfection of pRad51-Luc with combinations of plasmids encoding oncogenes, incubating the cells for 72 h, and then assaying for luciferase activity as described above. Each co-transfection by Amaxa Electroporation maintained a total of 4 µg of plasmid DNA, so as to not alter the efficiency of transfection. The co-transfection regimens were: (1) (Control); 3 µg pEGPF-N1 (Clontech), and 1 µg pRad51-Luc; (2) 1 µg pEGPF-N1, 1 µg pRad51-Luc, and pSG5 Large T (Addgene 9053); (3) 2 µg pEGPF-N1, 1 µg pRad51-Luc, and 1 µg pRas-V12 (Addgene 1786); (4) 1 µg pEGPF-N1, 1 µg pRad51-Luc, 1 µg pRas-V12, and 1 µg pSG5 Large T; (5) 1 µg pRad51-Luc, 1 µg pRas-V12, 1 µg pSG5 Large T, and 1 µg pBabe Small T (Addgene 8583).
Statistical analysis
All transfections were repeated at least 3 times. Mean and standard deviations were calculated using Microsoft Excel and GraphPad. Statistical significance (P value) was determined using GraphPad t test calculator.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of the Gorbunova Lab for their help and support in completing this project by their technical assistance and stimulating conversations. This work was supported by the Messersmith Fellowship to C.H. and National Institutes of Health grants to V.G.
References
- 1.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–4. [PubMed] [Google Scholar]
- 2.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 3.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T, et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia. 2011;13:864–73. doi: 10.1593/neo.11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955–60. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 5.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 6.Treré D, Ceccarelli C, Migaldi M, Santini D, Taffurelli M, Tosti E, Chieco P, Derenzini M. Cell proliferation in breast cancer is a major determinant of clinical outcome in node-positive but not in node-negative patients. Appl Immunohistochem Mol Morphol. 2006;14:314–23. doi: 10.1097/00129039-200609000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Wu X, Roth JA, Zhao H, Luo S, Zheng YL, Chiang S, Spitz MR. Cell cycle checkpoints, DNA damage/repair, and lung cancer risk. Cancer Res. 2005;65:349–57. [PubMed] [Google Scholar]
- 8.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schild D, Wiese C. Overexpression of RAD51 suppresses recombination defects: a possible mechanism to reverse genomic instability. Nucleic Acids Res. 2010;38:1061–70. doi: 10.1093/nar/gkp1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hine CM, Seluanov A, Gorbunova V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc Natl Acad Sci U S A. 2008;105:20810–5. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–53. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- 12.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–25. [PubMed] [Google Scholar]
- 13.Hannay JA, Liu J, Zhu QS, Bolshakov SV, Li L, Pisters PW, Lazar AJ, Yu D, Pollock RE, Lev D. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007;6:1650–60. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 14.Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, Lüttges J, Kalthoff H, Stürzbecher HW. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–5. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 15.Maacke H, Opitz S, Jost K, Hamdorf W, Henning W, Krüger S, Feller AC, Lopens A, Diedrich K, Schwinger E, et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;88:907–13. doi: 10.1002/1097-0215(20001215)88:6<907::AID-IJC11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Collis SJ, Tighe A, Scott SD, Roberts SA, Hendry JH, Margison GP. Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res. 2001;29:1534–8. doi: 10.1093/nar/29.7.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kauffmann A, Rosselli F, Lazar V, Winnepenninckx V, Mansuet-Lupo A, Dessen P, van den Oord JJ, Spatz A, Sarasin A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27:565–73. doi: 10.1038/sj.onc.1210700. [DOI] [PubMed] [Google Scholar]
- 18.Mitra A, Jameson C, Barbachano Y, Sanchez L, Kote-Jarai Z, Peock S, Sodha N, Bancroft E, Fletcher A, Cooper C, et al. IMPACT Steering Committee and IMPACT and EMBRACE Collaborators Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology. 2009;55:696–704. doi: 10.1111/j.1365-2559.2009.03448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbano R, Copetti M, Perrone G, Pazienza V, Muscarella LA, Balsamo T, Storlazzi CT, Ripoli M, Rinaldi M, Valori VM, et al. High RAD51 mRNA expression characterize estrogen receptor-positive/progesteron receptor-negative breast cancer and is associated with patient’s outcome. Int J Cancer. 2011;129:536–45. doi: 10.1002/ijc.25736. [DOI] [PubMed] [Google Scholar]
- 20.Qiao GB, Wu YL, Yang XN, Zhong WZ, Xie D, Guan XY, Fischer D, Kolberg HC, Kruger S, Stuerzbecher HW. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br J Cancer. 2005;93:137–43. doi: 10.1038/sj.bjc.6602665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nogueira A, Catarino R, Coelho A, Araújo A, Gomes M, Medeiros R. Influence of DNA repair RAD51 gene variants in overall survival of non-small cell lung cancer patients treated with first line chemotherapy. Cancer Chemother Pharmacol. 2010;66:501–6. doi: 10.1007/s00280-009-1187-2. [DOI] [PubMed] [Google Scholar]
- 22.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, Ashworth A, Silva A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–24. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong V, Osterbur M, Capella C, Kim YE, Hine C, Gorbunova V, Seluanov A, Dewhurst S. Adenoviral vector driven by a minimal Rad51 promoter is selective for p53-deficient tumor cells. PLoS One. 2011;6:e28714. doi: 10.1371/journal.pone.0028714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hine CM, Seluanov A, Gorbunova V. Rad51 promoter-targeted gene therapy is effective for in vivo visualization and treatment of cancer. Mol Ther. 2012;20:347–55. doi: 10.1038/mt.2011.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasselbach L, Haase S, Fischer D, Kolberg HC, Stürzbecher HW. Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur J Gynaecol Oncol. 2005;26:589–98. [PubMed] [Google Scholar]
- 26.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 27.Yang SZ, Eltoum IA, Abdulkadir SA. Enhanced EGR1 activity promotes the growth of prostate cancer cells in an androgen-depleted environment. J Cell Biochem. 2006;97:1292–9. doi: 10.1002/jcb.20736. [DOI] [PubMed] [Google Scholar]
- 28.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 29.Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- 30.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–83. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Virolle T, Krones-Herzig A, Baron V, De Gregorio G, Adamson ED, Mercola D. Egr1 promotes growth and survival of prostate cancer cells. Identification of novel Egr1 target genes. J Biol Chem. 2003;278:11802–10. doi: 10.1074/jbc.M210279200. [DOI] [PubMed] [Google Scholar]
- 32.Ahuja D, Sáenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–45. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 33.Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. 2011;121:3623–34. doi: 10.1172/JCI46323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–41. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 35.Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. 2007;26:2048–57. doi: 10.1038/sj.onc.1210001. [DOI] [PubMed] [Google Scholar]
- 36.Popov B, Chang LS, Serikov V. Cell cycle-related transformation of the E2F4-p130 repressor complex. Biochem Biophys Res Commun. 2005;336:762–9. doi: 10.1016/j.bbrc.2005.08.163. [DOI] [PubMed] [Google Scholar]
- 37.Slupianek A, Hoser G, Majsterek I, Bronisz A, Malecki M, Blasiak J, Fishel R, Skorski T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell. 2001;8:795–806. doi: 10.1016/S1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 39.Schmutte C, Tombline G, Rhiem K, Sadoff MM, Schmutzler R, von Deimling A, Fishel R. Characterization of the human Rad51 genomic locus and examination of tumors with 15q14-15 loss of heterozygosity (LOH) Cancer Res. 1999;59:4564–9. [PubMed] [Google Scholar]
- 40.Yu Y, Kudchodkar SB, Alwine JC. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J Virol. 2005;79:6882–9. doi: 10.1128/JVI.79.11.6882-6889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayman TJ, Williams ES, Jamal M, Shankavaram UT, Camphausen K, Tofilon PJ. Translation initiation factor eIF4E is a target for tumor cell radiosensitization. Cancer Res. 2012;72:2362–72. doi: 10.1158/0008-5472.CAN-12-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu L, Johnson M, Sato M. Transcriptionally targeted gene therapy to detect and treat cancer. Trends Mol Med. 2003;9:421–9. doi: 10.1016/j.molmed.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lo HW, Day CP, Hung MC. Cancer-specific gene therapy. Adv Genet. 2005;54:235–55. doi: 10.1016/S0065-2660(05)54010-0. [DOI] [PubMed] [Google Scholar]