

A large number of neurodegenerative disorders are triggered by the aggregation of misfolded proteins called amyloids. The main pathogenic event in these proteinopathies is the accumulation of oligomers, soluble assemblies that can disrupt synaptic activity, alter intracellular signaling, and spread to neighboring cells. Thus, preventing the formation of oligomers is a common therapeutic strategy for many neurodegenerative disorders. A number of laboratories are focusing on developing specific anti-amyloid agents, but this deliberate approach may benefit only the most prevalent conditions, such as Alzheimer and Parkinson disease. Alternatively, it may be possible to target the conserved structure and pathobiology of amyloids with potentially widespread benefits. Interestingly, cells produce protein machines that prevent protein misfolding and aggregation and promote protein refolding: the molecular chaperones. The mighty Heat shock protein 70 (Hsp70) is the most abundant and efficient chaperone, binds a variety of substrates, and exerts numerous essential functions, from routine protein synthesis to stress response. Hence, Hsp70 is an ideal therapeutic target against the deleterious effects of amyloids. Unfortunately, Hsp70 is a poor pharmacologic target because its constitutive isoforms are highly expressed, it has high affinity for misfolded substrates, and it's easily activated by abundant ATP. The key, though, is tapping into an abundant source of Hsp70 exclusively activated under cellular stress: the inducible Hsp70. Unfortunately, stress-inducing conditions are not appropriate therapeutic methods for obvious reasons. In a recent paper, we efficiently induced Hsp70 by combining 2 compounds with different targets and pharmacologic mechanisms.1
A fascinating mechanism regulating the transcription of inducible Hsp70 offers an opportunity to highjack Hsp70 expression. The main transcriptional activator of inducible Hsp70 is heat shock factor 1 (HSF1), which under physiological conditions binds Hsp90, inhibiting its transcriptional activity (Fig. 1). Under stress, Hsp90 binds misfolded substrates, releasing HSF1, which induces the expression of several Heat shock proteins (HSPs). Here, the neurodegeneration field got an assist from cancer, because Hsp90 is a relevant target in cancer biology. Hsp90 allows certain tumors to stabilize otherwise lethal mutations in protein kinases, steroid hormone receptors, and transcription factors, all with oncogenic activity. Hsp90 inhibitors inactivate, destabilize, and degrade these oncogenic clients,2 limiting the survival of tumorous cells. The antibiotic geldanamycin, the first identified Hsp90 inhibitor, has shown some promise in cellular and animal tumor models but exhibits poor solubility and high toxicity. Several geldanamycin derivatives with improved solubility are now available but are still toxic due to the essential function of Hsp90. Although decades of research have led to a rational pharmacologic strategy to block the toxicity of amyloids, we still face serious obstacles for launching the protective HSP response.
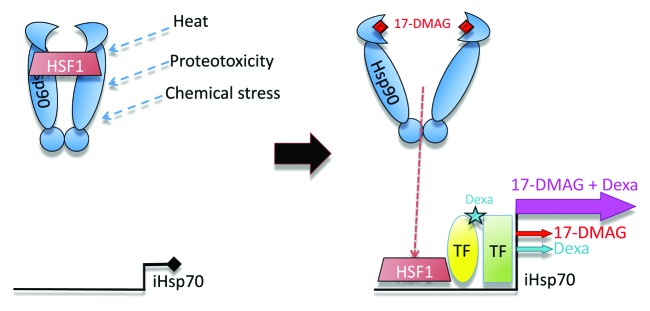
Figure 1. Synergistic activation of Hsp70. Left: inducible Hsp70 is not normally expressed, because HSF1 is bound to Hsp90. However, cellular stressors can release HSF1 from Hsp90. Right: 17-DMAG releases HSF1 to bind and activate the iHsp70 promoter, while dexamethasone increases the transcriptional activity of HSF1. Each drug has little effect alone, but their combination results in a potent, synergistic activation of iHsp70.
To optimize the pharmacological activation of Hsp70, we exploited a Drosophila model of prionopathies in which the hamster prion protein (PrP) progressively misfolds and induces neurotoxicity.3 We showed previously that co-expression of Hsp70 rescued both PrP misfolding and neurotoxicity, suggesting that a fraction of Hsp70 directly binds PrP at the membrane, prevents its misfolding, and promotes its degradation by unknown mechanisms.3 These results led us to study the potential protective activity of pharmacologic activators of Hsp70. We first fed flies a water-soluble geldanamycin derivative, 17-DMAG, but high doses of 17-DMAG had no effect on PrP accumulation. Instead of increasing the 17-DMAG dose, which could lead to toxicity, we searched for other compounds known to activate Hsp70. We found an FDA-approved glucocorticoid with anti-inflammatory activity, dexamethasone, which was described as an Hsp70 activator, among its many other activities. Dexamethasone is proposed to increase the affinity of HSF1 for the Hsp70 promoter, thus resulting in stronger transcriptional activity. However, dexamethasone alone had no effect on PrP levels. Remarkably, the combination of 17-DMAG and dexamethasone resulted in a significant reduction of the PrP steady-state levels that was accompanied by significant increases of inducible Hsp70 mRNA and protein (Fig. 1), but not constitutive Hsp70.1 This robust induction of Hsp70 reduced the levels of pathogenic PrP conformations and improved locomotor activity. Overall, we described a novel drug combination resulting in significant and synergistic induction of Hsp70 and reduction of PrP while avoiding the potential deleterious effects of higher doses of either drug.
Outlook
Quite remarkably, Hsp90 is a common therapeutic target in rather diverse conditions such as cancer and neurodegeneration. However, Hsp90 inhibitors seek different cellular effects in these almost opposite conditions: one of unregulated cell proliferation, the other of progressive neuronal loss. Inhibition of Hsp90 eliminates the protective activity of Hsp90 over oncogenic mutations, thus restricting the growth and viability of cancerous cells. In brain proteinopathies, inhibition of Hsp90 is intended to promote the expression of a protective downstream target: inducible Hsp70. Although neurodegenerative conditions can benefit from the development of safer and more effective Hsp90 inhibitors from the cancer field, these compounds alone may be of limited use due to their inherent cellular toxicity. Therefore, alternative therapeutic strategies may be based on the identification of additional drugs with small individual effects on Hsp70 induction. The combination of 2 or more such compounds could exhibit a synergistic Hsp70-inducing activity, leading to highly effective therapies with widespread clinical applications, including proteinopathies (Fig. 1).
Zhang Y, et al. PLoS One. 2014;9:e88522. doi: 10.1371/journal.pone.0088522.
References
- 1.Zhang Y, et al. PLoS One. 2014;9:e88522. doi: 10.1371/journal.pone.0088522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Carbonero R, et al. Lancet Oncol. 2013;14:e358–69. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Funez P, et al. PLoS Genet. 2009;5:e1000507. doi: 10.1371/journal.pgen.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]