

The tumor suppressor p53 is a well-characterized regulator of energy metabolism. p53 transcriptionally regulates a number of major metabolic regulators such as 5′ AMP-activated protein kinase (AMPK) and regulators of AMPK including the sestrins, mTOR through TSC1/TSC2, and glycolytic modulators such as TIGAR, hexokinase, and SCO2, which affect cellular oxidative stress.1 Moreover, p53 has 2 defined links to NAD+, a multifunctional cellular metabolite and coenzyme. First, the NAD+-dependent deacetylase sirtuin 1 (SIRT1) has been shown to modify the p53 response through deacetylation of the p53 C terminus.2 Second, p53 interacts with PARP1, an enzyme that uses NAD+ as a substrate to modify cellular proteins, including those involved in the DNA damage response.3
In this issue, Pan et al. have demonstrated that DNA damage-induced stabilization of p53 can transcriptionally activate the NAD+ synthetase NMNAT-2, which leads to increased NAD+ pools that sustain the DNA damage response.4 NMNAT-2 converts nicotinamide adenine mononucleotide (NMN) to nicotinamide adenine dinucleotide (NAD+) by the covalent addition of AMP in the energetic form of ATP. They also showed that p53 promotes NMNAT-2 expression, which is mediated by 2 p53 binding sites within the first intron of the NMNAT-2 gene and that could impact transcript variant selection. In the presence of sustained DNA damage, p53 depletion leads to significantly impaired NAD+ levels, most likely due to failed induction of NMNAT-2. Critically, depletion of NMNAT-2 impairs p53-mediated cell death, suggesting that NMNAT-2 is a regulator of p53 activity. So how could NAD+ affect p53 function?
SIRT1 utilizes NAD+ as a coenzyme for its deacetylase activity. SIRT1 is a class III histone deacetylase involved in stress responses, cellular metabolism, and aging.2 SIRT1 deacetylation of p53 lysine 382 (mouse K379) prevents p53 nuclear translocation, promoting transcription-independent apoptosis via BAX-mediated cytochrome c release.2 SIRT1 deacetylation of p53 also serves to suppress transcriptionally mediated cell cycle arrest and apoptosis as well as senescence.2 Because the availability of NAD+ affects SIRT1 activity, it is possible that a feedback loop exists in which p53, NMNAT-2, and SIRT1 respond to stressors and achieve co-regulation (see Fig. 1).
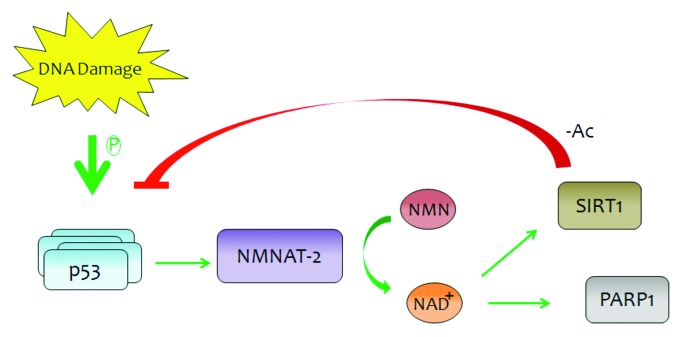
Figure 1. Damage-stabilized p53 transcriptionally activates NMNAT-2, leading to increased levels of NAD+. Increased availability of NAD+ may then increase SIRT1 and PARP1 activity, which can both directly and indirectly affect p53 response to cell stress. A putative negative feedback mechanism may exist whereby SIRT1 may downregulate p53 transcription of NMNAT-2 by limiting p53 nuclear translocation.2
p53 interacts with PARP1, an enzyme that uses NAD+ as a substrate to attach ADP-ribose polymers onto proteins, including p53, to promote the DNA damage response.3 In the presence of severe DNA damage, PARP1 can induce cell death via 2 defined mechanisms: (1) through parthanatos, a form of cell death that is mediated by accumulation of poly(ADP-ribose) polymers; and (2) through NAD+ utilization that depletes the cellular ATP level, resulting in AMPK-mediated necrotic cell death.3 However, during less severe DNA damage, PARP1 activity may be modulated so that p53 is the primary determinant of cellular fate. As p53 may modulate PARP1 activity by influencing cellular NAD+ levels through NMNAT-2, p53 could promote cell survival in the presence of recoverable DNA damage, trigger apoptosis in the presence of moderate DNA damage, or coordinate with PARP1 to mediate necrotic cell death in the presence of severe DNA damage.5 It is intriguing to consider that p53 may thus be a sensor for PARP1, another key regulator of cell fate.
The present study generates several questions and avenues for future research. First, it is interesting to consider a role for AMPK, as activated AMPK has been shown to increase SIRT1 activity by increasing the cellular levels of NAD+.6 Interestingly, AMPK has not only been shown to activate p53 by serine 15 phosphorylation, it also directly phosphorylatesand inactivates SIRT1, leading to inhibition of SIRT1-mediated p53 repression, so the role of AMPK in this pathway requires further investigation.7,8 Second, under which conditions does p53 regulate NAD+ to mediate cellular fate (by activating NMNAT enzymes), and under which conditions does NAD+ function to repress, bypass, promote, or alter p53-mediated cell fate decisions (by complex regulation of SIRT1, PARP1, AMPK, and other factors)? Third, does p53 regulate NAD+ in the absence of stress? Finally, it is important to consider how this relationship could be harnessed in p53-mediated diseases such as cancers, aging, and neurological disorders.
Pan LZ, et al. Cell Cycle. 2014;13:1041–8. doi: 10.4161/cc.28128.
References
- 1.Vousden KH, et al. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 2.Yi J, et al. Biochim Biophys Acta. 2010;1804:1684–9. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elkholi R, et al. Bioessays. 2014;36:46–51. doi: 10.1002/bies.201300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan LZ, et al. Cell Cycle. 2014;13:1041–8. doi: 10.4161/cc.28128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montero J, et al. Cell Death Differ. 2013;20:1465–74. doi: 10.1038/cdd.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee CW, et al. Cancer Res. 2012;72:4394–404. doi: 10.1158/0008-5472.CAN-12-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones RG, et al. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 8.Cantó C, et al. Nature. 2009;458:1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]