

Abstract
Small-molecule inhibitors of poly (ADP-ribose) polymerase (PARP) have shown considerable promise in the treatment of homologous recombination (HR)-defective tumors, such as BRCA1- and BRCA2-deficient breast and ovarian cancers. We previously reported that mantle cell lymphoma cells with deficiency in ataxia telangiectasia mutated (ATM) are sensitive to PARP-1 inhibitors in vitro and in vivo. Here, we report that PARP inhibitors can potentially target ATM deficiency arising in a solid malignancy. We show that ATM protein expression varies between gastric cancer cell lines, with NUGC4 having significantly reduced protein levels. Significant correlation was found between ATM protein expression and sensitivity to the PARP inhibitor olaparib, with NUGC4 being the most sensitive. Moreover, reducing ATM kinase activity using a small-molecule inhibitor (KU55933) or shRNA-mediated depletion of ATM protein enhanced olaparib sensitivity in gastric cancer cell lines with depletion or inactivation of p53. Our results demonstrate that ATM is a potential predictive biomarker for PARP-1 inhibitor activity in gastric cancer harboring disruption of p53, and that combined inhibition of ATM and PARP-1 is a rational strategy for expanding the utility of PARP-1 inhibitors to gastric cancer with p53 disruption.
Keywords: PARP-1 inhibitor, olaparib, ATM, p53, gastric cancer
Introduction
Gastric cancer is the second highest cause of cancer death worldwide, with almost 1 million new cases and 738 000 deaths per year.1 Gastric cancer is most common in East Asia, including Japan, Korea, and China, and is a significant health concern in North America and the European Union. Despite several therapeutic advances, including modified combination chemotherapy and chemo-radiation regimens,2,3 and, more recently, biological therapy targeting human epidermal growth factor 2 (HER2),4 outcomes in advanced gastric cancer remain poor, and the need to develop new treatment approaches is urgent.
Disruption of DNA repair genes by somatic mutation or epigenetic silencing has been documented in a variety of human cancers, and, consequently, DNA damage response pathways have emerged as potential therapeutic targets.5,6 One such DNA damage response gene, ataxia telangiectasia mutated (ATM), plays a critical role in cellular signaling in response to DNA double-strand breaks, and its alteration is associated with the development and progression of several types of human cancers.7-11 Moreover, expression of ATM and other DNA damage response genes correlates with clinic-pathological outcome in gastric cancer patients. Consequently, ATM has been proposed to be an independent prognostic12 and a predictive marker for individual therapy in advanced gastric cancer patients.13
The clinical significance of disrupted DNA repair pathways lies in the potential exploitation of synthetic lethality, best exemplified by the preferential toxicity of small-molecule PARP-1 inhibitors in BRCA-deficient tumors.14,15 Similar synthetic lethal interactions have been demonstrated between PARP inhibitors and a number of other genes related to DNA repair and the DNA damage response, including ATM.16-20 Moreover, inactivation of ATM together with PARP-1 in mice is embryonic lethal,21 and synthetic lethality between PARP and ATM has been reported in cells from patients with ataxia telangiectasia, a cancer predisposition syndrome associated with loss of both ATM alleles.22 Based on these observations, we postulated that PARP inhibitors might have therapeutic utility in ATM-deficient malignancies and, subsequently, demonstrated that the concept of PARP inhibitor synthetic lethality can be applied to mantle cell lymphoma (MCL) cell lines with loss of function of ATM.23 Moreover, MCL cell lines with disruption of p53 (mutant p53, deletion of p53, overexpression of dominant-negative p53, or inhibition by pifithrin) were more sensitive to the PARP-1 inhibitor olaparib than were MCL cells with wild-type p53.24 Furthermore, the ATM protein kinase inhibitor KU55933 sensitized MCL cells with disrupted p53 to olaparib, indicating that the combination of ATM inhibitors and PARP inhibitors has potential in the treatment of human malignancies with dysfunctional p53.24
Here, we extend these studies to ask whether olaparib might have utility in the treatment of gastric cancer harboring disruption of ATM. ATM deficiency has been reported in up to 60% of gastric cancer and is associated with poor prognosis.13 Like many other malignancies, gastric cancer also displays a high rate of p53 mutation.25 Therefore, we hypothesized that a significant fraction of gastric cancer cases might benefit from treatment with PARP-1 inhibitor, either with or without ATM inhibitor. We demonstrate that the concept of PARP inhibitor synthetic lethality is applicable to gastric cancer with low ATM protein expression and disruption or depletion of p53, and that the combination of PARP and ATM inhibitors has potential against ATM-proficient gastric cancer with p53 inactivation.
Results
ATM protein expression and DNA damage-induced signaling varies in gastric cancer cell lines
Whole-cell extracts were prepared from the gastric cancer cell lines STKM-2, KATO III, AGS, NUGC4, MKN1, MKN28, MKN45, and ISt-1 as well as control normal human lymphoblastoid cells (BT) and lymphoblastoid cells from an A-T patient (L3). Extracts were run on SDS PAGE and probed for ATM, DNA-PKcs, SMC-1, Ku80, and actin protein expression (Fig. 1A). Levels of ATM protein expression were normalized to SMC-1 and expressed relative to BT cells (Fig. 1B). ATM protein was expressed to varying degrees in all gastric cancer cell lines examined; however, ATM protein expression was significantly reduced in NUGC4, MKN45, and AGS compared with the other cell lines examined. Relative levels of ATM protein expression in the gastric cancer cell lines (compared with that in BT cells) were NUGC4 (13.8%), MKN45 (15.3%), AGS (42.8%), ISt-1 (67.9%), MKN28 (55.7%), STKM-2 (81.1%), KATOIII (127%), and MKN1 (67.8%), (Fig. 1B).
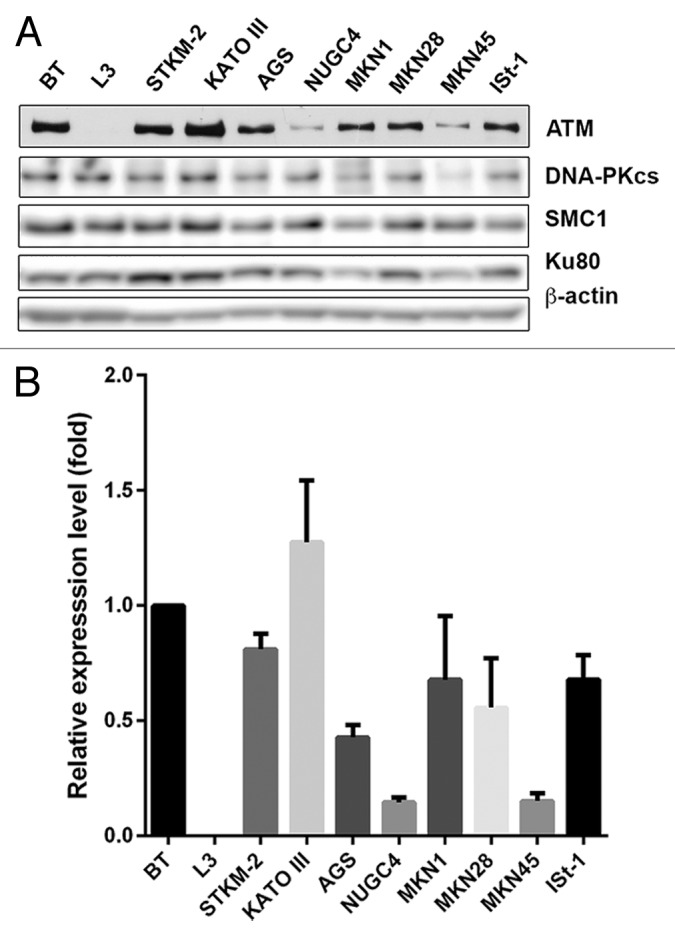
Figure 1. Relative levels of ATM in the gastric cancer cell lines STKM2, KATOIII, AGS, NUGC4, MKN1, MKN28, MKN45, and ISt-1 compared with a control lymphoblastoid cell line C35ABR (BT) and an A-T patient-derived lymphoblastoid cell line (L3). (A) Western blots were probed with antibodies to ATM, DNA-PKcs, SMC1, Ku80, and β−actin as indicated and visualized on a Fuji LAS 4000 imager. (B) ATM protein levels were quantitated, normalized to SMC1 expression and compared with expression in BT cells. n = 3. Error bars denote SEM.
To address ATM functionality, cell lines were exposed to 2 Gy ionizing radiation (IR) and harvested after 1 h. Extracts were prepared as described, and western blots were examined for ATM-dependent phosphorylation events. Consistent with the reduced protein expression levels, reduced ATM-1981 auto phosphorylation was observed in NUGC4 and MKN45 cell lines (Fig. 2), suggesting that these cell lines have reduced ATM function. DNA damage-induced p53 serine-15 phosphorylation was observed in STKM2, AGS, MKN1, MKN28, and ISt-1, but p53 was undetectable in KATOIII, consistent with the TP53-null phenotype in this cell line (Table S1). Both p53 phosphorylation and p53 protein levels were low in NUGC4 and MKN45 following IR treatment, consistent with defective or reduced ATM function in these cells (Fig. 2).
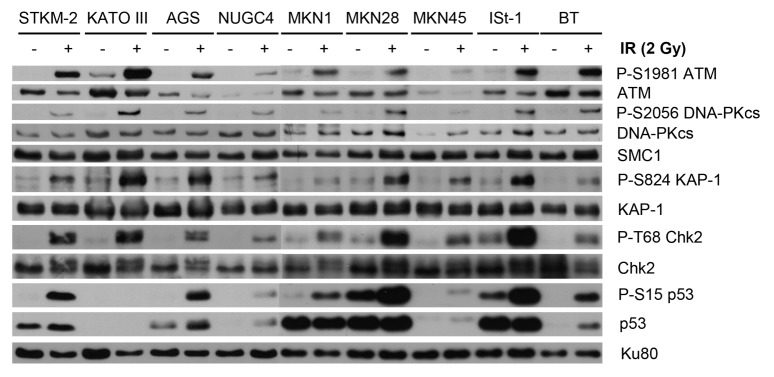
Figure 2. DNA damage induced signaling in the panel of gastric cancer cell lines. The panel of gastric cancer cell lines and BT control cells were exposed to 2 Gy IR and harvested following 1 h. Whole-cell extracts (25 µg total protein) were analyzed by SDS PAGE and immunoblotted for autophosphorylation of ATM on Ser1981 (P-S1981), phosphorylation of DNAPKcs on Ser2056 (P-S2056), phosphorylation of KAP-1 on Ser824 (P-S824), phosphorylation of Chk2 on Thr68 (P-T68), and phosphorylation of p53 on Ser15 (P-S15). Total ATM, KAP-1, Chk2, Ku80, and p53 protein levels are also shown. SMC1 is shown as a loading control.
It is well established that cells lacking ATM are highly radiation sensitive.26 As a further test of ATM functionality, the panel of gastric cancer cell lines was exposed to IR and survival was determined using the clonogenic survival assay. Consistent with a functional defect in ATM, the NUGC4 cell line was highly radiosensitive (less than 10% survival after 2 Gy IR; Fig. 3A). The AGS cell line was also highly radiosensitive (Fig. 3A), while MKN1, MKN45, and STKM2 cell lines had intermediate radiation sensitivity, and ISt-1, MKN28, and KATOIII were the most resistant (Fig. 3A). However, relative ATM protein expression levels only modestly correlated with sensitivity to IR, with a Spearman correlation coefficient of less than 0.55 at a variety of IR doses (from 1 to 6 Gy) (Fig. 3B; Fig. S1), suggesting that ATM status is not the only determinant of radiosensitivity in these cell lines.
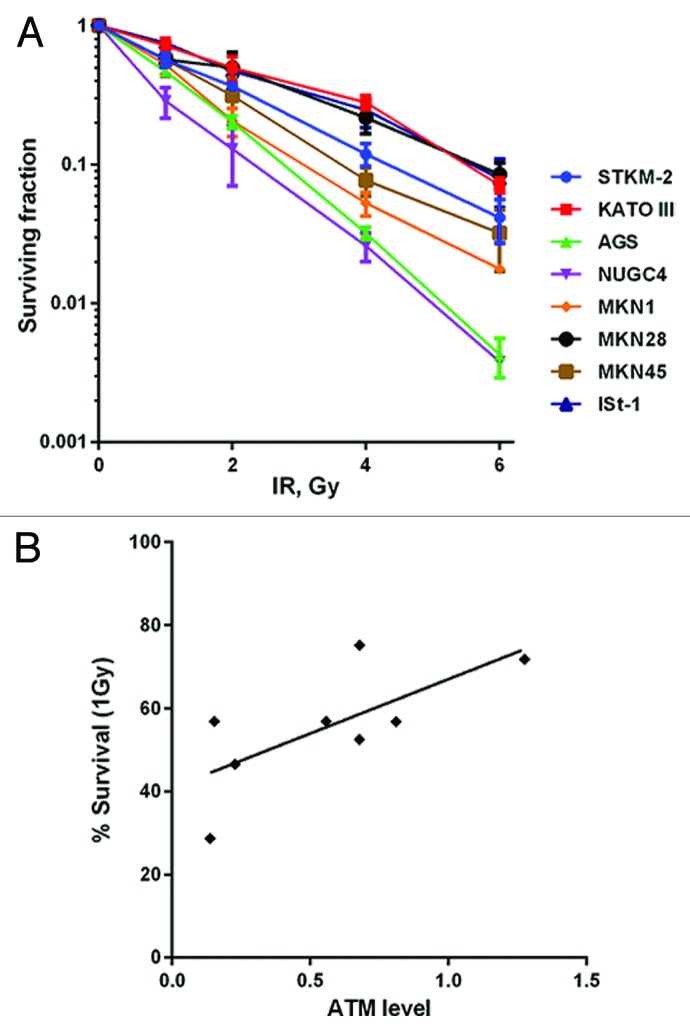
Figure 3. Clonogenic survival of gastric cancer cells after treatment with IR. (A) Cells were seeded then irradiated on plates 4 h later. Plates were incubated 10 d prior to staining. (B) The Spearman’s correlation coefficient between relative levels of ATM protein and cellular survival by IR was calculated to be 0.4935. Each point is in triplicate in 3 independent experiments. Error bars represent SEM.
Low ATM protein expression correlates with olaparib sensitivity
Next we determined the sensitivity of the cell lines to the PARP inhibitor olaparib (previously called AZD288127). Again, NUGC4 and AGS were the most sensitive of the cell lines examined (approximately 20% survival after 1 µM olaparib; Fig. 4A). ISt-1 and MKN45 had intermediate olaparib sensitivity, while MKN28, MKN1, STKM2, and KATO III were more resistant (Fig. 4A). The Spearman correlation at 1 µM olaparib was 0.8143 (P = 0.0022), indicating good correlation between ATM protein expression and olaparib sensitivity (Fig. 4B). Indeed, the correlation constant was at least 0.81 at all concentrations of olaparib tested (0.3–3 µM, data not shown). In contrast, protein levels of Mre11, Nbs1, Chk2, Rad51, XRCC1, 53BP1, and PNKP did not correlate with olaparib sensitivity (Figs. S2 and 3; Table S4).
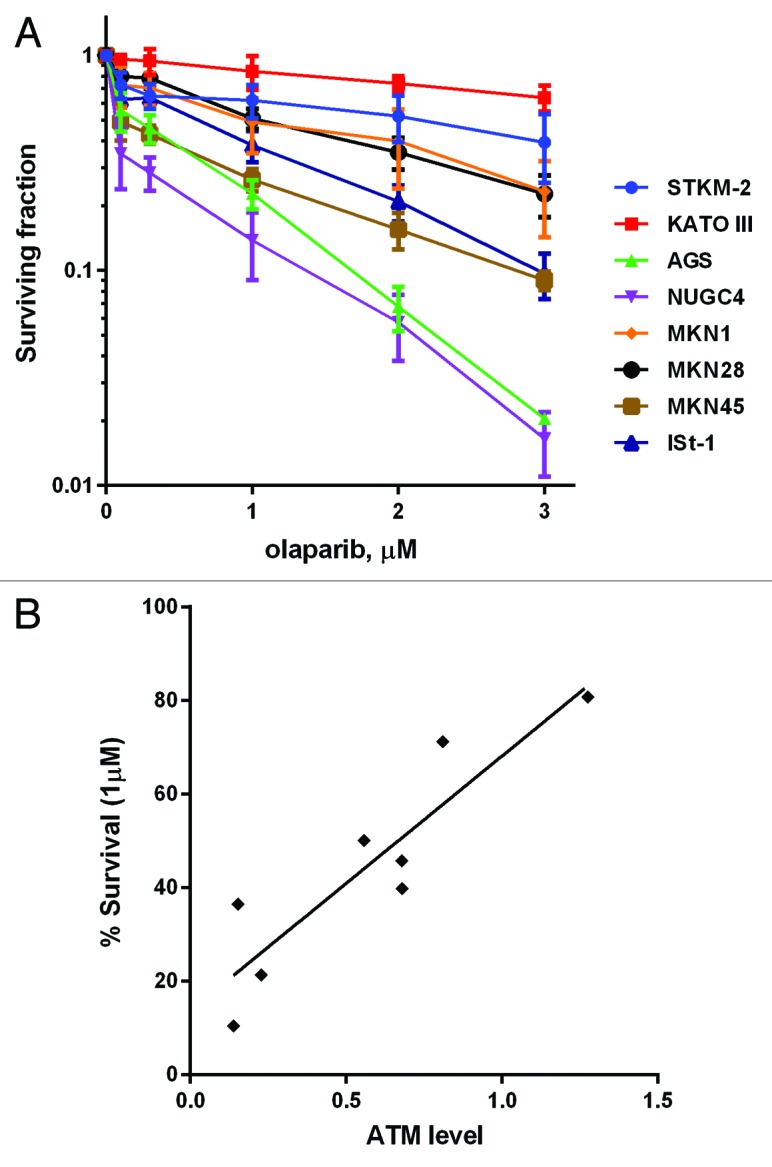
Figure 4. Clonogenic survival of gastric cancer cells after treatment with olaparib. (A) Cells were seeded and 4 h later olaparib as added to 0.1–3.0 µM as indicated. Plates were incubated 10 d prior to staining. (B) The Spearman correlation coefficient between relative levels of ATM protein and cellular survival by olaparib was calculated to be 0.8143 (P = 0.0022). Each point is in triplicate in 3 independent experiments. Error bars represent SEM.
Disruption of p53 enhances olaparib sensitivity in gastric cancer cell lines with low ATM protein expression
We have previously shown that ATM-deficient MCL cell lines with inactivation or loss of p53 are more sensitive to olaparib than their p53-proficient counterparts, and that inhibition of ATM in a mutant p53 background also enhances olaparib sensitivity.24 To determine whether p53 deficiency also enhanced olaparib sensitivity in gastric cancer, the ATM-proficient/p53-proficient (ATM+/p53+) gastric cancer cell line STKM2, and the ATM-proficient/p53-deficient (ATM+/p53del) gastric cancer cell line KATOIII (Figs. 1 and 2; Table S1) were incubated with increasing concentrations of the ATM inhibitor KU55933 or DMSO (control) in either the absence or presence of increasing concentrations of olaparib. Consistent with previous results, inhibition of both ATM (KU55933) and PARP (olaparib) in STKM2 (ATM+/p53+) had little effect on cellular viability, whereas inhibition of ATM significantly enhanced the toxicity of olaparib in the KATOIII (ATM+/p53 del) (Fig. 5A and B), supporting our previous findings that inhibition of ATM sensitizes cells with disruption of p53 to PARP inhibition.24
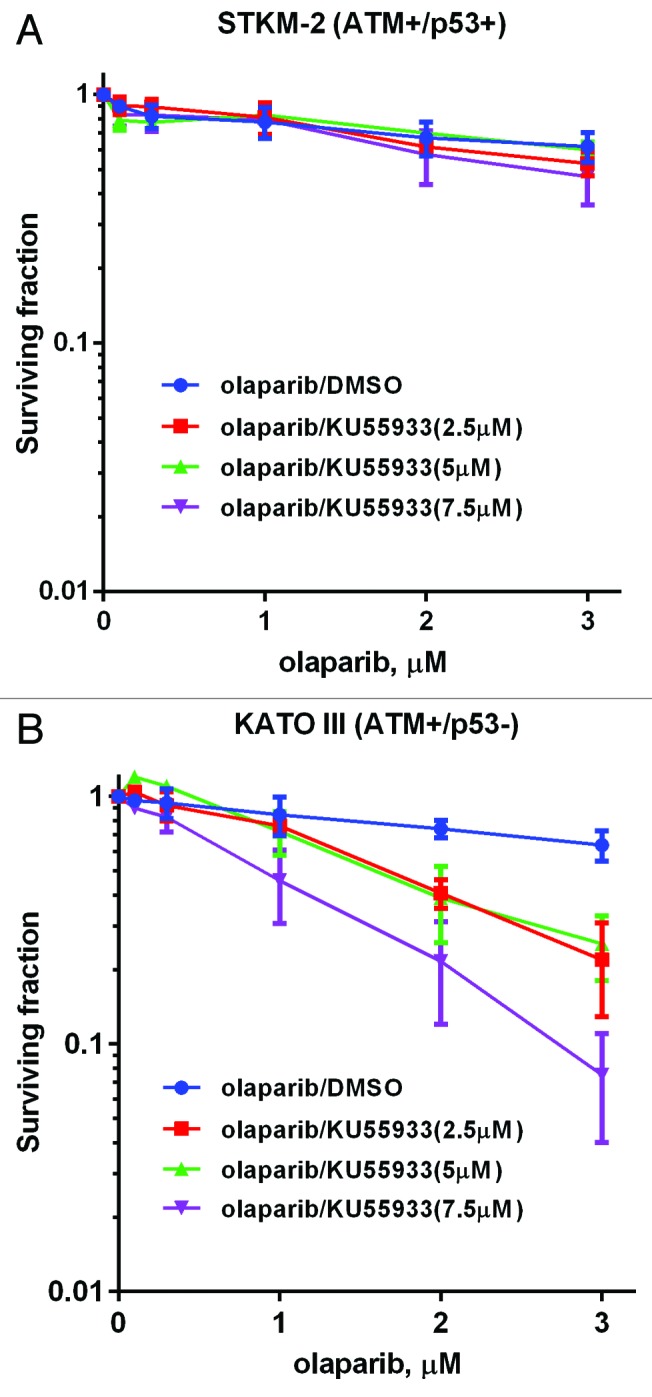
Figure 5. Sensitivity of STKM-2 (ATM+/p53+) and STKM-2 (ATM+/p53−) to olaparib in the absence and presence of ATM inhibitor KU55933. ATM-proficient gastric cancer cell lines (A) STKM2 (p53 wt) and (B) KATO III (p53 del) were incubated with 0.1–3 µM olaparib or vehicle control, in the presence of DMSO vehicle (blue) or increasing concentrations of the ATM inhibitor KU55933 (brown, 2.5; green, 5, purple, 7.5 µM). Survival was determined using clonogenic survival assays as described in Figure 3. Each point is in triplicate in 3 independent experiments. Error bars represent SEM.
Since STMK-2 and KATO III are non-isogenic tumor cell lines, we next sought to confirm these results by stably depleting ATM using shRNA. STMK-2 and KATO III cell lines with stable knockdown of ATM were first characterized for ATM protein levels and substrate phosphorylation. Both transfected cell lines lacked detectable ATM expression and ATM-P-S1981 phosphorylation, indicating substantial loss of ATM function (Fig. S4). Depletion of ATM induced radiosensitivity in both STKM2 and KATOIII cell lines, i.e., regardless of p53 status (Fig. 6A and B). In contrast, depletion of ATM enhanced olaparib sensitivity in KATOIII (p53 mutant) but not STKM-2 (wt-p53) (Fig. 6C and D), consistent with our previous findings that mutant p53 enhances sensitivity to olaparib in ATM-defective cells.24

Figure 6. Sensitivity of gastric cancer cell lines with stable knockdown of ATM to IR and olaparib. ATM-proficient gastric cancer cell lines STKM2 (p53 wt) and KATOIII (p53 mutant) were transfected with shATM-expressing vector, and were exposed to the indicated concentrations of IR (A and B) or olaparib (C and D). Survival was determined using clonogenic survival assay as described in Figure 3. Each point is in triplicate in 3 independent experiments. Error bars represent SEM.
To further confirm these findings, we depleted p53 in the parental STMK-2 cells using shRNA (STKM-2-shp53) and in STKM-2 cells that had previously been depleted for ATM (STKM-2-shATM/shp53). For these experiments, STKM-2 and STKM-2-shATM cells were infected with lentivirus expressing shp53 or control shRNA and stable cell lines isolated. As expected, STMK-2 cells expressing shATM were negative for ATM protein expression and IR-induced ATM-1981 phosphorylation but showed some IR-induced p53 serine 15 phosphorylation, possibly due to the activity of related kinases such as DNA-PKcs (Fig. S5, lane 4). Furthermore, STMK-2 cells expressing shp53 or STMK-2 cells expressing shATM/shp53 had considerably lower p53 expression and reduced serine 15 phosphorylation (Fig. S5, lanes 6 and 8). As predicted by our earlier results, STKM-2 cells with p53 depletion (STMK-shp53) were more sensitive to olaparib than STKM-2 control cells when combined with the ATM inhibitor KU55933 (Fig. 7A and B). Moreover, STKM-2 with shRNA depletion of both p53 and ATM (STKM-2-shATM/shp53) were more sensitive than STKM-2 cells with depletion of ATM alone (STKM-2 shATM) (Fig. 7C).

Figure 7. Olaparib sensitivity in gastric cancer cell lines with shRNA knockdown of ATM and p53. The ATM-proficient gastric cancer cell line STKM-2 (p53 wt) was infected with lentivirus vector encoding control shRNA (A) or shp53 (B) and incubated with 0.1–3 µM olaparib in the presence of DMSO vehicle or 7.5 µM of the ATM inhibitor KU55933. (C) ATM-knockdown STKM-2 cells were infected with lentivirus vector encoding control shRNA or shp53 and incubated with 0.1–3 µM olaparib. Survival was determined using clonogenic survival assay as described in Figure 3. Each point is in triplicate in 3 independent experiments. Error bars represent SEM.
Mechanism of olaparib induced cell death
We next examined the mechanism of olaparib-induced cell death in the gastric cancer cell lines. Olaparib induced auto phosphorylation of ATM (S1981) and DNA-PKcs (S2056) in STKM-2 and KATOIII cells, consistent with it inducing DNA damage (Fig. S5) as we reported previously in MCL.24 We next assayed for apoptosis using Annexin V/propidium iodide (PI) staining or Annexin V/SYTOX blue staining. Olaparib alone did not induce apoptosis in either STKM-2 or KATO III (Fig. S7). Although inhibition of both PARP-1 and ATM had little effect on STKM-2, apoptosis was induced by combination of PARP and ATM inhibitors on KATO III, which have deletion of both p53 alleles (Fig. S7D). Apoptosis was also induced by ATM knockdown in KATO III when treated with olaparib (Fig. S8). Furthermore, olaparib induced apoptosis in STKM-2 with depletion of p53 (STMK-2-shp53) when combined with the ATM inhibitor KU55933 (Fig. S9). Thus, olaparib induces apoptosis in gastric cancer cells when both ATM and p53 have been inactivated or inhibited.
To investigate the ability of olaparib to induce other forms of cell death in gastric cancer cells, KATOIII cells with shRNA-depleted ATM were treated with olaparib (5 µM, 72 h) then stained for the presence of large, multinuclear cells, indicative of aberrant mitoses and mitotic catastrophe.28 As shown in Figure S10, olaparib induced an increase in multinucleated cells in gastric cancer cells either depleted of ATM (Fig. S10A) or in cells in which ATM protein kinase activity had been inhibited by KU55933 (Fig. S10B), indicating that inhibition of PARP by olaparib induces genomic instability in ATM-deficient gastric cancer cells.
To further clarify the role of p53 in sensitivity to olaparib, we investigated the expression of p53-target genes induced by olaparib in STKM2 cells with stable knockdown of ATM, p53, or both ATM and p53, using quantitative RT-PCR (Fig. S11). Olaparib treatment of STKM2 control cells induced expression of p21 and PUMA, whereas no significant increases were observed in either gene in STKM-2 with knockdown of ATM and/or p53, indicating that p21 and PUMA are ATM/p53 target genes in these cells. Together, these results suggest that olaparib induces upregulation of ATM and p53-dependent cell cycle regulation and apoptotic pathways, and that in the absence of either ATM protein, or ATM activity, olaparib triggers multiple cell death pathways, including apoptosis and mitotic catastrophe.
Discussion
Here we show that ATM protein expression varies in gastric cancer cell lines, and that those with reduced expression of ATM protein expression are highly sensitive to the PARP inhibitor olaparib. In addition, we demonstrate that among the ATM-deficient gastric cancer cell lines tested, those with inactivation of p53 had heightened olaparib sensitivity. Furthermore, inactivation of ATM kinase activity with KU55933 enhanced sensitivity to olaparib in gastric cancer cells with deletion/depletion of p53.
PARP inhibitors are a promising next-generation class of anti-cancer drugs that target DNA damage response pathways.6,29 To ease their adoption into the clinical setting, predictive markers of their efficacy have been sought. Encouragingly, it has been reported that by exploiting synthetic lethality, PARP-1 inhibitors selectivity kill cancer cells with defects in the homologous recombination repair (HR) pathway while having little effect on normal cells. Specifically, breast cancer cell lines deficient in the HR and the DNA damage response factors BRCA1/BRCA2 are extremely sensitive to PARP-1 inhibitors. Early clinical trials demonstrating high response rates in BRCA1/2-deficient cancers27,30 suggest that this in vitro observation translates into the clinical setting. However, triple-negative (ER-, PR-, and HER2-negative) breast cancers, sometimes inferred as a surrogate for BRCA-deficient tumors, do not demonstrate such a response31 implying that well-delineated molecular markers of response to PARP inhibitors are critical for the pharmacological development of this class of drugs.
Several studies, including work from our laboratory, have shown that synthetic lethality with PARP-1 inhibitors can be extended to non-breast cancer cell lines with defects in other DNA repair and DNA damage response proteins, one of which is ATM.16-18 ATM is important for the maintenance of genome stability, likely through its roles in regulation of cell cycle checkpoints32 and repair of damage in heterochromatin.33 We have previously reported that ATM-deficient MCL cell lines have suppressed DNA damage signaling and are sensitive to PARP-1 inhibitors. Based on these results, we postulated that ATM is a potential predictive biomarker of PARP-1 inhibitor activity in malignancies other than MCL.23 Others have described a similar phenomenon in chronic lymphocytic leukemia (CLL).34 In addition to lymphoid malignancies, ATM alteration has been reported in many human carcinomas, including gastric cancer. Approximately, 15% of gastric cancers contain ATM gene alterations, and about 60% of gastric cancer showed decreased expression of ATM.13 Our results suggest that ATM may be an independent, predictive biomarker of PARP-1 inhibitor sensitivity in gastric cancer, and that targeting ATM-defective tumors by PARP-1 inhibition may be applicable in this disease. Indeed, a recent phase II trial has reported that olaparib plus paclitaxel led to significant improvement in overall survival in gastric cancer patients, with a larger benefit in those with low ATM levels.35
Our studies raise several questions regarding the mechanisms underlying ATM expression in gastric cancer cells. The most well-characterized phenotype associated with loss of ATM is ataxia-telangiectasia (A-T), an inherited disorder characterized by neurodegeneration, cancer predisposition, and radiation sensitivity.36 In A-T patients, nonsense, frame shift, and splicing mutations in ATM are found throughout the gene and often result in protein truncation.37,38 Mutation or deletion of ATM also occurs in a variety of lymphomas,39 in particular MCL;40 however, the cellular mechanisms that result in loss of ATM expression or function in these tumors remain unclear (see “Note Added in Proof”). ATM mutation has also been associated with increased risk of breast cancer.41 Multiple mechanisms for loss of ATM expression have been proposed, including epigenetic silencing and microRNA expression (ref. 42 and references therein). As a first step to understanding the basis of low ATM expression in the panel of gastric cancer cell lines used in this study, we interrogated the Novartis-Broad Institute Cancer Cell Line Encyclopedia (CCLE)43 and Catalogue of Somatic Mutations in Cancer (COSMIC)44 databases for reported mutations in ATM in gastric cancer cell lines. No ATM mutations were reported for AGS, MKN1, or NUGC4 cell lines, while a mis-sense mutation (S2581I) was reported in KATOIII; however, as shown here, this mutation does not affect ATM expression or function. No information was available for ISt-1, STKM-2, or MKN45 cell lines in either database; however, MKN45 has previously been reported to harbor an R2832H mutation in ATM (Table S1). Interestingly, a recent study suggests that mutations in ATM introns, targeted by microsatellite instability may account for low ATM expression in a subset of gastric cancer cell lines,45 but whether this is applicable to other gastric cancer cell lines or gastric cancer patients with low ATM protein expression46,47 remains to be determined.
Materials and Methods
Cell culture
ISt-1, KATOIII, NUGC-4, MKN-28, MKN-45, STKM-2, C35ABR (BT), and L3 cell lines were cultured in RPMI1640 (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Hyclone III, Invitrogen), while AGS was cultured in Ham's F-12K (Kaighn) medium (Invitrogen) supplemented with 10% FBS. BT and L3 are human lymphoblastoid cell lines from normal control and an A-T patient and have been described previously.24 All other cell lines used are gastric cancer cell lines (Table S1). All cell lines were cultured under an atmosphere of 5% CO2 at 37 °C.
Transfections
Stable transfection of KATOIII and STKM2 cells was performed with pSUPER.retro.puro vectors encoding short hairpin RNA (shRNA) to either green fluorescent protein (GFP, control) or ATM, kindly provided by Dr Y Shiloh, Tel Aviv University, Israel, as previously described.48 Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The transfected cells were selected by growth in medium containing 4 µg/ml puromycin (Sigma-Aldrich), and single-cell populations were isolated.
For knockdown p53 expression assay, STKM2 was infected with lentivirus vector encoding control or shp53 shRNA (Open Biosystems, Thermo Fisher Scientific, ca# V12070603 [control] or ca# V3LHS_404717, ca# V3LHS_333919, ca# V3LHS_333920 [p53]), under serum-free conditions at a multiplicity of infection (MOI) between 10 and 20. Cells were selected with 5μg/mL puromycin. ATM-knockdown STKM2 was also infected with lentivirus vectors expressing either control shRNA or shp53.
Immunoblots
Cells were lysed in an ice cold NET-N lysis buffer (150 mmol/L NaCl, 0.2 mmol/L EDTA, 50 mmol/L TRIS-HCl [pH 7.5], and 1% [v/v] NP-40) containing protein phosphatase and protease inhibitors (1 μmol/L microcystin-LR, 0.2 mmol/L phenylmethylsulfonyl fluoride, 0.1 μg/mL pepstatin, 0.1 μg/mL aprotinin, and 0.1 μg/mL leupeptin), and lysed on ice by sonication (2 × 5 s bursts). Total protein (50 μg; as determined by the Detergent-Compatible Protein Assay [Bio-Rad] using bovine serum albumin as standard) was resolved by SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked with 20% (w/v) skim milk powder in T-TBS buffer (20 mmol/L TRIS-HCl [pH 7.5], 500 mmol/L NaCl, and 0.1% [v/v] Tween 20) for 1 h and probed with antibodies to total proteins or phosphorylated proteins as indicated. Antibodies to DNA-PKcs and P-S824 KAP-149,50 were generated in house. Antibodies to ATM (Epitomics), p21 (Santa Cruz,), p53 (Santa Cruz), SMC-1 (Novus), KAP-1 (Abcam), βeta-actin (Abcam), Ku80 (Abcam), PARP (Cell Signaling), 53BP1 (Santa Cruz), MRE11 (Cell Signaling,), NBS1 (Cell Signaling), Rad51 (Santa Cruz), XRCC1 (Cell Signaling), p21 (Santa Cruz), GADD45αlpha (Abcam), PNKP (Bethyl), and phosphospecific antibodies to ATM (Epitomics), P-S15 p53 (Cell Signaling) and P-S2056 DNA-PKcs (Abcam) were purchased commercially.
Clonogenic survival assays
Cells were trypsinized, seeded in 4 mL medium and incubated overnight before treatment with increasing doses of olaparib (0.1, 0.3, 1, 2, 3 µM) or an equal volume of vehicle control (DMSO) to assay for their colony-forming ability. Colonies (greater than 50 cells) were fixed then stained with crystal violet as described previously51 and counted 10 d later. Survival curves were plotted using Prism 6.0 Software (GraphPad). Where indicated, cells were treated with 2, 4, or 6 Gy IR using a 137Cs source Gammacell 1000 tissue irradiator (MDS Nordion) at a dose rate of 3.7 Gy/min.
Annexin V Assays
Cells were exposed to olaparib (5 μmol/L) with or without KU55933 (10 μmol/L) and resuspended in Annexin V binding buffer (10 mmol/L HEPES [pH 7.5], 140 mmol/L NaCl, and 2.5 mmol/L CaCl2) before incubation with FITC Annexin V (GeneTex) and 5 μg/mL propidium iodide with RNase, or 1 μg/mL Sytox blue for 5 min and then analyzed by flow cytometry at the University of Calgary Flow Cytometry Facility.
WST-1 cell proliferation assay
Cells were seeded into 96-well plates at a density of 5 × 103 cells/well. Cell viability was measured after 24, 48, and 72 h after seeding using WST-1 Cell Proliferation Reagent (Roche) according to the manufacturer’s instructions.
Quantitative reverse transcription polymerase chain reaction
RNA was isolated and purified from using TRIzol (Invitrogen) and RNeasy RNA isolation kit (Qiagen), and 5 μg of RNA was reverse transcribed using the first-strand cDNA synthesis kit (BioChain). Real-time PCR using Sybr green (Invitrogen) was performed by 7500 Fast Real-Time PCR System (Applied Biosystems), and the data was analyzed using Applied Biosystems 7500 Software v2.0.3. Expression was normalized to GUSB and GAPDH. The sequences of primers used are provided in Table S2.
Statistical analysis
Statistical analysis for the correlation between the ATM levels and inhibition of cell growth by PARP-1 inhibitor/olaparib was performed using GraphPad Prism 6.0 Software using Spearman correlation. Comparison between each 2 groups was analyzed by the unpaired Student t test using Microsoft Office Excel software. P values of < 0.05 were considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note Added in Proof
Recently, Zhang et al. reported that mutation of genes, including ATM, in MCL, is related to epigenetics and open chromatin.52
Acknowledgments
We thank Dr Mark O’Connor (AstraZeneca) for helpful discussions and for providing olaparib for these studies, Drs Martin Lavin (Queensland Institute for Medical research, Australia) and Yossi Shiloh (Tel Aviv University, Israel) for cells lines (BT and L3) and shRNA reagents and Randal Johnston, University of Calgary for reagents. This work was funded by a grant from Alberta Innovates Health Solutions (#201000575) to D.G.B. and S.P.L.M., as well as funds from the National Cancer Institute of Canada (S.P.L.M.). S.P.L.M. acknowledges support from the Engineered Air Chair in Cancer Research, a Killam Annual Professorship, and Alberta Innovates-Health Solutions.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W, et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215–21. doi: 10.1016/S1470-2045(08)70035-4. [DOI] [PubMed] [Google Scholar]
- 3.Koizumi W, Tanabe S, Azuma M, Ishido K, Nishimura K, Sasaki T, Nakatani K, Higuchi K, Nakayama N, Katada C. Impacts of fluorouracil-metabolizing enzymes on the outcomes of patients treated with S-1 alone or S-1 plus cisplatin for first-line treatment of advanced gastric cancer. Int J Cancer. 2010;126:162–70. doi: 10.1002/ijc.24726. [DOI] [PubMed] [Google Scholar]
- 4.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. ToGA Trial Investigators Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 5.Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31:372–80. doi: 10.1016/j.tips.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 7.Teraoka SN, Malone KE, Doody DR, Suter NM, Ostrander EA, Daling JR, Concannon P. Increased frequency of ATM mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001;92:479–87. doi: 10.1002/1097-0142(20010801)92:3<479::AID-CNCR1346>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 8.Ai L, Vo QN, Zuo C, Li L, Ling W, Suen JY, Hanna E, Brown KD, Fan CY. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol Biomarkers Prev. 2004;13:150–6. doi: 10.1158/1055-9965.EPI-082-3. [DOI] [PubMed] [Google Scholar]
- 9.Safar AM, Spencer H, 3rd, Su X, Coffey M, Cooney CA, Ratnasinghe LD, Hutchins LF, Fan CY. Methylation profiling of archived non-small cell lung cancer: a promising prognostic system. Clin Cancer Res. 2005;11:4400–5. doi: 10.1158/1078-0432.CCR-04-2378. [DOI] [PubMed] [Google Scholar]
- 10.Austen B, Powell JE, Alvi A, Edwards I, Hooper L, Starczynski J, Taylor AM, Fegan C, Moss P, Stankovic T. Mutations in the ATM gene lead to impaired overall and treatment-free survival that is independent of IGVH mutation status in patients with B-CLL. Blood. 2005;106:3175–82. doi: 10.1182/blood-2004-11-4516. [DOI] [PubMed] [Google Scholar]
- 11.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdel-Fatah T, Arora A, Gorguc I, Abbotts R, Beebeejaun S, Storr S, Mohan V, Hawkes C, Soomro I, Lobo DN, et al. Are DNA repair factors promising biomarkers for personalized therapy in gastric cancer? Antioxid Redox Signal. 2013;18:2392–8. doi: 10.1089/ars.2012.4873. [DOI] [PubMed] [Google Scholar]
- 13.Kang B, Guo RF, Tan XH, Zhao M, Tang ZB, Lu YY. Expression status of ataxia-telangiectasia-mutated gene correlated with prognosis in advanced gastric cancer. Mutat Res. 2008;638:17–25. doi: 10.1016/j.mrfmmm.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 14.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 15.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 16.Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–6. doi: 10.1016/j.gde.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 19.Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G, et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011;71:2632–42. doi: 10.1158/0008-5472.CAN-10-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ménisser-de Murcia J, Mark M, Wendling O, Wynshaw-Boris A, de Murcia G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol Cell Biol. 2001;21:1828–32. doi: 10.1128/MCB.21.5.1828-1832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem. 2007;282:16441–53. doi: 10.1074/jbc.M608406200. [DOI] [PubMed] [Google Scholar]
- 23.Williamson CT, Muzik H, Turhan AG, Zamò A, O’Connor MJ, Bebb DG, Lees-Miller SP. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010;9:347–57. doi: 10.1158/1535-7163.MCT-09-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, Dean M, Tu L, Gilley D, Magliocco AM, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med. 2012;4:515–27. doi: 10.1002/emmm.201200229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fenoglio-Preiser CM, Wang J, Stemmermann GN, Noffsinger A. TP53 and gastric carcinoma: a review. Hum Mutat. 2003;21:258–70. doi: 10.1002/humu.10180. [DOI] [PubMed] [Google Scholar]
- 26.Jeggo P, Lavin MF. Cellular radiosensitivity: how much better do we understand it? Int J Radiat Biol. 2009;85:1061–81. doi: 10.3109/09553000903261263. [DOI] [PubMed] [Google Scholar]
- 27.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 28.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 29.Sandhu SK, Yap TA, de Bono JS. The emerging role of poly(ADP-Ribose) polymerase inhibitors in cancer treatment. Curr Drug Targets. 2011;12:2034–44. doi: 10.2174/138945011798829438. [DOI] [PubMed] [Google Scholar]
- 30.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 31.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 32.Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 33.Goodarzi AA, Jeggo P, Lobrich M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair (Amst) 2010;9:1273–82. doi: 10.1016/j.dnarep.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 34.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 35.Bang YJ, Im SY. Lee KWea. Olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer:A randomised, double-blind phase II study. J Clin Oncol. 2013 doi: 10.1200/JCO.2014.60.0320. [DOI] [PubMed] [Google Scholar]
- 36.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 37.Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst) 2004;3:1187–96. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, Gatti RA. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab. 2003;80:437–43. doi: 10.1016/j.ymgme.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Stankovic T, Stewart GS, Byrd P, Fegan C, Moss PA, Taylor AM. ATM mutations in sporadic lymphoid tumours. Leuk Lymphoma. 2002;43:1563–71. doi: 10.1080/1042819021000002884. [DOI] [PubMed] [Google Scholar]
- 40.Beà S, Salaverria I, Armengol L, Pinyol M, Fernández V, Hartmann EM, Jares P, Amador V, Hernández L, Navarro A, et al. Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood. 2009;113:3059–69. doi: 10.1182/blood-2008-07-170183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–22. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 42.Bueno RC, Canevari RA, Villacis RA, Domingues MA, Caldeira JR, Rocha RM, Drigo SA, Rogatto SR. ATM down-regulation is associated with poor prognosis in sporadic breast carcinomas. Ann Oncol. 2014;25:69–75. doi: 10.1093/annonc/mdt421. [DOI] [PubMed] [Google Scholar]
- 43.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HS, Choi SI, Min HL, Kim MA, Kim WH. Mutation at intronic repeats of the ataxia-telangiectasia mutated (ATM) gene and ATM protein loss in primary gastric cancer with microsatellite instability. PLoS One. 2013;8:e82769. doi: 10.1371/journal.pone.0082769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim HS, Kim MA, Hodgson D, Harbron C, Wellings R, O’Connor MJ, Womack C, Yin X, Bang YJ, Im SA, et al. Concordance of ATM (ataxia telangiectasia mutated) immunohistochemistry between biopsy or metastatic tumor samples and primary tumors in gastric cancer patients. Pathobiology. 2013;80:127–37. doi: 10.1159/000346034. [DOI] [PubMed] [Google Scholar]
- 47.Lee HE, Han N, Kim MA, Lee HS, Yang HK, Lee BL, Kim WH. DNA damage response-related proteins in gastric cancer: ATM, Chk2 and p53 expression and their prognostic value. Pathobiology. 2014;81:25–35. doi: 10.1159/000351072. [DOI] [PubMed] [Google Scholar]
- 48.Elkon R, Rashi-Elkeles S, Lerenthal Y, Linhart C, Tenne T, Amariglio N, Rechavi G, Shamir R, Shiloh Y. Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biol. 2005;6:R43. doi: 10.1186/gb-2005-6-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J. 2002;368:243–51. doi: 10.1042/BJ20020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–77. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 51.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 2010;30:1368–81. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Jima D, Moffitt AB, Liu Q, Czader M, Hsi ED, Fedoriw Y, Dunphy CH, Richards KL, Gill JI, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123:2988–96. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.