

Abstract
In hematopoietic malignancies, oncogenic alterations interfere with cellular differentiation and lead to tumoral development. Identification of the proteins regulating differentiation is essential to understand how they are altered in malignancies. Chronic myelogenous leukemia (CML) is a biphasic disease initiated by an alteration taking place in hematopoietic stem cells. CML progresses to a blast crisis (BC) due to a secondary differentiation block in any of the hematopoietic lineages. However, the molecular mechanisms of CML evolution to T-cell BC remain unclear. Here, we have profiled the changes in DNA methylation patterns in human samples from BC-CML, in order to identify genes whose expression is epigenetically silenced during progression to T-cell lineage-specific BC. We have found that the CpG-island of the ENGRAILED-2 (EN2) gene becomes methylated in this progression. Afterwards, we demonstrate that En2 is expressed during T-cell development in mice and humans. Finally, we further show that genetic deletion of En2 in a CML transgenic mouse model induces a T-cell lineage BC that recapitulates human disease. These results identify En2 as a new regulator of T-cell differentiation whose disruption induces a malignant T-cell fate in CML progression, and validate the strategy used to identify new developmental regulators of hematopoiesis.
Keywords: chronic myelogenous leukemia, blast crisis, T-cell development, engrailed-2, DNA methylation
Introduction
Hematopoiesis is a tightly regulated developmental process essential throughout life to generate all the cell lineages in the blood. This process is mainly driven by the equilibrium between transcription factors, epigenetic regulators, and extracellular signals that regulate the gene expression program of every one of the different blood cell types.1 Any deregulation of this delicate machinery leads to abnormal hematopoietic development. This is the case in the progression of chronic myelogenous leukemia (CML), a disease produced by the BCR–ABLp210 oncogene. As its name indicates, CML appears as a chronic disease lasting a median time of 5 y; in the absence of treatment, CML inevitably progresses to a blast crisis resembling an acute leukemia.2 Blast crises are mainly of the myeloid or B-cell types, and in both of them it has been shown that essential regulators of the normal myeloid (C/EBPβ) or B-cell (IKAROS) differentiation become disrupted in the progression of the disease.3,4
Interestingly, CML can also progress, with less frequency, to blast crises of a T-cell phenotype.5,6 However, nothing is known about the molecular biology of this type of leukemic progression and, so far, none of the known regulators of physiological T-cell development has been shown to be directly involved. In this project, we hypothesized that human T-cell blast crisis cells can be used as a starting material for the identification of unknown regulators of the normal T-cell fate differentiation, whose disruption would cooperate with the BCR-ABLp210 oncogene in establishing a malignant T-cell fate.
Since DNA methylation has been postulated as one of the main mechanisms involved in gene disruption during CML progression,7 in the present study we have used human samples from CMLs at the blast crisis stage (including an extremely infrequent T-cell blast crisis) and have performed an unbiased search for differentially methylated CpG islands in the disease progression. Our results have proven that methylation at the CpG island level is correlated with the progression from CML to blast crisis, and have allowed us to identify new genes involved in T-cell development. Indeed, we have found that the CpG island of the ENGRAILED-2 (EN2) gene is methylated in this progression. Afterwards, we have shown that En2 is expressed during T-cell development, both in mouse and in humans. Finally, we have been able to recapitulate the human disease and to confirm a role for En2 in T-cell normal and pathologic development by showing that, in the absence of En2 in a transgenic mouse model of CML, the development of T-cell blast crises is induced. Overall, these results identify En2 as a new regulator of T-cell differentiation whose disruption establishes a malignant T-cell fate in CML progression.
Results
Aberrant methylation affects genes involved in hematopoietic differentiation during the progression from CML to BC-CML
Genomic DNA from the different samples (see “Materials and Methods”; Table S1) was digested with MseI restriction enzyme to release intact CpG islands, and then loaded and fractionated in the MBD column. Non-methylated DNA was used as probe to screen a CpG islands library,8 and those library clones appearing as methylated in blast crises samples were sequenced and identified. In total, 539 clones were selected from the library, corresponding to islands that have become methylated in the progression to blast crisis. 217 clones (36.6% of the total) presented more than a 98% of homology with regions localized in genes with a known function. We classified these 217 genes according to their function after a literature search, defining 7 groups of functions (where a gene can be associated with more than one function) (Fig. 1; Table S2): (1) constitutive (i.e., housekeeping) genes; (2) genes involved in cellular differentiation; (3) genes controlling proliferation or cell cycle; (4) transcriptional regulators; (5) epigenetic modifiers; (6) genes described as being related to cancer; and (7) other functions (for example, specific functions in differentiated cell types). The remaining 322 clones (43.4%) appear in intergenic regions and do not seem to be related with any described gene. Similar findings have been previously described when this method has been used for the study of lung adenocarcinomas,9 and it is likely that these fragments correspond with what has been recently defined as CpG island shores.10,11 These results confirm that there is an aberrant methylation in blast crises. More importantly, this approach has allowed us to identify genes that were not previously described as playing a role in hematopoietic development (see below).

Figure 1. Functional classification of the genes identified in the screening. (1) Constitutively expressed genes; (2) genes involved in cellular differentiation; (3) genes controlling proliferation or cell cycle; (4) transcriptional regulators; (5) epigenetic modifiers; (6) genes described as being related to cancer; and (7) other functions.
ENGRAILED-2 CpG island is specifically methylated in T-cell blast crises and T-ALLs
To validate the screening, we chose a small sample among the most physiologically relevant genes identified and analyzed the methylation status of their CpG islands (either as identified from the library or by using the classical criteria for CpG island definition) by bisulfite sequencing in a wider set of samples of human CML, BC-CML, ALL, and also in selected human leukemic cell lines (data not shown). One of the interesting genes identified was ENGRAILED-2 (EN2), an homeobox-containing gene that acts mainly as a transcriptional repressor and plays an essential role in the patterning and development of essential structures of the central nervous system.12-15 Although EN2 has not been implicated in hematopoietic development so far, it has been previously shown that neuropoiesis and hematopoiesis share many common genetic pathways,16 and that there are many transcription factors that have essential functions in both developmental processes. Also, many of the genes and proteins that are functionally related to En2 in other organisms or systems (for example, Wnt, Pax5, or Hedgehog) also have important functions in the hematopoietic system. All these evidences supported the possibility of a role for EN2 in hematopoietic development and justified a deeper analysis of the changes affecting the gene in BC-CML. First, we proceeded to confirm by bisulfite sequencing that EN2 CpG island, as expected, appeared as totally unmethylated in healthy controls and in samples from chronic phase-CML (Fig. 2). Furthermore, it also appeared as unmethylated in non-T-cell blast crises and in B-ALL cases. However, dense methylation could be readily detected in several of the clones sequenced for the T-cell blast crisis sample and in human T-ALLs. Leukemic cell lines were used as positive controls for dense methylation, because, independently of their origin, it is well known that cell lines in culture tend to methylate any non-essential gene.17 This T-cell tumor specificity of EN2 CpG island methylation pointed out to a potential role of EN2 in T-cell development and T-cell leukemogenesis.

Figure 2. Methylation of EN2 CpG island in human leukemias. (A) Scheme of hEN2 gene, showing the predicted CpG island (according to ENSEMBL, in purple) and the region analyzed by bisulfite sequencing (blue), covering 68 CpG pairs, and situated approximately 300 bp upstream of EN2 ATG start codon. (B) Methylated status of CpG pairs in the selected area (open circles, unmethylated, closed circles, methylated), as analyzed by bisulfite conversion and sequencing of individual clones belonging to the indicated sample types (n = number of different samples).
Engrailed-2 is expressed during T-cell development in mice and humans
To gain further insight into any possible role of En2 in T-cell development, we first proceeded to determine if En2 was expressed during the different stages of T-cell differentiation. We could demonstrate that En2 is expressed at low levels in sorted mouse CD4+CD8+ double positive (DP) thymocytes, and also in CD8+ single positives (CD8-SP), both in the thymus and in the spleen (Fig. 3). Expression of EN2 mRNA could also be detected in human DP thymocytes (Fig. S1A). On the contrary, expression of the only other Engrailed orthologs present in mammals, En1, was not detected at any stage, therefore excluding any potential functional redundancy in the thymus (Fig. S1B).
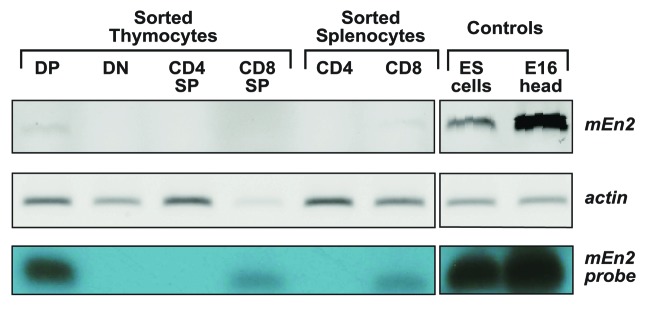
Figure 3.En2 is expressed in mouse T-cell development. RT-PCR analysis of the indicated sorted stages of T-cell development shows that En2 is expressed in CD4+ CD8+ DP and CD8+ SP thymocytes, and in CD8+ T cells in the spleen. The identity of the En2 transcript was confirmed by using as positive controls cDNAs from mouse ES cells and from E16 mouse embryo head, and by using a sequenced En2 probe to hybridize a Southern blot of the amplified PCR products (lower lane).
Having shown that En2 is expressed at specific stages of T-cell differentiation, we set out to analyze the main hematopoietic compartments of En2 knockout mice in comparison with those from WT littermates, in order to study any potential alteration in T-cell development due to the absence of En2. However, a detailed flow cytometry study did not reveal significant differences between En2−/− mice and wild-type controls in any of the main hematopoietic cell types and organs, including the thymus (data not shown).
Absence of En2 confers a competitive developmental advantage during T-cell differentiation
Non-stringent developmental alterations can be masked in a non-competitive setting when defects are minor. Therefore, the best way of revealing these developmental defects, when not directly visible in the constitutive knockout mouse, is to use a competitive transplantation setup in which mutant cells must compete with normal WT cells. Therefore, to gain insight into the role played by En2 in T-cell development, we performed competitive bone marrow transplantations in which we injected a 1:1 ratio of En2−/− cells, together with WT syngeneic cells, into lethally irradiated Rag1−/− recipients. Mutant cells could be distinguished from WT ones by using 2 different forms of the pan hematopoietic marker CD45 (Ly5.2 in En2−/− cells and Ly5.1 in WT cells). Controls were performed in parallel, in which Ly5.2+ cells from WT littermates were injected 1:1 with the competitor to exclude any potential effect of the Ly5.1 vs. Ly5.2 background in the competition. Results are shown in Figure 4, representing the percentage of contribution of Ly5.2+ cells (from either WT or En2−/− donor littermates) to each of the different lineages and hematopoietic tissues shown. In the thymus, En2−/− cells contribute with higher percentages to all the main developmental stages of T-cell development, from CD4−CD8− double negatives (DN), through CD4+CD8+ double positives (DP) to single positives CD8+ or CD4+. This increased contribution of En2−/− cells to T-cell development can also be appreciated in the periphery, both in peripheral blood and, to a lesser degree, in the spleen (Fig. 4). These results suggest that the absence of En2 confers a developmental advantage during the course of T-cell generation.
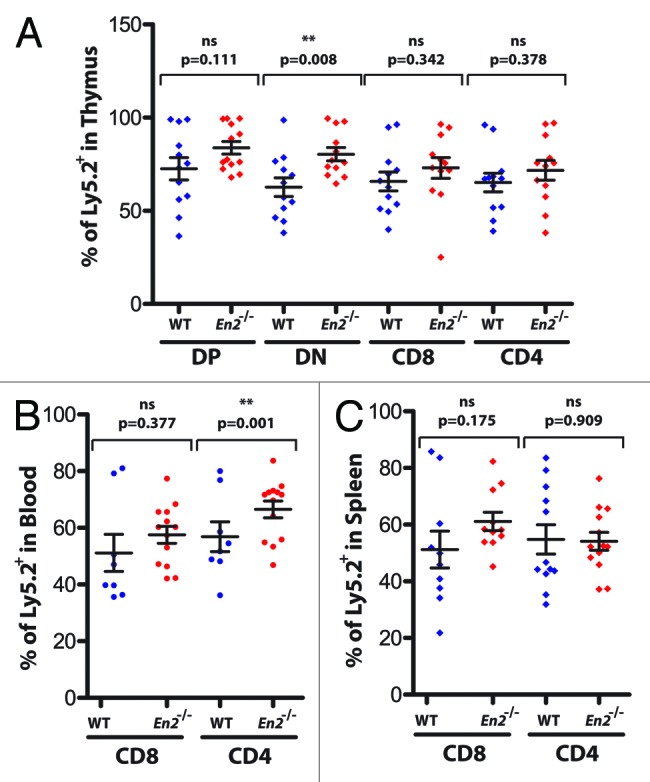
Figure 4. Advantage of En2−/− cells in a competitive bone marrow transplantation setting. Four millions cells in a 1:1 mixture of either WT(Ly5.2+):WT(Ly5.1+) or En2−/−(Ly5.2+):WT(Ly5.1+) were injected into lethally irradiated Rag1−/− recipients. Results represent the percentage of contribution of Ly5.2+ cells from either WT (blue dots) or En2−/− (red dots) littermates to each one of the different indicated lineages in the hematopoietic tissues shown: (A) thymus, (B) blood, and (C) spleen. Each dot represents an individual mouse. Statistics were performed using Student t test with the GraphPad Prism 5 program.
Changes in global gene-expression patterns in En2−/− T cells
To increase our understanding of the possible molecular role(s) of En2 during T-cell development, we compared by microarray analysis the gene expression patterns of En2−/− vs. WT sorted CD4+CD8+ and CD8+ cells (the 2 populations in which we had detected En2 expression). The results, summarized in Figure S2, show that, although the level of the changes is small, there are consistent differences in the patterns of gene expression between En2−/− and WT cells. Furthermore, some of the genes whose levels are changed are known to be involved specifically in T-cell development; for example, Slfn3, which is known to be differentially regulated during T-cell differentiation and whose overexpression interferes with the maturation of DP thymocytes,18-20 or Ezh2, a well-known epigenetic regulator, or Hmgb1, involved in V(D)J rearrangements because of its capacity for interacting with RAG proteins.21
Lack of En2 specifically triggers CD8+ T-cell blast crisis in a transgenic model of CML
In order to confirm, in a definitive manner, that En2 disruption would cooperate with the BCR–ABLp210 oncogene in establishing a malignant T-cell fate during the progression of CML, we sought to reproduce in the mice the same characteristics of the human disease, using the same molecular alteration that we had previously identified in the screening of the human samples. To this aim, we crossed a well-established transgenic mouse model of CML, Sca1-BCRABLp210 mice,22 into an En2−/− genetic background.23 In normal conditions, Sca1-BCRABL p210 mice develop CML that, with time, evolves into blast crises that can be either myeloid or B lymphoid in nature, but the CMLs developing in Sca1-BCRABLp210 mice only very rarely give rise to T-cell crises.22 This is similar to humans, since, as mentioned, in human CML patients only a very small percentage of blast crises are of the T-cell type. However, in Sca1-BCRABLp210, En2−/− mice, a large percentage (69.5%, 16 out of 23 mice analyzed) of the animals develop, with time, a T-cell leukemia characterized by the massive accumulation of CD8+ T cells in the blood, in the thymus, and in other peripheral hematopoietic organs, like the spleen or the LNs (Figs. 5 and 6A). These cells also infiltrated other non-hematopoietic organs, like the lungs (Fig. 6B and C). Also, the mice consistently developed cutaneous lesions in the abdomen, in the form of erythematous rashes that resemble those described in human adult T-cell leukemia/lymphoma (ATLL) patients24 (Fig. 6D). In the Sca1–BCRABLp210, En2−/− sick mice, the lesions present with minimal cytological atypia (data not shown), similarly to in human ATLL patients, where these lesions appear in approximately 50% of the cases and can range between erythematous rashes, papules, or nodules, among others, always with an unclear categorization.25 These results identify En-2 as a new regulator of T-cell differentiation whose disruption establishes a malignant T-cell fate in CML progression.

Figure 5. Lack of En2 leads to alterations in T cell development in the presence of the human BCR-ABLp210 oncogene. Flow cytometric analysis of the indicated hematopoietic organs, showing the alterations appearing in T-cell development in aged Sca1-BCRABLp210, En2−/− mice in comparison to WT controls. The massive accumulation of CD8+ cells can be clearly appreciated in the different hematopoietic compartments. Figures are representative from 16 mice analyzed.

Figure 6. T-cell blast crises triggered by the absence of En2 in a CML transgenic model. (A and B) Leukemic T cells in Sca1-BCRABLp210, En2−/− mice infiltrate hematopoietic (spleen, A) and non-hematopoietic (B) tissues. Representative hematoxylin and eosin (H&E) staining analysis of the spleen (A) and lung (B) of leukemic Sca1-BCRABLp210, En2−/− mice and control WT mice. Spleen panels show lymphoid tumor cells disrupting the normal architecture of the tissue. In the lung, the lymphoid tumor cells are infiltrating the lamina propria and the epithelium, giving rise to lymphoepithelial lesions. Magnifications are 40×. (C) The infiltrating lymphocytes in the lung are CD8+ T cells. Three-fold increase in the percentage of CD8+ T cells in the lungs of Sca1-BCRABLp210, En2−/− leukemic mice. (D) Erythematous rashes in the skin of leukemic mice.
Discussion
In this study, using human samples from BC-CML, we have studied the pattern of DNA methylation in the last stage of the disease progression in order to identify genes whose expression is epigenetically repressed during tumoral evolution. The number of clones identified in our screening roughly correlates with other previous studies using different “non-omic” approaches like restriction landmark genomic scanning that show that an average of 600 CpG islands become aberrantly methylated in human tumors.26 Our results show that there is an aberrant differential methylation in BC-CML, and that this process involves both bona fide gene-associated CpG islands, and also less defined CpG-rich regions that could correlate with CpG island shores. Among the CpG islands, several correspond to genes known to be involved in differentiation, and whose methylation could lead to loss of function and consequent developmental block of the affected lineage(s). Among them, we have focused our attention on ENGRAILED-2 (EN2), since it has been previously shown that neuropoiesis and hematopoiesis share many common genetic pathways, and that there are many transcription factors that have essential functions in both developmental processes.16 These and other evidences (see “Results”) supported the possibility of a role for EN2 in hematopoietic development. The first confirmation of this possibility comes from the previously unreported detection of expression of En2 during different stages of T-cell development, both in the mouse and in humans, therefore strongly reinforcing the idea of the participation of this gene in T lymphopoiesis. The lack of an apparent T-cell phenotype in En2-deficient mice, as determined by flow cytometry, is nevertheless not necessarily surprising; first, there are examples of cases in which knockout mice do not show any obvious phenotype, and this is of course not an indication that the gene is not fulfilling any function. Second, the low levels of En2 expression detected during T-cell development suggest that the gene effect might be subtle or modulatory and, therefore, not easily revealed just by gene targeting. Indeed, competitive bone marrow transplantation experiments showed that there is an increased contribution of En2−/− cells to T-cell development, both in the thymus and in the periphery, suggesting that the absence of En2 confers a developmental advantage during the course of T-cell generation. In fact, microarray analysis of purified En2−/− T-cell populations suggests that En2 might be regulating, in a subtle manner, the levels of different genes participating in T-cell development, like Slfn3, Ezh2, or Hmgb1.
Finally, to go full circle with our hypothesis, we attempted recapitulating in the mice the human T-cell blast crisis phenotype, by crossing a CML mouse model into an En2−/− background. Almost 70% of Sca1-BCRABLp210, En2−/− mice developed T-cell leukemia with accumulation of CD8+ T cells in the blood, and leukemic infiltration of non-hematopoietic organs like the lungs or the skin. It has previously been described that, in an FVB background, Sca1-BCRABLp210 mice develop a thymoma characterized by the accumulation of CD44+CD25−CD8−CD4− (double-negative-1, DN1) early progenitors,27 but with little involvement of more differentiated T cells and no signs of skin lesions. This accumulation of DN1 cells could also be found in the Sca1–BCRABLp210, En2−/− sick mice (data not shown). Nevertheless, in the mixed B6CBA genetic background used in this study, Sca1–BCRABLp210, En2+/+ mice generally presented with the initially published B or myeloid blast crises,22 while involvement of mature T cells was only detected in some cases. As an additional proof of the involvement of En2 in T-cell development, Sca1–BCRABLp210, En2+/− mice with a single normal copy of the En2 gene also developed a T-cell blast crisis, although with slightly less penetrance (57%, 8 out of 14) when compared with the Sca1-BCRABLp210, En2−/− animals. These facts confirm that the absence of En2 cooperates with the BCR-ABLp210 oncogene in the development of a malignant T-cell fate.
Our findings were prompted by gene identification in a single disease subject of an extremely rare form of BC-CML. When we assayed for phenocopy in a mouse model, we could recapitulate the human disease. Our results have led to new insights into the basic biology of T-cell development and the mechanism of progression from CML to BC-CML in general, and to T-cell BC in particular. Because of the small number of patients and the high heterogeneity among them, causative gene discovery in rare subtypes of tumors, like T-cell BC, has been challenging. However, with approaches such as the one described here, these limitations can be broken, and new opportunities are at hand to understand malignant progression and its underlying genetic and developmental mechanisms, especially when coupled with phenocopy validation in appropriate models.
Materials and Methods
Mice
Sca1–BCRABLp210, En2−/− mice were obtained by breeding Sca1–BCRABLp210 transgenic mice22 into an En2−/− background;23 they were genotyped as previously described and maintained in a B6CBA background. Rag1−/− mice28 were used as recipients for bone marrow transplantations (BMT). C57Bl6/J Ly5.1+ mice (B6.SJL-Ptprca Pep3b/BoyJ strain) were used as WT competitors for BMT experiments. All animal experiments were performed in accordance with the guidelines of the corresponding local Committees on Animal Research.
Human samples
Samples (total peripheral blood mononuclear cells) were collected prior to therapy after informed consent was given, in accordance with the ethical committee guidelines and the Declaration of Helsinki. Samples are listed in Table S1. For the screening of the CpG island library, DNA from samples BC#1, BC#2, BC#3, and BC#6 were used as probes after MBD column separation (see below).
Separation of methylated CpG islands on the MBD column
The recombinant protein HMBD, bearing the methyl-CpG binding domain of MeCP2 linked to a poly-His tail, was produced in E. coli, as previously described, purified, and coupled to Ni2+–NTA agarose onto a Pharmacia HR 5/10 FPLC column.8 This column was used to separate differentially methylated CpG islands from BC–CML samples as follows. Fifty–100 μg of DNA obtained from whole blood from patients with blast crisis and a large percentage of blasts were digested with MseI to release intact CpG islands, and this digested DNA was loaded into the column.8 Non-bound DNA (10–20% of total load) elutes in the first ml of the flow-through; weakly bound DNA (i.e., non-methylated CpG islands) elutes at the beginning of the 0.4 M NaCl wash (fractions 2–3); and methylated DNA elutes in the last 2/3 of the linear salt gradient. Weakly bound DNA (i.e., non-methylated CpG islands) was pooled and used to probe the CpG islands library.
CpG islands library screening
The CpG islands library, cloned in the NdeI site of pGEM5Zf(-) and transformed into the strain XL1-blue of E. coli (available at the HGMP) was grown in LB–Amp plates to obtain the representative number of 60 000 independent clones. Then colonies were transferred to Hybond filters (Amersham) and allowed to regrow on the plate. Colonies were lysed on the filter, and their denatured DNA was UV-cross-linked to the membrane. The membranes were probed with 32P-labeled DNA from the fractions of non-methylated CpG islands. Superimposing the autoradiography films from these hybridizations to the LB–Amp plates, clones that had not been labeled with several probes made of DNA coming from different patients (i.e., clones corresponding to islands that were not present in the probe because they were methylated and had been retained in the column) could be selected. These clones (approx. 550) were picked individually and sequenced from both sides with T7 and SP6 sequencing primers in an automatic sequencer. The sequences were then compared against GenBank database using the BLAST program.
Bisulfite methylation analysis
For bisulfite conversion, 4 μg of genomic DNA were digested with MseI (TTAA recognition site), which cuts rarely within CpG islands and releases them almost intact, then phenol extracted, and, after ethanol precipitation, dissolved in TE at a concentration of 2 μg DNA in 5 μl TE. DNA was bisulfite-treated as previously described.29 Five microliters of this DNA were used per PCR reaction. Nested PCR reactions were performed with the indicated oligonucleotide primers (see below), at an annealing temperature of 50 °C for 35 cycles each, in the presence of 2.5 mM MgCl2. PCR products were cloned into pBluescript and several clones (usually 5 to 10) sequenced to identify the bisulfite-mediated cytosine-to-uracil(thymine) conversions.
Human ENGRAILED-2 CpG island bisulfite analysis
The sense strand of bisulfite-modified human EN2 CpG island (Fig. 2) was amplified by nested PCR, as described above, using for the first PCR the oligonucleotides 5′ggtttagtty ggttyggt3′ and 5′accaaaacta aaatcctaca aatcacc3′, and for the second PCR, 5′gggttttggt ttygygtttt ttatttat3′ and 5′aaatctaaac aatcrcaata atccracraa a3′.
RT-PCR analysis of gene expression
RNA was prepared from FACS-sorted cell populations using the TRIzol reagent (GIBCO-BRL). CDNA was synthesized with random hexamers and the Reverse Transcription System (Promega). Expression of mouse En2 transcript was detected using 5′GTTCCTTGGA TGGAGTGC3′ and 5′CAAAGTGTTC TTGTTGCC3′ primers. Expression of mouse En1 transcript was detected using 5′CCTCGAAGCC CTCGGACAGT 3′ and 5′TCCCTGGGCC ATGAGGTGCA3′ primers. Expression of human EN2 transcript was detected using 5′TGGACGGGTC GCTCAAGG3′ and 5′CCCTGTGCCA TGAGGTGC3′ primers. PCR products were separated on agarose gels and visualized by staining with ethidium bromide. PCR products were also cloned, sequenced, and, once verified, used as a Southern blot 32P-labeled probe to detect low expression levels of the studied transcripts.
Competitive bone marrow transplantations
Single-cell suspensions were prepared from the bone marrow of either En2−/− (Ly5.2+), WT (Ly5.2+), or WT competitor (Ly5.1+) mice. Donor T cells were removed from the BM samples by using anti-CD4-PE, anti CD8-PE (BD-PharMingen), and anti-PE Beads and magnetic columns (Miltenyi Biotech). Cells were counted using a Scepter Cell Coulter Counter (Millipore), washed in PBS, and injected into the tail vein of 8–12-wk-old lethally γ-irradiated Rag1–/– recipients. The total amount of injected cells was of 4 × 106 in a 1:1 mixture of either WT(Ly5.2+):WT(Ly5.1+) or En2−/−(Ly5.2+):WT(Ly5.1+) cells. Recipient mice were analyzed after more than 2 mo, and the percentage of contribution of Ly5.2+ cells was evaluated in the different blood cell types in the main hematopoietic organs.
Flow cytometry
Flow cytometry was performed as previously described.22,30-32 Briefly, nucleated cells were obtained from the different hematopoietic organs. Contaminating red blood cells were lysed with RCLB lysis buffer, and the remaining cells were then washed in PBS with 1% FCS. Cell suspensions were incubated first with purified anti-mouse CD32/CD16 (PharMingen) prior to the addition of other antibodies to block binding through Fc receptors. After staining, all cells were washed once in PBS with 1% FCS containing 2 mg/ml propidium iodide (PI) to allow dead cells to be excluded from both analyses and sorting procedures. The samples and the data were analyzed in a FACSCantoII or a FACSCalibur using either CellQuest (Becton Dickinson) or FlowJo (Tree Star) softwares. Specific fluorescence of the fluorochromes was used, as well as known forward and orthogonal light-scattering properties of mouse cells, to establish gates. For each analysis a total of at least 50 000 viable (PI-) cells were assessed. For cell sorter separation, CD8a and CD4 lineage markers were used to purify cells from the thymus and spleen of both En2−/− or control WT mice by fluorescence-activated cell sorting (FACS) (FACSVANTAGE; Becton Dickinson). Purity of the sorted cells was over 98%, as determined by FACS reanalysis. FACS analysis of lung infiltration by tumoral CD8+ cells was performed as described above for hematopoietic tissues.
Microarray hybridization
Sorted purified cells were harvested with TRIzol Reagent (Invitrogen) and the RNA was extracted according to the manufacturer's instructions. As a last step of the extraction procedure, the RNA was purified with the RNeasy Mini-kit (Qiagen). Before cDNA synthesis, RNA integrity from each sample was confirmed on Agilent RNA Nano LabChips (Agilent Technologies). The sense cDNA was prepared from 300 ng of total RNA using the Ambion® WT Expression Kit. The sense strand cDNA was then fragmented and biotinylated with the Affymetrix GeneChip® WT Terminal Labeling Kit (PN 900671). Labeled sense cDNA was hybridized to the Affymetrix Mouse Gene 1.0 ST microarray according to the manufacturer protocols and using GeneChip® Hybridization, Wash, and Stain Kit. Genechips were scanned with the Affymetrix GeneChip® Scanner 3000, as previously reported.31,33
Microarray data analysis
Both background correction and normalization were done using RMA (Robust Multichip Average) algorithm.34 After quality assessment and outlier detection, 5 En2−/− and 4 WT CD8+ samples were compared for the CD8 experiment, and 2 En2−/− and 3 WT CD4+CD8+ (DP) samples were compared for DP experiment. A filtering process was performed to eliminate low expression probe sets. Applying the criterion of an expression value greater than 64 in 3 samples in the CD8 experiment and greater than 64 in 2 samples in the DP experiment for each experimental condition (En2−/−, WT), 27151 and 27166 probe sets were selected for statistical analysis. R and Bioconductor were used for preprocessing and statistical analysis. LIMMA (Linear Models for Microarray Data) was used to find out the probe sets that showed significant differential expression between experimental conditions. Genes were selected as significant using a criteria of P value < 0.01 and FC > 1.56. Functional enrichment analysis of Gene Ontology (GO) categories was performed using standard hypergeometric test. The biological knowledge extraction was complemented through the use of Ingenuity Pathway Analysis (Ingenuity Systems, http://www.ingenuity.com), which database includes manually curated and fully traceable data derived from literature sources. Raw gene expression microarray files were submitted to GEO and are available under the accession number GSE47612.
Histological analysis
Mice were subjected to standard necropsy. Major organs were examined under the dissecting microscope, and samples of each organ were processed into paraffin, sectioned, and examined histologically. Tissue samples were taken from homogenous and viable portions of the resected sample by the pathologist and fixed within 2–5 min of excision. Haematoxylin-eosin-stained sections of each tissue were reviewed by a single pathologist.22,30-32
Supplementary Material
Acknowledgments
We are indebted to all members of C.C. and I.S.G. groups for useful discussions and for their critical reading of the manuscript. We are indebted to patients who gave samples. We thank Alexandra Joyner for the En2−/− mouse. We thank Adrian Bird for the pET6HMBD plasmid. We thank Berta Raposo and Silvia Andrade for help with cell sorting. We thank Angela Aznar and Lourdes Ortiz for support with gene expression microarrays. We are grateful to the Animal Care Facility staff members for their valuable help. Research at C.C.’s lab was partially supported by FEDER, Fondo de Investigaciones Sanitarias, CSIC P.I.E., Junta de Castilla y León, and from an institutional grant from the Fundación Ramón Areces. Research in ISG group is partially supported by FEDER and by MICINN (SAF2012-32810), by MEC OncoBIO Consolider-Ingenio 2010 (Ref. CSD2007-0017), by NIH grant (R01 CA109335-04A1), the ARIMMORA project (FP7-ENV-2011, European Union Seventh Framework Program), by Junta de Castilla y León (BIO/SA06/13) and by “Proyecto en red de investigación en células madre tumorales” supported by Obra Social Kutxa y Consejería de Sanidad de la Junta de Castilla y Leon. C.V.D.’s research is supported by Junta de Castilla y León (proyecto de investigación en biomedicina SAN/39/2010). J.A.M.C.’s research is supported by the Instituto de Salud Carlos III (ISCIII), grants FIS-PI12/00202 and RTICC-RD12/0036/0063. All Spanish funding is co-sponsored by the European Union FEDER program. I.S.G. is an API lab of the EuroSyStem project and a partner of DECIDE European network. F.A.-J. and E.C.S. were supported by Spanish Ministry of Science and Innovation fellowships. E.C.-S. was a “Residencia de Estudiantes” Fellow. A.T.N. was the recipient of a “Beca de Postgrado de la Fundación Ramón Areces/UAM.”
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
F.A.J., E.C.S., A.T.N., and C.V.D. performed experiments, analyzed the data and wrote the paper. I.G.H., E.A.E., V.S., J.A.M.C., and O.B. performed experiments and analysis of data. M.G. provided human samples and analyzed data. C.C. and I.S.G. designed the study, analyzed the data and wrote the paper. All the authors reviewed the manuscript and agree with its content.
Glossary
Abbreviations,
- ATLL
adult T cell leukemia/lymphoma
- BC
blast crisis
- CML
chronic myelogenous leukemia
- HSC
hematopoietic stem cell
- PBS
phosphate buffer saline
- RCLB
red cell lysis buffer
References
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–53. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 3.Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, Iervolino A, Condorelli F, Gambacorti-Passerini C, Caligiuri MA, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 4.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 5.Allouche M, Bourinbaiar A, Georgoulias V, Consolini R, Salvatore A, Auclair H, Jasmin C. T cell lineage involvement in lymphoid blast crisis of chronic myeloid leukemia. Blood. 1985;66:1155–61. [PubMed] [Google Scholar]
- 6.Dorfman DM, Longtine JA, Fox EA, Weinberg DS, Pinkus GS. T-cell blast crisis in chronic myelogenous leukemia. Immunophenotypic and molecular biologic findings. Am J Clin Pathol. 1997;107:168–76. doi: 10.1093/ajcp/107.2.168. [DOI] [PubMed] [Google Scholar]
- 7.Jelinek J, Gharibyan V, Estecio MR, Kondo K, He R, Chung W, Lu Y, Zhang N, Liang S, Kantarjian HM, et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS One. 2011;6:e22110. doi: 10.1371/journal.pone.0022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nat Genet. 1994;6:236–44. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- 9.Shiraishi M, Chuu YH, Sekiya T. Isolation of DNA fragments associated with methylated CpG islands in human adenocarcinomas of the lung using a methylated DNA binding column and denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1999;96:2913–8. doi: 10.1073/pnas.96.6.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condron BG, Patel NH, Zinn K. Engrailed controls glial/neuronal cell fate decisions at the midline of the central nervous system. Neuron. 1994;13:541–54. doi: 10.1016/0896-6273(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 13.Davis CA, Joyner AL. Expression patterns of the homeo box-containing genes En-1 and En-2 and the proto-oncogene int-1 diverge during mouse development. Genes Dev. 1988;2(12B):1736–44. doi: 10.1101/gad.2.12b.1736. [DOI] [PubMed] [Google Scholar]
- 14.Davis CA, Noble-Topham SE, Rossant J, Joyner AL. Expression of the homeo box-containing gene En-2 delineates a specific region of the developing mouse brain. Genes Dev. 1988;2:361–71. doi: 10.1101/gad.2.3.361. [DOI] [PubMed] [Google Scholar]
- 15.Marie B, Bacon JP, Blagburn JM. Double-stranded RNA interference shows that Engrailed controls the synaptic specificity of identified sensory neurons. Curr Biol. 2000;10:289–92. doi: 10.1016/S0960-9822(00)00361-4. [DOI] [PubMed] [Google Scholar]
- 16.Terskikh AV, Easterday MC, Li L, Hood L, Kornblum HI, Geschwind DH, Weissman IL. From hematopoiesis to neuropoiesis: evidence of overlapping genetic programs. Proc Natl Acad Sci U S A. 2001;98:7934–9. doi: 10.1073/pnas.131200898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62:503–14. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- 18.Condamine T, Le Luduec JB, Chiffoleau E, Bériou G, Louvet C, Heslan M, Tilly G, Cuturi MC. Characterization of Schlafen-3 expression in effector and regulatory T cells. J Leukoc Biol. 2010;87:451–6. doi: 10.1189/jlb.0609410. [DOI] [PubMed] [Google Scholar]
- 19.Geserick P, Kaiser F, Klemm U, Kaufmann SH, Zerrahn J. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol. 2004;16:1535–48. doi: 10.1093/intimm/dxh155. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz DA, Katayama CD, Hedrick SM. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity. 1998;9:657–68. doi: 10.1016/S1074-7613(00)80663-9. [DOI] [PubMed] [Google Scholar]
- 21.Dai Y, Wong B, Yen YM, Oettinger MA, Kwon J, Johnson RC. Determinants of HMGB proteins required to promote RAG1/2-recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol Cell Biol. 2005;25:4413–25. doi: 10.1128/MCB.25.11.4413-4425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pérez-Caro M, Cobaleda C, González-Herrero I, Vicente-Dueñas C, Bermejo-Rodríguez C, Sánchez-Beato M, Orfao A, Pintado B, Flores T, Sánchez-Martín M, et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009;28:8–20. doi: 10.1038/emboj.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyner AL, Herrup K, Auerbach BA, Davis CA, Rossant J. Subtle cerebellar phenotype in mice homozygous for a targeted deletion of the En-2 homeobox. Science. 1991;251:1239–43. doi: 10.1126/science.1672471. [DOI] [PubMed] [Google Scholar]
- 24.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon, France: International Agency for Research on Cancer, 2008. [Google Scholar]
- 25.Sawada Y, Hino R, Hama K, Ohmori S, Fueki H, Yamada S, Fukamachi S, Tajiri M, Kubo R, Yoshioka M, et al. Type of skin eruption is an independent prognostic indicator for adult T-cell leukemia/lymphoma. Blood. 2011;117:3961–7. doi: 10.1182/blood-2010-11-316794. [DOI] [PubMed] [Google Scholar]
- 26.Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P, Lang JC, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 27.García-Ramírez I, Ruiz-Roca L, Martín-Lorenzo A, Blanco O, García-Cenador MB, García-Criado FJ, Vicente-Dueñas C, Sánchez-García I. Genetic background affects susceptibility to tumoral stem cell reprogramming. Cell Cycle. 2013;12:2505–9. doi: 10.4161/cc.25544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–77. doi: 10.1016/0092-8674(92)90030-G. [DOI] [PubMed] [Google Scholar]
- 29.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–7. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romero-Camarero I, Jiang X, Natkunam Y, Lu X, Vicente-Dueñas C, Gonzalez-Herrero I, Flores T, Garcia JL, McNamara G, Kunder C, et al. Germinal centre protein HGAL promotes lymphoid hyperplasia and amyloidosis via BCR-mediated Syk activation. Nat Commun. 2013;4:1338. doi: 10.1038/ncomms2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vicente-Dueñas C, Fontán L, Gonzalez-Herrero I, Romero-Camarero I, Segura V, Aznar MA, Alonso-Escudero E, Campos-Sanchez E, Ruiz-Roca L, Barajas-Diego M, et al. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci U S A. 2012;109:10534–9. doi: 10.1073/pnas.1204127109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vicente-Dueñas C, Romero-Camarero I, González-Herrero I, Alonso-Escudero E, Abollo-Jiménez F, Jiang X, Gutierrez NC, Orfao A, Marín N, Villar LM, et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J. 2012;31:3704–17. doi: 10.1038/emboj.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beltran E, Fresquet V, Martinez-Useros J, Richter-Larrea JA, Sagardoy A, Sesma I, Almada LL, Montes-Moreno S, Siebert R, Gesk S, et al. A cyclin-D1 interaction with BAX underlies its oncogenic role and potential as a therapeutic target in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2011;108:12461–6. doi: 10.1073/pnas.1018941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.