

Abstract
Timely and proper cellular response to DNA damage is essential for maintenance of genome stability and integrity. B-cell lymphoma/leukemia 10 (BCL10) facilitates ubiquitination of NEMO in the cytosol, activating NFκB signaling. Translocation and/or point mutations of BCL10 associate with mucosa-associated lymphoid tissue lymphomas and other malignancies. However, the mechanisms by which the resulting aberrant expression of BCL10 leads to cellular oncogenesis are poorly understood. In this report, we found that BCL10 in the nucleus is enriched at the DNA damage sites in an ATM- and RNF8-dependent manner. ATM-dependent phosphorylation of BCL10 promotes its interaction with and presentation of UBC13 to RNF8, and RNF8-mediated ubiquitination of BCL10 enhances binding of BCL10 and UBC13 to RNF168. This allows mono-ubiquitination on H2AX by RNF168 and further poly-ubiquitination by the RNF8/RNF168-containing complex. Depletion of BCL10 compromised homology recombination-mediated DNA double-strand break (DSB) repair because of insufficient recruitment of BRCA1, RAD51, and the ubiquitinated DNA damage response factors. Taken together, our results demonstrate a novel function of BCL10 in delivering UBC13 to RNF8/RNF168 to regulate ubiquitination-mediated DSB signaling and repair.
Keywords: BCL10, DNA damage, RNF168, RNF8, ubiquitination
Introduction
Our genome is constantly attacked by a variety of environmental agents and endogenous factors.1 On average, one human cell encounters more than 6 × 104 naturally occurring DNA damage events per day, including single-strand breaks, depurination of bases, alkylation, oxidation, deamination, intra-strand cross-links, and DNA double-strand breaks (DSBs).2 Though a DSB occurs rarely, it is the most dangerous form of DNA damage, since one unrepaired DSB is sufficient to induce cell death. Fortunately, a highly efficient network of DNA damage response (DDR) has evolved to cope with these DNA damaging events. This network starts with sensing the damage, transducing and amplifying the damage signal, and ends with activating checkpoints to halt the cell cycle, allowing time for repairing the damaged DNA, or inducing apoptosis when the damage is irreparable.2-4 Failure of the DDR network is directly linked to genome instability, a hallmark of cancer cells, whereas an increase of DNA repair capacity at the advanced stages of cancer contributes significantly to resistance toward chemo- and radio-therapy.5
The delicate and complex network of DSB signaling and repair largely depends on post-translation modifications (PTM)6 and reversible phosphorylation and ubiquitination in particular. Phosphorylation promotes the first wave of recruitment of DDR factors to the DSB sites. Phosphorylation of histone H2AX at serine-139 (γ-H2AX) by the phosphatidyl inositol 3′ kinase-related kinases (PIKK) provides a platform for enrichment of DDR factors on the damage sites.7,8 Mediator of DNA damage checkpoint 1 (MDC1) serves as an important checkpoint mediator that directly binds to γ-H2AX during the early response, and amplifies the damage signal by recruiting ATM in a NBS1-dependent manner.9,10 The ATM-mediated phosphorylation of MDC1 promotes its oligomerization11,12 and binding to the ring finger protein RNF8, an E3 ubiquitin ligase. RNF8 is the first E3 ligase recruited to the DSB sites by MDC1 and is required for assembly of repair proteins at the sites of DNA damage. RNF8 couples with the ubiquitin-conjugating enzyme UBC13 to catalyze the K63-linked ubiquitination of histone H2A and H2AX, thus promoting the formation of ionizing radiation-induced foci (IRIF) of 53BP1, BRCA1-RAP80, and FK2 and enforcing the G2/M DNA damage checkpoint.13-16 Recent work by Mattiroil et al. unexpectedly revealed that RNF8-dependent recruitment of another E3 ligase RNF168 initiates the mono-ubiquitination on K13 and K15 of H2AX, enabling RNF8 and other E3 ligases to add K63-linked polyubiquitination on H2AX and other DDR factors.17 RNF8 also recruits additional E3 ligases in the DDR. These include the BRCA1/BARD1 complex, the HECT-type E3 HERC2, RAD18, and RNF169. RNF8 also couples with UBCH8, promoting K48-mediated autoubiquitination and proteasome-mediated degradation.18-21
Different E2s allow synthesis of ubiquitin conjugates of different topologies. UBC13 is a ubiquitin-conjugating enzyme mainly for K63-linked ubiquitination. It coordinates with RNF8 and RNF168 to modulate the DDR.15,22-25 Nevertheless, the outstanding puzzles include how UBC13 and RNF168 are recruited by RNF8. UBC13/RNF8 ubiquitin ligases control focus formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Its molecular mechanism is largely speculative.
UBC13 also is involved in NFκB signaling.26-28 The transcription factor NFκB is a critical target of T-cell receptor (TCR) signaling, playing a central role in driving entry into the cell cycle via stimulating transcription of numerous effector genes. The adaptor protein B-cell lymphoma/leukemia 10 (BCL10) plays a key role in transmitting signals from TCR to NFκB. The formation of the CARMA1–BCL10–MALT1 (CBM) complex in the cytosol is one of the essential steps in the NFκB signaling pathway. BCL10 and MALT1 interact constitutively with each other. CARMA1 is recruited to this BCL10-MALT1 complex in an inducible manner, depending on site-specific phosphorylation of CARMA1.29-31 Following CBM complex formation, the NFκB essential modulator (NEMO) is ubiquitinated by UBC13-TRAF6, and this K63-linked ubiquitin chain functions as an anchor for the recruitment of the IκB-kinase complex, which is subsequently activated. The activated IκB–kinase complex phosphorylates IκB proteins and thereby targets these proteins for proteasomal degradation. NFκB is subsequently liberated and translocates into the nucleus, where it supports the expression of various NFκB target genes.26,32 Defects in BCL10 are a cause of malignant mesothelioma (MESOM), an aggressive neoplasm of the serosal lining of the chest. The translocation t(1;14)(p22;q32) involving BCL10 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma).33
In this study, we uncovered a novel function of BCL10 in the DDR. ATM-dependent phosphorylation and subsequent RNF8-mediated ubiquitination of BCL10 in the nucleus promotes RNF8/RNF168-mediated ubiquitination through presenting UBC13 to RNF8/RNF168 and HR-mediated DSB repair through facilitating recruitment of BRCA1, RAP80, and ubiquitinated DDR factors.
Results
BCL10 is enriched at the DNA damage sites in an ATM-dependent manner
The adaptor protein BCL10 promotes K63-linked ubiquitination of NEMO through recruiting the E2 enzyme UBC13 for the E3 ligase TRAF6, leading to NFκB activation.26 These events mainly occur in the cytosol. In a chromatin fractionation assay, we found that BCL10 was present both in the cytosol fraction and in the chromatin-enriched fraction (Fig. 1A). Given that UBC13 is an important E2, which coordinates with RNF8 to transduce the ubiquitination-mediated signaling and to repair damaged DNA in DDR, we sought to explore if and how the nuclear fraction of BCL10 is involved in this process. We first determined if endogenous BCL10 localized to DNA damage sites. Immunofluorescence staining in HeLa cells using a BCL10 antibody revealed that weak staining was present in the cytosol, and a diffused staining pattern was present in the nucleus of proliferating cells under unperturbed conditions (Fig. 1B). When cells were treated with etoposide for 1 h, BCL10 formed discrete foci within the nucleus, and these foci colocalized with the γ-H2AX foci (Fig. 1B). BCL10 focus formation was also observed in cells when treated with bleomycin (data not shown). To exclude the possibility of antibody cross reactivity, we found that etoposide-induced BCL10 foci diminished when BCL10 expression was depleted by siRNA (data not shown). When cells were pre-treated with different PIKK inhibitors, the ATM-specific inhibitor KU55933 efficiently reduced etoposide-induced focus formation of BCL10, whereas pretreatment of cells with NU6027, an ATR-specific inhibitor, or NU7026, a DNA-PKcs-specific inhibitor, did not compromise etoposide-induced focus formation of BCL10 (Fig. 1C). Furthermore, inhibition of ATM expression by siRNA compromised etoposide-induced focus formation of BCL10 in HeLa cells (Fig. 1D). These results demonstrated that BCL10 is enriched on the DNA damage sites in an ATM-dependent manner.

Figure 1. BCL10 is a chromatin-associated protein and accumulates at DSB site in an ATM- dependent manner. (A) BCL10 was present in the chromatin-enriched fraction. 293T cells were treated with bleomycin or mock-treated, and then subjected to chromatin fractionation. S2, cytosol; S3, nuclear soluble; P3, chromatin-enriched fraction. (B) BCL10 formed discrete nuclear foci in response to etoposide treatment. HeLa cells were subjected to DMSO/Etoposide treatment for 1 hour and immunostained with mouse anti-γ-H2AX monoclonal antibody and rabbit anti-BCL10 polyclonal antibodies. (C) DNA damage-induced focus formation of BCL10 was compromised by the ATM-specific inhibitor KU55933. HeLa cells were treated with DMSO or PIKK inhibitors 1.5 h before etoposide was added and processed for immunostaining as described in (B). (D) Depletion of ATM compromised DNA damage-induced focus formation of BCL10. HeLa cells were transfected with control siRNA (si-CTR) or ATM-specific siRNA (si-ATM), treated 2 d after transfection with DMSO or etopside for 1.5 h, and fixed for immunostaining with antibodies as indicated. The efficiency of ATM knockdown was determined by immunoblotting with antibodies against ATM and β-actin.
ATM phosphorylates BCL10 in response to DNA damage
We asked if BCL10 is a substrate of ATM. Co-immunoprecipitation assays revealed that endogenous BCL10 was present in the ATM immunocomplex, and HA-BCL10 in the FLAG-ATM immunocomplex in 293T cells (Fig. 2A). We examined the amino acid sequence of the BCL10 polypeptide and identified the only 91TQ motif as a potential ATM/ATR/DNA-PK phosphorylation site. We performed in vitro kinase assays using ATM kinase immunoprecipitated from 293T cells treated with etoposide for 1 h. As a positive control, ATM efficiently phosphorylated bacterially produced kinase-dead CHK2, and pretreatment of the ATM immunocomplex with ATM specific inhibitor KU55933 essentially diminished the incorporation of 32P-ATP into kinase-dead CHK2 by ATM (Fig. 2B). As expected, wild-type BCL10 was phosphorylated by ATM, and both pretreatment of the ATM immunocomplex with KU55933 or phosphorylation-defective mutation of BCL10 abolished the incorporation of 32P-ATP into BCL10 or BCL10 (T91A) by ATM, respectively (Fig. 2B). We then raised a rabbit polyclonal antibody against a pT91 phosphopeptide containing 11 amino acids of BCL10. The specificity of this antibody was demonstrated by the fact that it was reactive to FLAG-BCL10, but not FLAG-BCL10(T91A), immunoprecipitated from 293T cells after etoposide treatment, and this reactivity disappeared when the immunocomplex was pre-treated with phosphatase CIP (Fig. 2C). Furthermore, this antibody was weakly reactive to endogenous BCL10 immunoprecipitated from 293T cells under unperturbed condition, and this reactivity strongly increased upon etoposide treatment (Fig. 2D). Taken together, these results demonstrated that BCL10 is phosphorylated at T91 by ATM in response to etoposide treatment.
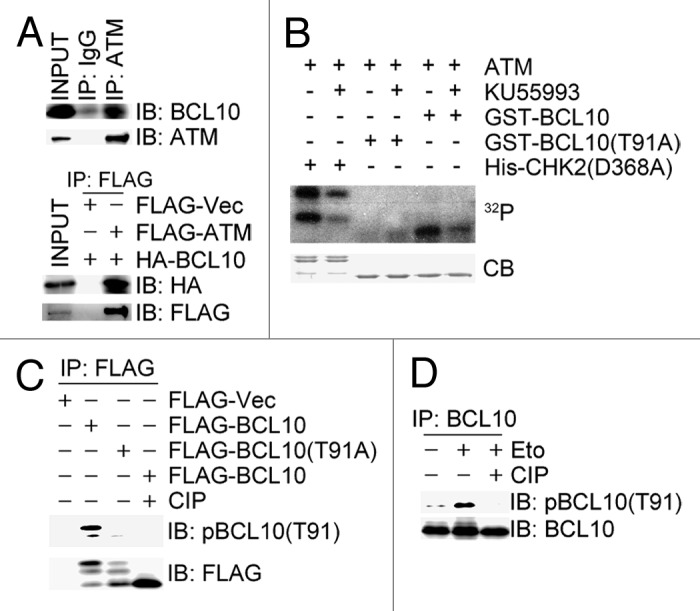
Figure 2. ATM phosphorylates BCL10 on T91 in response to DNA damage. (A) BCL10 interacted with ATM. Total cell lysates were extracted from 293T cells or 293T cells co-transfected with FLAG-ATM and HA-BCL10 and subjected to immunoprecipitation and immunoblotting with antibodies as indicated. (B) ATM phosphorylated BCL10 in vitro. ATM kinase complex was immunoprecipitated from 293T cells treated with etoposide for 1 h. In the in vitro kinase assays, bacterially produced GST-CHK2 (D368A) or GST-BCL10 (WT orT91A) was incubated with ATM kinase complex in the presence of γ-32P ATP with or without ATM inhibitor KU55933. The proteins were resolved by SDS-PAGE and detected by autoradiography and Coomassie blue staining (CB). (C) ATM phosphorylated BCL10 on T91. 293T cells were transfected with FLAG vector, FLAG-BCL10, or FLAG-BCL10(T91A) and treated 48 h later with etoposide for 1 h. Total cell lysates were extracted for immunoprecipitation with an anti-FLAG antibody. One of the duplicate FLAG-BCL10 immunoprecipitates was treated with CIP at 30 °C for 1 h. Phosphorylation of BCL10 was detected by immunoblotting with affinity-purified phospho-specific antibodies for pT91-BCL10. (D) Endogenous BCL10 was phosphorylated on T91 in response to DNA damage. Total cell lysates were extracted from 293T cells before and 1 h after etoposide treatment and immunoprecipitated with an anti-BCL10 antibody. Half of BCL10 immunocomplexes were incubated with CIP at 30 °C for 1 h. The immune-complexes were resolved by SDS-PAGE and detected with antibodies as indicated.
Phosphorylated BCL10 presents UBC13 to RNF8 and is ubiquitinated by RNF8
RNF8 catalyzes K63-linked ubiquitination, and primarily initiates ubiquitination-mediated signaling in DDR, while UBC13 serves as one of the major conjugating enzymes for RNF8.15 We wanted to investigate the potential link between BCL10 and RNF8/UBC13. Co-immunoprecipitation assays using total cell lysates from 293T cells revealed that endogenous BCL10 was present in the immunocomplex of RNF8 (Fig. 3A), and HA-BCL10 was present in both the FLAG–RNF8 immunocomplex (Fig. 3B) and the FLAG–UBC13 immunocomplex (data not shown). The phosphorylation-defective mutant BCL10(T91A) failed to interact with RNF8 (Fig. 3B), and the FHA domain mutant RNF8(R42A) reduced its binding with BCL10 (Fig. 3C), suggesting that the TQXF motif in BCL10 mediates binding to RNF8, and the intact FHA domain in RNF8 is partially responsible for binding BCL10. Inhibition of BCL10 expression by siRNA compromised the interaction between RNF8 and UBC13, whereas this interaction was restored by the expression of the siRNA-resistant form of BCL10, but not by the phosphorylation-defective mutant BCL10(T91A) (Fig. 3D). These findings demonstrated that the ATM-mediated phosphorylation on T91 of BCL10 is required for recruitment of both BCL10 and UBC13 by RNF8, and, put in another way, BCL10 presents UBC13 to RNF8, and this presentation is dependent on BCL10 phosphorylation on T91 by ATM.

Figure 3. BCL10 functions as an assembly factor for the RNF8–Ubc13 complex in a phosphorylation-dependent manner. (A) BCL10 interacted with RNF8. Total cell lysates were extracted from 293T cells, immunoprecipitated and immunoblotted with antibodies as indicated. (B) ATM-mediated phosphorylation of BCL10 on T91 was required for its binding to RNF8. 293T cells were co-transfected with FLAG-RNF8 and HA-BCL10 or HA-BCL10(T91A). Total cell lysates were extracted 48 h later, immunoprecipitated, and immunoblotted with antibodies as indicated. (C) The FHA domain of RNF8 was necessary for its binding to BCL10. 293T cells were co-transfected with HA-BCL10 and FLAG-RNF8 or the FHA domain mutant FALG-RNF8(R42A). Total cell lysates were extracted 48 h later, immunoprecipitated, and immunoblotted with antibodies as indicated. (D) Depletion of BCL10 by siRNA reduced the interaction between RNF8 and UBC13. BCL10-depleted 293T cells were co-transfected with HA-UBC13, FLAG-RNF8, and siRNA-resistant MYC-BCL10res or MYC-BCL10(T91A)res. Total cell lysates were extracted 48 h after co-transfection, immunoprecipitated, and immunoblotted with antibodies as indicated.
We then explored if BCL10 is a substrate of the E3 ligase RNF8. A recent report revealed that BCL10 was poly-ubiquitinated on K31 and K63 with K63-linked ubiquitin chains in response to T-cell activation, creating binding sites for NEMO.34 We found that, when wild-type BCL10 was co-transfected with HA-ubiquitin in 293T cells, it was efficiently ubiquitinated (Fig. 4A). When RNF8 expression was depleted by siRNA, BCL10 ubiquitination was diminished, in which K63-linked ubiquitination was severely decreased, whereas K48-linked ubiquitination was not obviously compromised (Fig. 4A). The phosphorylation-defective mutant BCL10(T91A) was not efficiently modified with K63-linked ubiquitin chains in vivo (Fig. 4B). We thus concluded that BCL10 is ubiquitinated mainly with K63-linked ubiquitination by RNF8.

Figure 4. K63-linked ubiquitination of BCL10 is mediated by RNF8–UBC13 complex. (A) BCL10 is modified with K63-linked ubiquitins. RNF8-depleted or mock-depleted 293T cells were co-transfected with FLAG-Vec or FLAG-BCL10 along with HA-ubiquitin, HA-ubiquitin K48 only (HA-Ub[K48[), or HA-ubiquitin K63 only (HA-Ub[K63]). Total cell lysates extracted 48 h after transfection were subjected to immunoprecipitation with an anti-FLAG antibody and immunoblotting with antibodies as indicated. (B) BCL10(T91A) was not ubiquitinated in vivo. 293T cells were transiently co-transfected with HA-ubiquitin and FLAG-Vec, FLAG-BCL10, or FLAG-BCL10(T91A). Immunoprecipitates with an anti-FLAG antibody were probed with antibodies as indicated. (C) Depletion of RNF8 reduced etoposide-induced focus formation of BCL10. HeLa cells were transfected twice with either RNF8 siRNAs or a non-targeting control siRNA (si-CTR). Forty-eight hours after the second transfection, cells were treated with etoposide for 1 h before they were fixed with 4% paraformaldehyde. Immunofluorescence staining with anti-BCL10 and anti-γ-H2AX was performed as described in the Methods. The efficiency of RNF8 knockdown was determined by immunoblotting with antibodies as indicated. Scale bars: 20 μM. (D) Depletion of UBC13 reduced etoposide-induced focus formation of BCL10. Experiments were performed as described in (C) except that UBC13 was depleted in HeLa cells.
We sought to determine the epistatic status of BCL10, UBC13, and RNF8. When the RNF8 (Fig. 4C) or UBC13 (Fig. 4D) expression was depleted by siRNA, etoposide-induced focus formation of BCL10 was compromised. This suggests that recruitment of BCL10 to the damage sites requires both RNF8 and UBC13.
Phosphorylated and ubiquitinated BCL10 presents UBC13 to RNF168
A recent report demonstrated that RNF8-dependent RNF168-mediated mono-ubiquitination of γ-H2AX on K13 and K15 required an adaptor protein X, which should be previously ubiquitinated by RNF8.17 ATM-dependent phosphorylation and RNF8-mediated ubiquitination of BCL10 prompted us to speculate that BCL10 could be the protein X. To test this hypothesis, we first examined if BCL10 physically associated with RNF168. Indeed, both endogenous (Fig. 5A) and epitope-tagged (Fig. 5B) BCL10 interacted with RNF168, and this interaction increased upon bleomycin treatment in 293T cells. The interaction between RNF168 and the phosphorylation-defective mutant BCL10(T91A) was greatly diminished, while the interaction between RNF168 and the ubiquitination-defective mutant BCL10(2KR) was slightly decreased (Fig. 5B). Second, depletion of BCL10 reduced binding of UBC13 to RNF168 (Fig. 5C). However, it was unexpected that inhibition of BCL10 expression did not compromise focus formation of RNF168 (Fig. S2D and S2E); depletion of RNF168, on the contrary, impaired focus formation of BCL10 upon etoposide treatment (Fig. 5D). Third, BCL10 failed to associate with the RNF168 mutant with deletion of its ubiquitin-binding domains (UBDs) (Fig. 5E). Fourth, inhibition of UBC13 expression reduced BCL10 ubiquitination and, thus, binding to RNF168 (Fig. S2A and S2B), co-transfection of wild-type HA-ubiquitin with BCL10 increased binding of BCL10 to RNF168, whereas this increase disappeared when BCL10 was co-transfected with a ubiquitin mutant UbΔG, in which the C-terminal glycines were removed, blocking Ub chain elongation (Fig. S2C). These results demonstrate that BCL10 is not the protein X, because it is not required for RNF168 recruitment to the DNA damage site. RNF8-mediated ubiquitination of BCL10 instead promotes its binding to and delivering E2s to RNF168.

Figure 5. Phosphorylated and ubiquitinated BCL10 is stabilized on the damage sites through binding to and presenting UBC13 to RNF168. (A) BCL10 interacted with RNF168. Total cell lysates were extracted from 293T cells with mock treatment or bleomycin treatment for 1 h and immunoprecipitated with an anti-RNF168 antibody. Immunoprecipitates were resolved by SDS-PAGE and probed with antibodies as indicated. (B) Both phosphorylation and ubiquitination of BCL10 promoted its interaction with RNF168. 293T cells were co-transfected with HA-RNF168 and FLAG-BCL10, FLAG-BCL10(T91A), or FLAG-BCL10(2KR). Total cell lysates were extracted 48 h after transfection and subjected to immunoprecipitation with an anti-HA antibody and immunoblotting with antibodies as indicated. (C) Depletion of BCL10 compromised the interaction between RNF168 and UBC13. BCL10-depleted 293T cells were co-transfected with HA-UBC13 and FLAG-RNF168. Total cell lysates were extracted 48 h after transfection and subjected to immunoprecipitation with an anti-HA antibody followed by immunoblotting with antibodies as indicated. (D) Depletion of RNF168 compromised etoposide-induced focus formation of BCL10. HeLa cells were transfected twice with si-RNF168 or si-CTR. Forty-eight hours after the second transfection, cells were treated with etoposide 1 h before they were fixed with 4% paraformaldehyde. Immunofluorescence staining with anti-BCL10 was performed as described in the Methods. The efficiency of RNF168 knockdown was determined by immunoblotting with antibodies as indicated. (E) UBDs of RNF168 were required for BCL10 binding. 293T cells were co-transfected with HA-BCL10 and FLAG-RNF168 or FLAG-RNF168ΔUBD. Total cell lysates were extracted 48 h after transfection and subjected to immunoprecipitation with an anti-FLAG antibody followed by immunoblotting with antibodies as indicated.
BCL10 promotes RNF8/RNF168-mediated ubiquitination in response to DNA damage
Both ATM-dependent phosphorylation and RNF8-mediated ubiquitination of BCL10 facilitate presentation of UBC13 to both RNF8 and RNF168 at the damage sites. We expected that BCL10 is required for RNF8/RNF168-mediated ubiquitination of downstream substrates in response to DNA damage. Indeed, knockdown of BCL10 compromised etoposide-induced FK2 foci (Fig. 6A) but not γ-H2AX foci (data not shown). FK2 antibody recognizes all types of mono- and poly-ubiquitinated proteins, but not free ubiquitin. H2AX is phosphorylated by ATM and ubiquitinated by RNF8/RNF168, providing a platform for the recruitment of repair factors. We found that bleomycin treatment increased RNF168-mediated mono-ubiquitination and subsequently RNF8/RNF168-mediated polyubiquitination of H2AX. Depletion of BCL10 compromised H2AX ubiquitination both under unperturbed conditions and under bleomycin treatment (Fig. 6B). These results demonstrate that BCL10 promotes RNF8/RNF168-mediated ubiquitination in DDR.
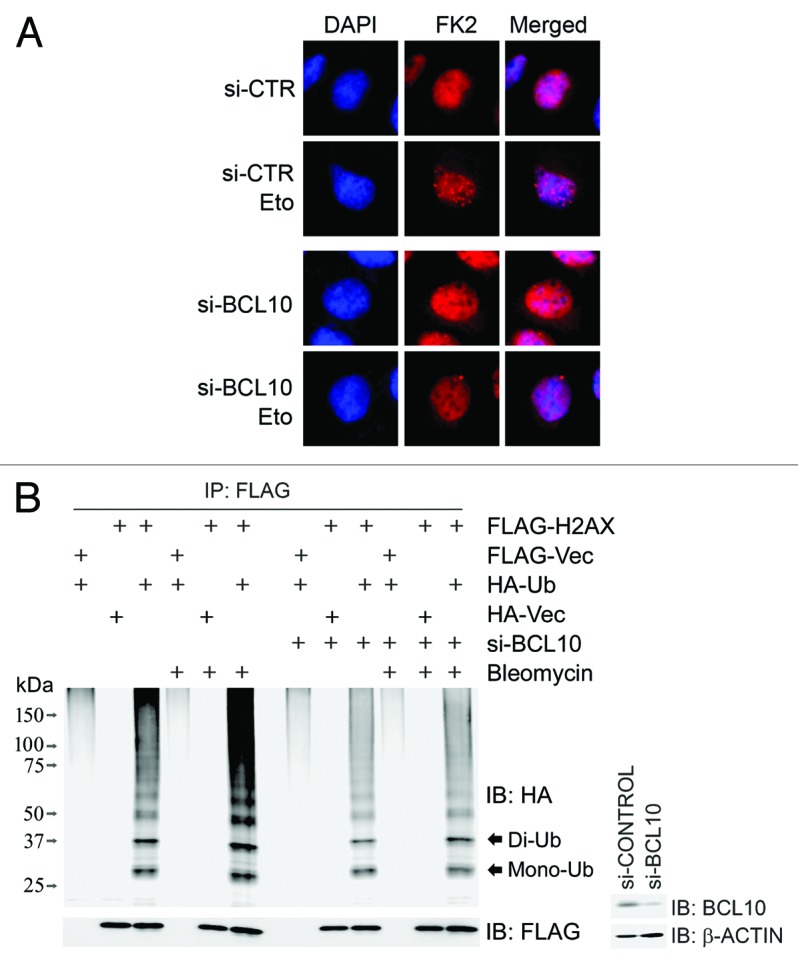
Figure 6. BCL10 promotes RNF8/RNF168-mediated ubiquitination in response to DNA damage. (A) Inhibition of BCL10 expression reduced DNA damage-induced FK2 foci. BCL10 or mock-depleted HeLa cells were treated with or without etoposide 1 h before fixed with 4% paraformaldehyde and subsequently immunostained with an anti-FK2 antibody. (B) Depletion of BCL10 compromised γ-H2AX mono- and poly-ubiquitination after DNA damage. BCL10-depleted or mock-depleted 293T cells were co-transfected with FLAG-H2AX and HA-Ub. Transfectants were treated 48 h after transfection with etoposide or DMSO for 1 h, and total cell lysates were extracted for immunoprecipitation with an anti-FLAG antibody and immunoblotting with antibodies as indicated. The efficiency of BCL10 knockdown was determined by immunoblotting with antibodies as indicated. Mono-Ub, mono-ubiquitinated H2AX; di-Ub: di-ubiquitinated H2AX.
BCL10 is required for recruitment of HR machinery onto the DNA damage sites
The BRCA1-A complex subunit RAP80 is a ubiquitin-binding protein that specifically recognizes and binds K63-linked ubiquitinated proteins, such as histones H2A and H2AX at DNA lesions, targeting the BRCA1-A complex and BRCA1-dependent HR factors (e.g., RAD51) to the DSB sites.35,36 Given that BCL10-dependent assembly of the RNF8/RNF168 ligase complex is essential for K63-linked ubiquitination of H2AX, we reasoned that BCL10 is required for the recruitment of HR machinery, including RAP80 and BRCA1, to the DNA damage sites. When BCL10 expression in U2OS cells was inhibited by siRNA (Fig. 7A), etoposide-induced focus formation of RAP80 (Fig. 7B and C) and BRCA1 (Fig. 7D and E) was impaired, suggesting a defect in the recruitment of these HR factors to the DNA damage sites. To eliminate the off-target effects of BCL10 siRNA, we generated a siRNA-resistant expression construct of BCL10, namely BCL10res, which, when expressed, was not targeted by the BCL10 siRNA (Fig. 7A). When expression of endogenous BCL10 was inhibited by siRNA in U2OS cells, expression of BCL10res efficiently restored the etoposide-induced focus formation of RAP80 (Fig. 7B and C) and BRCA1 (Fig. 7D and E), whereas expression of BCL10(T91A)res or BCL10(2KR)res failed to restore etoposide-induced BRCA1 foci (Fig. S3A and S3B). These results demonstrate that BCL10 is required for efficient recruitment of HR factors RAP80 and BRCA1 to the DSB sites.

Figure 7. BCL10 promotes recruitment of HR machinery to the DNA damage site. (A) U2OS cells were stably transfected with HA-BCL10res or HA-Vec. HA-Vec transfectants were further transfected with si-CTR or si-BCL10, while HA-BCL10res transfectants were further transfected si-BCL10. Total cell lysates were extracted for immunoblotting with antibodies as indicated. (B and C) Depletion of BCL10 expression compromised focus formation of RAP80. Transfectants described in (A) were treated with etoposide 1 h before fixed with 4% paraformaldehyde and subsequently immunostained with an anti-RAP80 antibody (B). Percentage of cells with > = 5 foci per cell was shown in (C). (D and E) Depletion of BCL10 expression compromised focus formation of BRCA1. Experiments were performed as described in (B and C), except that antibody used for immunostaining was specific for BRCA1. Percentage of cells with ≥ 5 foci per cell was shown in (D). (F and G) Depletion of BCL10 expression impaired recruitment of RAD51 onto the DNA damage sites. Experiments were performed as described in (A–C), except that antibody for immunostaining was specific for RAD51. The quantification of the percentage of cells with ≥ 5 foci was shown in (G).
BCL10 promotes HR-mediated DSB repair
As BCL10 promoted recruitment of HR-mediated repair factors such as BRCA1 and RAP80 to the DNA damage site (Fig. 7), we reasoned that BCL10 might play a role in the HR-mediated DSB repair. When the expression of BCL10 was inhibited by siRNA in U2OS cells (Fig. 7A), the etoposide-induced focus formation of RAD51 was diminished (Fig. 7F), and this defect was rescued upon expression of BCL10res (Fig. 7F and G), whereas expression of BCL10(T91A)res or BCL10(2KR)res failed to restore etoposide-induced RAD51 foci (Fig. S3C and S3D). To directly examine the role of BCL10 in DSB repair, we used the GFP-based chromosomal reporters DR-GFP in DRU2OS cells and EJ5-GFP in U2OS cells to measure HR and total NHEJ efficiency, respectively. When BCL10 expression was inhibited by siRNA, both HR- and NHEJ-mediated DSB repair was compromised (Fig. 8A; Fig. S4).
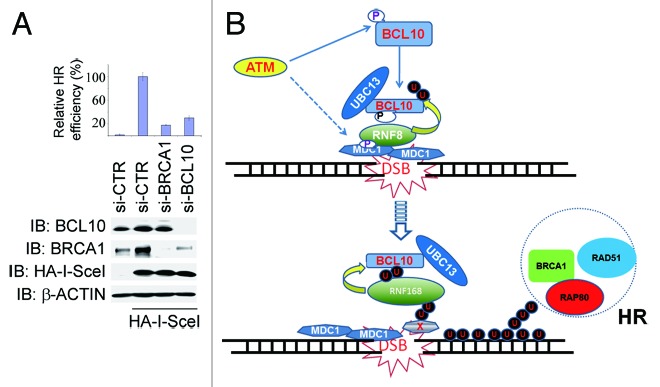
Figure 8. BCL10 promotes HR-mediated DSB repair. (A) Depletion of BCL10 expression compromised HR-mediated DSB repair efficiency. Mock, BRCA1, or BCL10-depleted DRGFP U2OS cells were transfected with HA-I-SceI. Total cell lysates were extracted 48 h after transfection for immunoblotting with antibodies as indicated, cells prepared in parallel were harvested for flow cytometry analysis, and percentage of GFP-positive cells representing HR-mediated DSB repair efficiency was determined. Histogram showed the relative HR efficiency. Experiments were performed in triplicates, and error bars indicated standard deviation. (B) A proposed working model of BCL10 in response to DNA damage. Details are described in “Discussion”.
BCL10 constitutively associates with MALT1 and forms a complex with CARMA1, namely CBM, facilitating the activation of the NFκB pathway in the cytosol.30,37 We sought to determine if BCL10 in the nucleus requires MALT1 to execute its function in DSB signaling and repair. We found that BCL10, but not RNF8, was present in the MALT1 immunocomplex (Fig. S5A), whereas BCL10, but not MALT1, was present in the RNF8 immunocomplex (Fig. S5B). This suggests that the BCL10-RNF8-containing complex is likely independent of the BCL10–MALT1 complex. Furthermore, we found that inhibition of MALT1 expression did not affect the etoposide-induced focus formation of BCL10, RAP80, BRCA1, or RAD51 (Fig. S5C), and inhibition of MALT1 expression (Fig. S5D) did not have an impact on HR-mediated DSB repair (Fig. S5E). These results suggest that BCL10’s function in DDR is independent of the CBM complex.
Discussion
Our study uncovers a novel function of BCL10 independent of the CBM complex and sheds light on the molecular details on how BCL10 regulates RNF8/RNF168-mediated ubiquitination through presenting UBC13 to RNF8/RNF68 in DSB signaling and repair (Fig. 8B). Upon DNA damage, ATM phosphorylates the residue T91 of BCL10, promoting binding of BCL10 to RNF8 and simultaneously presenting UBC13 to RNF8. The RNF8/UBC13-containing complex, in turn, ubiquitinates BCL10. Ubiquitinated BCL10 thereafter binds to the UBDs of RNF168 and presents UBC13, and potentially other E2s, to RNF168. This confers E3 ligase activity of RNF168 for mono-ubiquitination on γ-H2AX and then the RNF8/RNF168 ligase activity for poly-ubiquitination on γ-H2AX. Fully ubiquitinated γ-H2AX decorates the chromatin region surrounding the DSB site, serving as a platform for further depositing other factors required for DSB signaling and repair. This model predicts that BCL10 is required for enrichment of HR machinery and efficient HR-based DSB repair, and indeed this is the case (Figs. 7 and 8). We show that histone ubiquitination during the DSB pathway is initiated by RNF8 on BCL10, and both phosphorylated and ubiquitinated BCL10 delivers UBC13 to RNF168 to activate RNF168. This finding is further supported by the structure-based evidence that RNF168 does not stably associate with UBC13. BCL10 provides a docking mechanism for presenting UBC13 to RNF8 first, and RNF168 subsequently. However, the molecular details how phosphorylated and ubiquitinated BCL10 is released from RNF8 and presents E2s to RNF168 warrant further investigation.
Recently it has been proposed that an unknown substrate X of RNF8 binds to RNF168; this binding is necessary for localization of RNF168 to the DNA damage site and subsequent H2AX mono-ubiquitination.17 Apparently BCL10 is not the X protein required for RNF168 recruitment onto DNA damage sites; instead, BCL10 ubiquitination by RNF8 may promote binding of BCL10 to RNF168 and further stabilize BCL10 in the DNA damage site. Furthermore, BCL10 is required for RNF168 activity for delivering UBC13 and potentially other E2s. Therefore, RNF168 function in DDR requires the X protein for recruitment to the DNA damage site and BCL10 for supplying E2s.
NFκB signaling pathway, together with the cell cycle checkpoints and DNA repair, is activated in response to DNA damage.38,39 However, signaling cascades leading to DNA damage-induced NFκB activation are poorly understood.40 It is believed that DNA damaging agents cause ATM activation via induction of DSB and PIASy (protein inhibitor of activated STAT y)-dependent SUMOylation of NEMO. ATM subsequently phosphorylates NEMO, which results in cIAP1 (cellular inhibitor of apoptosis protein-1)-dependent monoubiquitination of NEMO. NEMO monoubiquitination is critical for NEMO export from nucleus to cytosol.39,40 To a certain degree, post-translational modifications of BCL10 are similar to those of NEMO. BCL10 is phosphorylated on T91 by ATM in response to DNA damage, and T91 phosphorylation and/or T91-dependend phosphorylation takes up the majority of BCL10 phosphorylation (Fig. 2C). Furthermore, phosphorylation of BCL10 on T91 is essential for its localization on chromatin, since BCL10(T91A) mutant could not be detected on the chromatin-enriched fraction, and ubiquitination-defective mutant BCL10(2KR) cannot efficiently be retained on the chromatin-bound fraction (data not shown).
BCL10 in the form of CBM complex in the cytosol promotes NEMO ubiquitination by TRAF6, leading to NFκB activation.26 However, BCL10 function in DDR mainly occurs on chromatin. MALT1, a constitutive partner of BCL10, apparently is not important in DDR, because depletion of MALT1 expression did not have an obvious impact on recruitment of DSB repair factors onto the damage sites and HR-mediated DSB repair (Fig. S5). Therefore, it would be very intriguing to find out if: (1) ATM-dependent phosphorylation of BCL10 modulates NEMO mono-ubiquitination and subsequent translocation from nucleus to cytosol; (2) BCL10 shuttles between nucleus and cytosol, and this shuttling is dependent on its post-translational modifications; and (3) BCL10 nuclear export, along with mono-ubiquitinated NEMO export, is required for DNA damage-induced NFκB activation. If indeed BCL10 shuttles between cytosol and nucleus, apparently ATM-dependent phosphorylation is not beneficial to BCL10 shuttling, because BCL10(T91A) was not present in the chromatin-enriched fraction (data not shown), suggesting that different PTMs may be required for this process.
It is not surprising that depletion of BCL10 compromises NHEJ-mediated DSB repair as well. It has been reported that RNF8 regulates NHEJ through modulating K48-chained KU80 ubiquitination and stability.41 UBCH8 couples with RNF8 to promote K48-linked ubiquitination;18 this warrants further investigation if BCL10 is a mediator to deliver UBCH8 to RNF8 and enables its ligase activity.
HERC2 (HECT domain and RCC1-like domain-containing protein 2) acts as another mediator presenting UBC13 to RNF8. It is recruited to sites of DNA damage in response to ionizing radiation and facilitates the assembly of UBC13 and RNF8, promoting DNA damage-induced formation of K63-linked ubiquitin chains, regulating ubiquitin-dependent retention of repair proteins on damaged chromosomes. HERC2 is also involved in the maintenance of RNF168 levels.19,42 We found that depletion of BCL10 did not have an impact on the protein levels of RNF168 (unpublished observation). The interaction between BCL10 and UBC13 does not change upon treatment with different DNA damaging agents, and is not modulated by T91 phosphorylation (data not shown). This suggests that BCL10 may constitutively associate with UBC13. However, it is not clear if and how HERC2 interacts with UBC13. Thus, it would be of great interest to reveal the functional interplay between BCL10 and HERC2 in modulating DNA damage-induced RNF8-mediated ubiquitination.
A chromosomal aberration involving BCL10 is recurrent in low-grade MALT lymphoma. MALT lymphoma takes up about 7–8% of all B-cell lymphomas. There are 2 translocations specifically associated with MALT lymphoma, namely, t(1;14)(p22;q32) and t(14;18)(q32;q21), leading to upregulation of BCL10 and MALT1 gene expression, respectively.33 The well-known function of BCL10 is to play a central role in the stimulation of immune responses triggered by T/B-cell antigen receptors. It executes these functions mainly in complex with MALT1 and CARMA1 in the cytosol.29,30,37 However, presence of BCL10 in the nucleus and its importance have long been underscored. It has been reported that BCL10 nuclear expression in both primary cutaneous marginal zone B-cell lymphomas and oral squamous cell carcinomas correlates with a significant decrease of patient survival.43,44 We now report that BCL10 modulates RNF8/RNF168-mediated ubiquitination and subsequent HR-mediated DSB repair. It is reasonable to speculate that BCL10 expression may enhance DSB repair capacity and thus confer resistance to DNA damage-based chemotherapeutic agents, exhibiting poor overall survival in patients.
Materials and Methods
Reagents, antibodies, and cell lines
The ATM inhibitor KU55933 (a final concentration of 10 μM was used throughout this research) was purchased from Selleck, and the DNA-PKcs inhibitor NU7026 (a final concentration of 10 μM was used), the ATR inhibitor NU6027 (a final concentration of 10 μM was used), and etoposide (a final concentration of 10 μM was used) were purchased from Sigma.
Rabbit polyclonal antibodies anti-BCL10, anti-MALT1, anti-DNA-PKs, anti-BRCA1, anti-MYC, anti-HA, and anti-RAP80 were from Bethyl; mouse monoclonal antibodies anti-γ-H2AX, rabbit polyclonal antibodies anti-RNF168 (ABE367) for IF, anti-RNF168 (06-1130) for IP, and anti-ubiquitin (FK2) were from Millipore; rabbit monoclonal antibody anti-GST (A00865) was from GenScript; mouse monoclonal antibody anti-FLAG (M2) and rabbit polyclonal antibody anti-BCL10 (MK-17 for IF) were from Sigma; mouse monoclonal antibody anti-BRCA1 (D-9) and anti-RNF168 (B-11) for IB were from Santa Cruz. Rabbit polyclonal anti-MDC1 antibody was described before.11 Rabbit polyclonal antibodies against RNF8, RAD51, and UBC13 were gifts from Drs Michael SY Huen, Jun Huang, and Wei Xiao, respectively. The rabbit polyclonal anti-pT91-BCL10 antibody was raised against the phospho-peptide IRREK(pT)QNFLI and affinity-purified (AbMart).
All cell lines were cultured in high-glucose Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C.
Plasmids
Human cDNA clones encoding BCL10, RNF8, RNF168, H2AX, Ubiquitin (Ub), and UBC13 were all generated by PCR from HeLa cells and subcloned into the mammalian expression vector pcDNA3.0 with an N-terminal epitope of 3 copies of HA or FLAG. The rescue expression constructs that were siRNA-resistant contained 4–6 silent point mutations within the siRNA targeting sites. MBP-RNF8 was a kind gift from Dr Michael SY Huen. BCL10 coding region was subcloned from pcDNA-HA-BCL10 into pGEX-4T-1 for producing GST-BCL10 in E. coli. The phosphorylation-defective mutant BCL10(T91A), the ubiquitination-defective mutant BCL10(2KR), the Ub mutants (K63-linked only mutant, K48-linked only mutant, and the chain elongation-defective mutant UbΔG, in which the 2 C-terminal glycine residues were removed) were generated using Quick Change Site Directed Mutagenesis Kit (Genetech). All the constructs were confirmed by DNA sequencing.
siRNA transfections
The mixture of 4 predesigned OnTarget plus siRNA oligonucleotide duplexes specific for ATM, DNA-PKs, BRCA1, and RNF8 were purchased from Dharmacon. The siRNA oligonucleotide duplexes against BCL10 (siRNA sequence: CACAGAACTT CCTGATACA), UBC13 (siRNA sequence: GGCTATATGC CATGAATAA), RNF168 mixture (siRNA#1: CTTTAAAGAT GCAGTTGAA; siRNA#2: GTGGAACTGT GGACGATAA; siRNA#3: GGAACTGAGA AGAGAATAT), and MALT1 mixture (siRNA#1: CCTCGGCAAA CCTGTCTAA; siRNA#2: GGAAGTGAAT GTTGGGAAA; siRNA#3: CAAACAGGCT CTAGAGATT) were purchased from RiboBio (Guangzhou RiboBio Co, Ltd). The gene-specific siRNA or the non-target siRNA control (si-CTR) was transfected into cells at a final concentration of 20 nM using RNAimax (Invitrogen) according to the manufacturer’s instructions.
Chromatin fractionation
Chromatin fractionation was performed essentially as described.45 Briefly, 3 × 106 cells were washed with PBS and resuspended in 200 μl of buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 Mm MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, and protease inhibitor mixture [Roche Molecular Biochemicals]). Triton X-100 was added to a final concentration of 0.1%, and the cells were incubated for 5 min on ice. Nuclei were collected in the pellet (P1) by low speed centrifugation (1500 g, 5 min, 4 °C). The supernatant (S1) was further clarified by high-speed centrifugation (13 000 g, 2 min, 4 °C) to remove cell debris and insoluble aggregates, resulting in the cytosol fraction S2. Nuclei were washed once with buffer A without Triton then lysed in 200 μl of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease inhibitor mixture). After a 10-min incubation on ice, soluble nuclear proteins (S3) were separated from chromatin by centrifugation (2000 g, 5 min). Isolated chromatin (P3) was washed once with buffer B and spun down at high speed (13 000 g, 1 min).
Immunoprecipitation and immunofluorescence
For immunoprecipitation assays, total cell lysates were prepared in NETN lysis buffer (100 mM NaCl, 20 mM Tris-Cl [pH 8.0], 0.5 mM EDTA, 0.5% [v/v] Nonidet P-40 [NP-40], 1 × cocktail protease inhibitor) and incubated with a desired antibody and appropriate protein A/G-agarose beads at 4 °C overnight with gentle agitation. Beads were washed 3 times with lysis buffer lacking protease inhibitor, and immunocomplexes were eluted by boiling in SDS sample buffer for 5 min. For calf intestine phosphatase (CIP) treatment, the immunocomplex were mock-treated or treated with CIP at 30 °C for 1 h. For immunofluorescence staining with anti-FK2, RAP80, BRCA1, and RAD51, cells cultured on coverslips were washed once with PBS, incubated in 4% paraformaldehyde for 12 min, and permeabilized in 0.5% Triton solution for 5 min at room temperature. Fixed cells were blocked with 2% BSA at room temperature for 30 min and incubated with primary antibody for 60 min, then washed 3 times with PBS and incubated with secondary antibody for 30 min. After washing with PBS 3 times, cells were stained with DAPI for 2 min to visualize nuclear DNA. The coverslips were mounted onto glass slides with anti-fade solution and visualized using a fluorescence microscope. For immunofluorescence staining with anti-BCL10, similar procedures were used, except that cells were permeabilized in cytoskeleton buffer (10 mM PIPES, pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100 [v/v]) at room temperature for 5 min.
In vivo ubiquitination assays
For in vivo ubiquitination assays for H2AX, mock, or BCL10-depleted 293T cells were co-transfected with FLAG-H2AX and HA-Ub, transfectants were pelleted 48 h after transfection, washed in PBS once, resuspended, and sonicated in denaturing buffer (20 mM Tris, pH 7.5; 50 mM NaCl, 0.5% NP-40; 0.5% sodium deoxycholate; 0.5% SDS; 1 mM EDTA) containing protease inhibitors. Cell extracts were subjected to immunoprecipitation with anti-FLAG agarose beads (Sigma) under denaturing conditions and immunoblotting with antibodies as indicated.14
In vitro kinase assays
For ATM kinase assay, total cell lysates were prepared in TGN buffer (50 mM Tris, pH 7.5; 50 mM glycerophosphate; 150 mM NaCl; 10% glycerol; 1% Tween-20; 1 mM NaVO4; 1 mM DTT; 1× cocktail protease inhibitor) from 293T cells treated with etoposide for 1 h and immunoprecipitated with an anti-ATM antibody. The immune-complex was washed with TGN buffer twice, LiCL buffer (50 mM TRIS-HCl buffer, pH 7.5, containing 0.5 M LiCl) twice, and ATM kinase buffer (10 mM HEPES, pH 7.5; 50 mM Glycerophosphate; 50 mM NaCl; 10 mM MgCl2; 10 mM MnCl2; 5 μM ATP; and 1 mM DTT) twice.46 The beads were equally split into 2 parts and resuspended in 30 μl of kinase buffer; ATM inhibitor KU55933 was added to one part 1 h before kinase assay. Two micrograms of GST-BCL10, GST-BCL10(T91A), or GST-CHK2(D368A) and 10 μCi 32P-ATP were added and incubated at 30 °C for 30 min. The reaction was stopped by adding 30 μl of SDS sample buffer and boiled for 5 min. Samples were resolved in SDS-PAGE and radioactive signal was captured by autoradiography.
HR- or NHEJ-mediated DSB repair assay
HR-mediated double-strand break (DSB) repair assay was performed essentially as described.47 Briefly, DRGFP U2OS cells with a single copy of DR-GFP were transfected twice with si-CTR or gene-specific siRNA, and further transfected with HA-I-SceI 24 h after second siRNA transfection. Cells were harvested 72 h after HA-I-SceI transfection and subjected to flow cytometry analysis. The percentage of GFP-positive cells, which indicated HR-mediated DSB repair efficiency, was determined. The mean values were obtained from 3 independent experiments.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Eric W McIntush from the Bethyl Laboratories for antibodies against BCL10, Dr Jun Huang from Zhejiang University for RAD51 antibody, Dr Wei Xiao from Capital Normal University for UBC13 antibody, and Dr Michael Huen from Hongkong University for RNF8 antibody and MBP-RNF8 expression construct. We thank other members of the Xu laboratory for help. This work was supported by the National Natural Science Foundation of China (31130017 and 31071190), the 973 projects2013CB911002 and 2010CB911904, Funding Project for Academic Human Resources Development in Institutions of Higher Learning under the Jurisdiction of Beijing Municipality (PHR20110508), and Research Fund for the Doctoral Program of Higher Education of China (20101108110002) to X.X.
References
- 1.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) 2010;9:1219–28. doi: 10.1016/j.dnarep.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Friesner JD, Liu B, Culligan K, Britt AB. Ionizing radiation-dependent gamma-H2AX focus formation requires ataxia telangiectasia mutated and ataxia telangiectasia mutated and Rad3-related. Mol Biol Cell. 2005;16:2566–76. doi: 10.1091/mbc.E04-10-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 10.Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008;9:795–801. doi: 10.1038/embor.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Luo S, Zhao H, Liao J, Li J, Yang C, Xu B, Stern DF, Xu X, Ye K. Structural mechanism of the phosphorylation-dependent dimerization of the MDC1 forkhead-associated domain. Nucleic Acids Res. 2012;40:3898–912. doi: 10.1093/nar/gkr1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jungmichel S, Clapperton JA, Lloyd J, Hari FJ, Spycher C, Pavic L, Li J, Haire LF, Bonalli M, Larsen DH, et al. The molecular basis of ATM-dependent dimerization of the Mdc1 DNA damage checkpoint mediator. Nucleic Acids Res. 2012;40:3913–28. doi: 10.1093/nar/gkr1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 15.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A. 2007;104:20759–63. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–95. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Lok GT, Sy SM, Dong SS, Ching YP, Tsao SW, Thomson TM, Huen MS. Differential regulation of RNF8-mediated Lys48- and Lys63-based poly-ubiquitylation. Nucleic Acids Res. 2012;40:196–205. doi: 10.1093/nar/gkr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Lukas C, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nature cell biology 2010; 12:80-6; sup pp 1-12. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Feng W, Jiang J, Deng Y, Huen MS. Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J Biol Chem. 2012;287:27715–22. doi: 10.1074/jbc.M112.373530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, Mailand N. Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J Cell Biol. 2012;197:189–99. doi: 10.1083/jcb.201109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat Struct Mol Biol. 2006;13:915–20. doi: 10.1038/nsmb1148. [DOI] [PubMed] [Google Scholar]
- 23.McKenna S, Moraes T, Pastushok L, Ptak C, Xiao W, Spyracopoulos L, Ellison MJ. An NMR-based model of the ubiquitin-bound human ubiquitin conjugation complex Mms2.Ubc13. The structural basis for lysine 63 chain catalysis. J Biol Chem. 2003;278:13151–8. doi: 10.1074/jbc.M212353200. [DOI] [PubMed] [Google Scholar]
- 24.Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 25.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 26.Zhou H, Wertz I, O’Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature. 2004;427:167–71. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 27.Fukushima T, Matsuzawa S, Kress CL, Bruey JM, Krajewska M, Lefebvre S, Zapata JM, Ronai Z, Reed JC. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci U S A. 2007;104:6371–6. doi: 10.1073/pnas.0700548104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61. doi: 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 29.Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2010;2:a003004. doi: 10.1101/cshperspect.a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–59. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 31.Rueda D, Thome M. Phosphorylation of CARMA1: the link(er) to NF-kappaB activation. Immunity. 2005;23:551–3. doi: 10.1016/j.immuni.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Thome M, Weil R. Post-translational modifications regulate distinct functions of CARMA1 and BCL10. Trends Immunol. 2007;28:281–8. doi: 10.1016/j.it.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Willis TGJD, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska I, et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell. 1999;96:35–45. doi: 10.1016/S0092-8674(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 34.Wu CJ, Ashwell JD. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:3023–8. doi: 10.1073/pnas.0712313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci U S A. 2009;106:3166–71. doi: 10.1073/pnas.0807485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007;67:6647–56. doi: 10.1158/0008-5472.CAN-07-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X, Wang D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin Immunol. 2004;16:429–35. doi: 10.1016/j.smim.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 38.Wu ZH, Miyamoto S. Many faces of NF-kappaB signaling induced by genotoxic stress. J Mol Med (Berl) 2007;85:1187–202. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- 39.Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ. 2006;13:773–84. doi: 10.1038/sj.cdd.4401843. [DOI] [PubMed] [Google Scholar]
- 40.Miyamoto S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–30. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng L, Chen J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat Struct Mol Biol. 2012;19:201–6. doi: 10.1038/nsmb.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danielsen JR, Povlsen LK, Villumsen BH, Streicher W, Nilsson J, Wikström M, Bekker-Jensen S, Mailand N. DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J Cell Biol. 2012;197:179–87. doi: 10.1083/jcb.201106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gallardo F, Bellosillo B, Espinet B, Pujol RM, Estrach T, Servitje O, Romagosa V, Barranco C, Boluda S, García M, et al. Aberrant nuclear BCL10 expression and lack of t(11;18)(q21;q21) in primary cutaneous marginal zone B-cell lymphoma. Hum Pathol. 2006;37:867–73. doi: 10.1016/j.humpath.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Chang HH, Kuo MY, Cheng SJ, Chiang CP. Expression of BCL10 is significantly associated with the progression and prognosis of oral squamous cell carcinomas in Taiwan. Oral Oncol. 2009;45:589–93. doi: 10.1016/j.oraloncology.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Xu X, Stern DF. NFBD1/KIAA0170 is a chromatin-associated protein involved in DNA damage signaling pathways. J Biol Chem. 2003;278:8795–803. doi: 10.1074/jbc.M211392200. [DOI] [PubMed] [Google Scholar]
- 46.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 47.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.