

Abstract
Developing anti-viral therapies targeting HIV-1 transcription has been hampered by the limited structural knowledge of the proteins involved. HIV-1 hijacks the cellular machinery that controls RNA polymerase II elongation through an interaction of HIV-1 Tat with the positive transcription elongation factor P-TEFb, which interacts with an AF4 family member (AFF1/2/3/4) in the super elongation complex (SEC). Because inclusion of Tat•P-TEFb into the SEC is critical for HIV transcription, we have determined the crystal structure of the Tat•AFF4•P-TEFb complex containing HIV-1 Tat (residues 1–48), human Cyclin T1 (1–266), human Cdk9 (7–332), and human AFF4 (27–69). Tat binding to AFF4•P-TEFb causes concerted structural changes in AFF4 via a shift of helix H5′ of Cyclin T1 and the α-310 helix of AFF4. The interaction between Tat and AFF4 provides structural constraints that explain tolerated Tat mutations. Analysis of the Tat-binding surface of AFF4 coupled with modeling of all other AF4 family members suggests that AFF1 and AFF4 would be preferred over AFF2 or AFF3 for interaction with Tat•P-TEFb. The structure establishes that the Tat-TAR recognition motif (TRM) in Cyclin T1 interacts with both Tat and AFF4, leading to the exposure of arginine side chains for binding to TAR RNA. Furthermore, modeling of Tat Lys28 acetylation suggests that the acetyl group would be in a favorable position for H-bond formation with Asn257 of TRM, thereby stabilizing the TRM in Cyclin T1, and provides a structural basis for the modulation of TAR RNA binding by acetylation of Tat Lys28.
Keywords: P-TEFb, AFF4, HIV, Tat, crystal structure
Introduction
The 2010 UNAIDS AIDS Epidemic Update estimated over 33 million people were infected with the human immunodeficiency virus (HIV-1) and, unfortunately, most of the 2 to 3 million newly infected individuals each year have a dismal prognosis.1 Approximately 1.5 million United States citizens are currently infected with the virus and about 500 000 have died of acquired immunodeficiency syndrome (AIDS) caused by HIV-1. The death rate from AIDS in the United States has declined due to tremendous progress made in developing anti-HIV drugs. A few of these drugs block viral entry, but most block 1 of the 3 virally encoded enzymes; reverse transcriptase, protease, or integrase.2 To date no effective vaccines have been developed and there is no practical cure.1 Removal of the antiviral treatment leads to rapid re-establishment of active viral infection due to latently infected cells that become activated and release virus.3 Patients whose viral titers are held to a very low level by current drug regimens can still infect others and suffer from ailments caused by the remaining viruses or the drugs themselves.4 Another serious problem for a number of infected individuals is that a number of strains of HIV have become resistant to the current cadre of anti-HIV drugs.5
HIV transcription is the most promising stage of the viral life cycle inhibition for which no drugs exist. Not only is transcription required for viral gene expression, but the RNA produced is the genetic material packaged into propagating virions. Because the viral Tat protein is the major transactivator of HIV transcription6 and is essential for viral replication,7 it is the most likely target for drugs that would specifically block HIV transcription. Importantly, compared with the current drugs, blocking the function of Tat would halt viral replication at a very early stage and stop production of viral particles in infected individuals. Thus, it would eliminate the toxicity of the viral particles and block the transmission of the virus. In fact, if an effective inhibitor of HIV transcription was developed it could be considered a functional cure for AIDS.4
HIV Tat hijacks the machinery that controls RNA polymerase II elongation which plays an important role in regulating much of cellular transcription.8-10 The positive transcription elongation factor P-TEFb plays the central role in this process by causing promoter proximal paused RNA polymerase II to enter productive elongation.11 HIV Tat interacts with P-TEFb comprised of Cdk9 and Cyclin T1.12,13 The RNA binding domain of Tat mediates recruitment of the Tat•P-TEFb complex to the nascent HIV transcript, TAR, leading to activation of the HIV LTR and robust HIV gene expression.13 Tat can also extract P-TEFb from the 7SK snRNP which normally functions to sequester P-TEFb in an inactive conformation until the kinase activity of the factor is needed.14-16 Tat can bind to 7SK RNA15,16 and when overexpressed in cells can be found associated with the snRNP.17 P-TEFb is also a component of the super elongation complex (SEC) comprised of an AF4 family member AFF1 or AFF4, AF9/ENL, ELL1/2/3, and EAF1/2.18-22 Tat has also been found in the SEC presumably through its interaction with P-TEFb.17,23-25
Although important aspects of the function of Tat in recruiting P-TEFb to TAR remain to be determined, the interaction between Tat, P-TEFb, and the SEC is critical.6 A structure of HIV-1 Tat bound to P-TEFb demonstrated that Tat interacts mainly with Cyclin T1 and forms an extensive interface burying 3500 Å2 of surface area.26 The interaction of P-TEFb with the SEC is mediated by the N-terminal domain of AFF1/4 referred to as Cyclin T1 binding site (CBS)23,27 and the Alber group has solved a structure of P-TEFb bound to the CBS of AFF4.28 In silico superimposition of the 2 structures suggested that both Tat and AFF4 might be able to associate with P-TEFb at the same time.28 We describe here a structure obtained from the crystallization of a complex formed by HIV Tat, and human AFF4 and P-TEFb. The Tat•AFF4•P-TEFb complex reveals extensive interactions between AFF4 and Tat that could provide a target for development of compounds that could disrupt the complex and thereby inhibit HIV transcription.
Results
Crystal structure determination
For the crystallization of a Tat•AFF4•P-TEFb complex we produced a large variety of Cyclin T1, Cdk9, Tat and AFF4 constructs with different N-terminal and C-terminal truncations co-expressed in baculovirus infected insect cells. In our previously reported structure of the Tat•P-TEFb complex26 residues 252–261 of Cyclin T1 were difficult to assign unambiguously due to potential competition between Cys261 and the histidines from the C-terminal His6-tag on Cdk9 for 1 of the 2 bound zinc atoms. To avoid this problem we fused the His6-tag to the N-terminus of Cyclin T1 with a cleavage site and removed the tag during purification. The Tat•AFF4•P-TEFb complex comprised of HIV-1 Tat (1–48), human Cyclin T1 (1–266), human Cdk9 (7–332), and human AFF4 (27–69) produced the best initial crystals in the form of tiny needles (Fig. S1A). Gradual variation of crystallization conditions only marginally improved the size and shape of crystals. However, addition of yttrium ions changed the crystal morphology to well-diffracting plate crystals (Fig. S1B) that contained equal molar amounts of the 4 proteins (Fig. S1C). Diffraction dataset was collected at the Argonne National Laboratory Advanced Photon Source. The structure was solved using molecular replacement and refined at 2.9 Å resolution to an Rfree of 26.7%. Inspection of crystal packing revealed that yttrium ions included in crystallization conditions mediated intermolecular interactions (Fig. S2). The crystal contains 2 independent Tat•AFF4•P-TEFb molecules in an asymmetric unit with rmsd of 0.69 Å for 652 matching α-carbons. Final refinement statistics are provided in Table 1.
Table 1. Data collection and refinement statistics.
| Data collection | |
|---|---|
| Space group | C2 |
| Cell dimensions: | |
| a (Å) | 166.862 |
| b (Å) | 186.739 |
| c (Å) | 108.661 |
| β (°) | 120.24 |
| Resolution (Å)* | 50–2.8 (2.85–2.80) |
| Unique reflections | 67551 |
| Rmerge (%)* | 8.3 (45.3) |
| I /σ(I) | 18.1 (2.2) |
| Completeness (%) | 95.0 (87.1) |
| Redundancy | 2.9 (2.1) |
| Temperature (K) | 100 |
| Mosaicity (°) | 0.22–0.49 |
| Refinement | |
| Resolution (Å) | 41.9–2.9 (3.08–2.9) |
| No. reflections | 60890 |
| Rwork / Rfree | 0.225/0.267 |
| No. atoms/B-factors (Å2) | |
| Protein | 10758/58.2 |
| Ions | 40/60.3 |
| Water | 100/27.6 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.4 |
| Ramachandran plot | |
| Favored (%) | 81.1 |
| Allowed (%) | 18.0 |
| Generously allowed (%) | 0.5 |
| Disallowed | 0.4 |
Values in parentheses are for the last shell.
Architecture of Tat•AFF4•P-TEFb complex
The Tat•AFF4•P-TEFb complex provides the link between promoter proximal paused Pol II on the HIV LTR and the SEC. Although the order of addition of Tat, P-TEFb, the SEC and the PAF1 complex to the elongation complex is not precisely known, the connectivity of the factors is illustrated in Figure 1A. The Tat•AFF4•P-TEFb complex is at the nexus. The 2 domains of Tat bridge from the nascent transcript (TAR) to P-TEFb associated with AFF4 in the SEC. The PAF1 complex contacts both Pol II and the SEC. The overall functional consequence of the Tat•AFF4•P-TEFb interaction is the recruitment of SEC to Pol II specifically engaged in transcription on the HIV-LTR. This leads to strong activation of transcription and productive HIV infection.
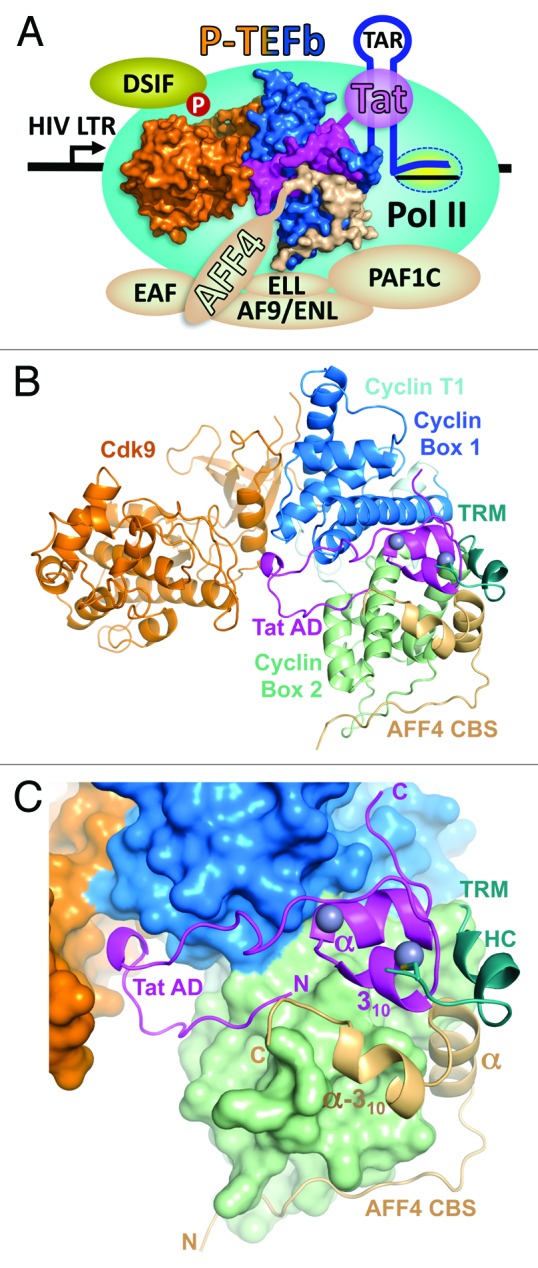
Figure 1. Overall view of Tat•AFF4•P-TEFb complex. (A) Functional context of the Tat•AFF4•P-TEFb complex. P-TEFb comprised of Cdk9 (orange) and Cyclin T1 (blue) provides a platform for the docking of HIV-1 Tat (magenta) and the AFF4 subunit (beige) of the super elongation complex (SEC). The SEC has 1 of each of these proteins: AFF1/4, EAF1/2, ELL1/2/3, and AF9/ENL. The nascent transcript (TAR) synthesized by Pol II engaged in the HIV LTR interacts with the RNA binding domain of HIV-1 viral protein Tat. Tat binds to Cyclin T1 and the N-terminus of the AFF4 subunit of the SEC which interacts with the PAF1 complex (PAF1C). PAF1C associates with Pol II further stabilizing the interaction of P-TEFb with the transcription complex. P-TEFb phosphorylates DSIF causing the transition into productive elongation and the synthesis of HIV mRNAs. (B) Cartoon representation of Tat•AFF4•P-TEFb complex. (C) A close-up view of activation domain (AD) of Tat and AFF4 CBS positioning on the surface of P-TEFb. In panel (B) the TRM of Cyclin T1 is drawn as cartoon and was not included in calculation of P-TEFb surface. Zinc ions of Tat are drawn as balls.
The Tat•AFF4•P-TEFb complex has an extended shape with Cdk9 on one side and Tat•AFF4•Cyclin T1 on other side (Fig. 1B). The N-terminal activation domain of Tat together with the CBS of AFF4 and Tat-TAR recognition motif (TRM) residues 249–261 of Cyclin T1 complement the V-shaped structure of cyclin boxes to form a more globular Cyclin T1 structure. The activation domain of Tat is bound between the cyclin boxes 1 and 2 (Fig. 1C). The N-terminal half of the activation domain is an extended U-shaped coiled-coil that is laid on Cyclin T1 surface and interacts with the T-loop of Cdk9. The C-terminal region of the activation domain is predominantly helical. It coordinates 2 zinc ions and is bound between the cyclin boxes. The N-terminal coiled-coil and first helix (α-helix) of the AFF4 CBS conforms to the surface of Cyclin T1 Cyclin Box 2 and with its second helix (α-310-helix) packs onto the hydrophobic depression formed by Tat and Cyclin T1 (Fig. 1C). The Cyclin T1 TRM is folded into an extended coil with a short one-turn α-helix HC in the middle. These residues lie at the junction of Tat and AFF4 and form a link between the cyclin boxes and the second zinc finger of Tat by providing Cys261 for zinc coordination (Fig. 1C). Interaction with P-TEFb results in a buried surface area of 3810 Å2 for Tat and 3264 Å2 for AFF4. The extensive interactions of AFF4 and Tat with P-TEFb bury a total of 6757 Å2, indicating that P-TEFb plays an important role in induction of both AFF4 and Tat folds. Indeed, the CBS of AFF4 does not possess a hydrophobic core and has only 1 well-defined intramolecular H-bond between its Lys40 and Asp60 residues. As seen in the Tat•P-TEFb structure,26 the tertiary structure of Tat is stabilized by 2 zinc ions, 6 intramolecular hydrogen bonds and an extended exposed hydrophobic patch that docks on Cyclin T1.
Comparison with P-TEFb, Tat•P-TEFb and AFF4•P-TEFb complexes
Comparison of Tat•AFF4•P-TEFb structure with the crystal structures of P-TEFb (PDB code 3BLH),29 Tat•P-TEFb (3MI9)26, and AFF4•P-TEFb (4IMY)28 provide a complete picture of conformational changes occurring during formation of ternary and quaternary complexes. The binding of either Tat or AFF4 affects the conformation and position of Cyclin T1 helices H5′ and HC (Figs. S3A and S3B). The α-helix in Tat displaces the HC helix of cyclin T1 and shifts the C-terminal portion of helix H5′ in Cyclin T1. This results in nearly complete straightening of the “banana” shape of helix H5′ seen in the P-TEFb alone structure. Complete straightening of helix H5′ occurs for the accommodation of AFF4 α-helix during AFF4 binding. In summary, comparisons of P-TEFb structure with Tat•P-TEFb and AFF4•P-TEFb structures point to a cooperative action of Tat and AFF4 in displacing the HC helix of Cyclin T1 and changing the conformation of the H5′ helix.
Superimposition of Cyclin T1 subunits in Tat•P-TEFb and in AFF4•P-TEFb shows some overlap in positions of 310-helix in Tat and α-310-helix in AFF4, as well as steric hindrance between the C-terminal portions of α-helix in AFF4 and helix H5′ in Cyclin T1 (Fig. 2A). This indicates that additional conformational changes are necessary to release the steric hindrance between Tat and AFF4. Indeed, Tat binding to AFF4•P-TEFb shifts the α- and α-310-helices in AFF4 up to 3 Å: first, by fixing helix H5′ in Cyclin T1 in a slightly bent conformation that pushes the C-terminal portion of the α-helix in AFF4; and, second, by pushing the α-310-helix of AFF4 with Phe32 from the 310-helix of Tat (Fig. 2A). Dislocation of the AFF4 α-helix results in a switch in the conformation of the preceding loop (residues 42–45), while dislocation of the α-310-helix places Asp60 side-chain at a position that leads to formation of an intramolecular H-bond with Lys40 (Fig. 2B). Only AFF4 residues N-terminal to Lys40 were not substantially affected by Tat binding.

Figure 2. Structural changes induced by Tat binding to AFF4•P-TEFb or AFF4 binding to Tat•P-TEFb. (A) AFF4 structural changes induced by Tat binding to AFF4•P-TEFb. Red arrows indicate the directions of shifts in AFF4. (B) Tat binding results in closer positioning of AFF4 Lys40 and Asp60 side chains enabling the formation of H-bond. (C) H-bonds between AFF4 and Cyclin T1 that present in Tat•AFF4•P-TEFb structure and absent in structure of AFF4•P-TEFb. (D) Sulfate-mediated H-bonds between AFF4 and Cyclin T1 in the structure of Tat•AFF4•P-TEFb. (E) H-bond between AFF4 Tyr59 and Cyclin T1 Asp169 in the structure of AFF4•P-TEFb that is substituted by sulfate-mediated H-bonds in Tat•AFF4•P-TEFb. For the comparison, the locations of sulfate ion and Asp169 from the superimposed structure of Tat•AFF4•P-TEFb are also shown. (F) Structural changes of Tat induced by AFF4 binding to Tat•P-TEFb.
The structure of Tat•AFF4•P-TEFb revealed additional AFF4•Cyclin T1 interactions that are absent in the AFF4•P-TEFb structure. For example, AFF4 Arg51 forms H-bonds with His239 and Glu246 of Cyclin T1 (Fig. 2C). Furthermore, a sulfate ion re-enforces the AFF4•Cyclin T1 interaction by bridging H-bonds from Tyr59 and Lys63 of AFF4 and Ser167 and Trp210 of Cyclin T1 (Fig. 2D). The AFF4•P-TEFb structure is lacking this sulfate-mediated interaction. Instead, a single H-bond is formed between Tyr59 in AFF4 and Asp169 in Cyclin T1 (Fig. 2E). Elimination of this H-bond by Asp169Ala substitution increases the KD of AFF4 binding 4-fold28 pointing to sensitivity of AFF4 binding to changes around the sulfate binding site. Cyclin T1 Ser167 with a closely located sulfate ion resembles a phosphorylated serine raising the possibility that phosphorylation of Ser 167 might enhance AFF4 binding.
Contrary to significant structural changes in AFF4 caused by Tat binding, the binding of AFF4 induces only local shifts in Tat at 2 positions. First, Asp68 of AFF4 forms a H-bond with the main-chain at Lys19 of Tat, shifting it by 1.1 Å. Second, Phe32 of Tat is moved 0.9 Å toward the hydrophobic surface formed by Leu56, Met62, Phe65, and Ile66 of AFF4, and by Phe176 and Trp207 of Cyclin T1 (Fig. 2F).
Tat•AFF4 interface
AFF4 significantly enhances Tat-binding to P-TEFb by burying 4374 Å2 of surface area between Tat and AFF4•P-TEFb compared with 3810 Å2 buried between Tat and P-TEFb. Residues 55–69 in CBS of AFF4 that include the C-terminus of the α-helix and the α-310-helix are involved in interactions with Tat. Met55, Leu56, Met62, Phe65 and Ile66 of AFF4 significantly expand the hydrophobic surface of Cyclin T1 to bury hydrophobic residues (Met1, Phe32, Phe38, and Leu43) and interact with hydrophobic portions of Lys28 and Lys29 of Tat (Fig. 3A and B). Gly67, Asp68, and Arg69 from AFF4 form 3 main-chain to side-chain H-bonds and 1 main-chain to main-chain H-bond with Tat (Fig. 3C).

Figure 3. Tat•AFF4 interface. (A) AFF4 hydrophobic residues in contact with Tat hydrophobic residues. Only AFF4 residues are labeled for clarity. Tat and AFF4 main-chains are drawn as ribbons. The interacting side chains are drawn as balls-and-sticks. Cyclin T1 is displayed in pale cyan surface representation. The darker (cyan) surface areas correspond to hydrophobic residues interacting with Tat. (B) Similar to panel (A) with the exception of only Tat residues labeling and AFF4 surface representation. Darker orange surface area corresponds to AFF4 hydrophobic residues shown in panel (A). (C) H-bonds between Tat and AFF4.
Structural constraints for Tat mutations
Tat tolerates up to 40% sequence variation without loss of transcriptional activity.30 Previously, based on the crystal structure of Tat•P-TEFb, we explained how mutations in the activation domain of Tat can avoid the disruption of Tat•P-TEFb complex.26 Similar analysis of the AFF4-interacting surface of Tat shows that among the eight interacting residues of Tat 3 are invariant residues (Met1, Lys28, and Leu43), one is a residue with a randomly occurring Phe38Leu polymorph, 2 are residues with predominantly functionally equivalent substitutions (Glu2Asp and Phe32Tyr/Trp/Leu), and 2 residues (Lys19 and Lys29) are with a variety of substitutions (Fig. 4A).

Figure 4. Structural constraints for Tat mutation imposed by AFF4. (A) Color-coded AFF4-interacting residues of Tat: invariant residues are in deep gray, residue with a randomly occurring polymorph is in slate, residues with predominantly functionally equivalent substitutions are in pink, and residues with a variety of substitutions are in green. AFF4 surface is shown in light orange. Darker orange surface area corresponds to Tat-interacting hydrophobic residues of AFF4. (B) Tat Phe32Tyr substitution results in a H-bond to main-chain oxygen of AFF4 Leu56. (C) Tat Phe32Trp substitution inserts tryptophan side chain into the hydrophobic cavity surrounded by Tat, Cyclin T1 and AFF4 residues. (D) Tat Phe32Leu substitution shows an empty space on the left of Leu32 side chain. (E) Tat Phe38Leu substitution places Leu38 side chain close to the side chain of Cyclin T1 Phe176. (F) Tat Glu2Asp substitution results in a H-bond formation between Asp2 and Asp 68 of AFF4. (G) Lys29 of Tat tolerates a variety of substitutions. This panel displays an example of Lys29 substitution to a bulky tryptophan side chain.
The substitutions at Tat positions 32 and 38 are of high interest since they are buried in the hydrophobic core at the interface of Tat, Cyclin T1 and AFF4. Because all the substitutions are for hydrophobic residues, the major issue is whether they can cause steric hindrance preventing Tat binding. Modeling of Phe32Tyr substitution shows that Tyr32 could stabilize the complex because the hydroxyl group of Tyr32 is in an ideal position to provide a H-bond to main-chain oxygen of AFF4 Leu56 (Fig. 4B). Indeed, the highest preference of Tat at this position is for Tyr, followed by Phe, Trp and Leu. The Trp side chain can fit into the hydrophobic cavity (Fig. 4C) with small (less than 1 Å) adjustments in side-chain position of Cyclin T1 Phe176, explaining less preference for Trp. The least preferable residue at position 32 is Leu because it freely fits the hydrophobic cavity, but participates in fewer intermolecular interactions (Fig. 4D). The low occurrence of Phe38Leu substitution is, first, due to formation of fewer intermolecular interactions with Leu38 compared with Phe38; and second, the fitting of Leu side chain requires approximately 1 Å shift in side-chain position of Cyclin T1 Phe176 (Fig. 4E).
The side chain of Lys19 in Tat is on a solvent exposed surface and does not participate in intermolecular interactions. Only its main-chain is H-bonded with AFF4 (Fig. 3C). Consequently, Lys19 substitutions are not expected to have an impact on Tat•AFF4 interaction. The Glu2Asp substitution also will have minimal impact because Asp2 retains a H-bond with the main-chain of Asp68 in AFF4 (Fig. 4F). Finally, Lys29 protrudes into the exposed surface of protein. Modeling of amino acid substitutions at this position shows no interference with Tat•AFF4 interactions (Fig. 4G). This analysis, together with the reported analysis of Tat polymorphs in the structure of Tat•P-TEFb complex,26 demonstrates that Tat polymorphs are selected in such way that enables Tat binding to AFF4•P-TEFb.
Tat-binding surface of AF4 family members
P-TEFb-mediated physical interactions have been reported for Tat with AFF1/4 within SEC.23,27 Other AF4 family members, AFF2 and AFF3 bind P-TEFb, ENL/MLLT1, and AF9/MLLT3 to form SEC-like 2 (SEC-L2) and SEC-like 3 (SEC-L3) complexes.31 Amino acid sequence alignment of 38-residue in the CBS of AFF4 with other AF4 family members shows 46% identity (18 positions) (Fig. 5A). Among eight Tat-interacting residues of AFF4, including 5 hydrophobic residues (Met55, Leu56, Met62, Phe65, and Ile66) and 3 hydrophilic residues (Gly67, Asp68, and Arg69), only Leu56 is identical.

Figure 5. Analysis of Tat binding by AF4 family members. (A) Amino acid sequence alignment of AFF4 CBS with AFF1, AFF2 and AFF4 sequences. Conserved sequences are marked in yellow. Tat-interacting hydrophobic sequences of AFF4 that are not conserved are marked in magenta. Corresponding residues in other AF4 family members are marked in magenta if they are the same as in AFF4 or gray if they are different than in AFF4. (B) Modeling of Met62Val, Phe65Leu and Ile66Leu substitutions. Original residues are in light orange and substituted residues are in blue. (C) Possible effect of Met55Thr substitution in AFF2 and AFF3. The Cyclin T1 domains of AFF4•P-TEFb and Tat•AFF4•P-TEFb were superimposed. Then the AFF4 domain from AFF4•P-TEFb with a modeled Met55Thr substitution was displayed together with Tat•AFF4•P-TEFb. Met55Thr shifts AFF4 α-helix toward the Cyclin T1 helix H5′ and forms a hydrogen bond with Glu246 of Cyclin T1. As result, the AFF2 or AFF3 conformation will be closer to the AFF4 conformation in AFF4•P-TEFb structure. This would cause a conflict between the α-310-helix of AFF2 or AFF3 and 310-helix of Tat. Red arrows indicate the directions of shifts caused by Met55Thr substitution.
Arg69 substitutions are not expected to have a negative impact on the AFF4•Tat interaction because only the main-chain of Arg69 is involved in a H-bond with Tat. The only negative effect of Asp68Thr substitution is expected to be disruption of a H-bond with Tat. However, analysis of Asp68 and Arg69 substitutions alone are not informative since substitution of preceding Gly67 with either Ser in AFF1 or Thr in AFF2, and AFF3 inevitably changes its main-chain φ and ψ torsion angles, resulting in a different conformation of entire C-terminal region of the CBS. Nevertheless, AFF1 still forms a complex with AFF4•P-TEFb,27 indicating that rearrangement of the AFF4 Gly67, Asp68, and Arg69, or its equivalents is not critical for Tat binding. Modeling of Met62Val, Phe65Leu, and Ile66Leu substitutions indicate that they can be tolerated without loss of interaction with Tat (Fig. 5B).
The most interesting difference between the 4 family members is in the position corresponding to Met55 in AFF4. AFF1 is identical, but AFF2 and AFF3 have a substitution of Thr for Met. Met55 in AFF4 faces the N-terminal part of Cyclin T1 helix H5′ and is among the major determinants of AFF4 α-helix positioning relative Cyclin T1 helix H5′ (Fig. 2A). Met55Thr substitution would be expected to shift the AFF4 α-helix toward the Cyclin T1 helix H5′ and possibly form a hydrogen bond with the side chain of Cyclin T1 Glu246 (Fig. 5C). As a result, the AFF4 conformation with Thr55 would be closer to its conformation in AFF4•P-TEFb structure that is not as favorable for Tat docking (Fig. 5C). This suggests that AFF1 and AFF4 would be preferred over AFF2 and AFF3 for interaction with Tat•P-TEFb. Two studies support of this idea. The double mutant Met55Ala/Leu56Ala in AFF428 and the equivalent Met60Ala/Leu61Ala mutant in AFF127 were shown to severely impact the binding of AFF1/4 to P-TEFb. Moreover, wild-type AFF1, but not the Met60Ala/Leu61Ala mutant, was shown to rescue binding of the weakly interacting Tat Cys22Gly mutant with P-TEFb.27
Acetylation of Tat Lys28 and implications for TAR binding
Acetylation plays an important role in regulation of HIV transcription.32-39 In particular, D’Orso and Frankel have found that acetylation of Tat Lys28 by PCAF modulates the assembly of Tat•TAR•P-TEFb complex.33 They also found that Asn257 in the TRM of Cyclin T1 is essential for the recognition of acetylated Tat. On the other hand, the photocrosslinking and protein footprinting studies by Rana and colleagues revealed that the TRM interacts with the loop in TAR RNA.40 This provides a plausible mechanism for the modulation of TAR RNA recognition through stabilization of the TRM in Cyclin T1 by interaction of acetylated Lys28 of Tat with Asn257 in the TRM.
The TRM of Cyclin T1 was disordered within the high-resolution Tat•P-TEFb structure;26 however, the structure of Tat•AFF4•P-TEFb resolved the folding of the TRM (Fig. 6A). Cys261 in the TRM coordinates to the second zinc ion of Tat, and TRM residues Leu252 and Ile255 participate in van-der Waals interactions with the residues of the Tat Zn-finger domain. One inter-subunit H-bond is formed between the side chain of Tat Lys28 and main-chain oxygen of Asn257. Furthermore, Leu252, Lys253, and Trp256 in the TRM participate in van-der Waals interactions with the C-terminus of the α-helix in AFF4.
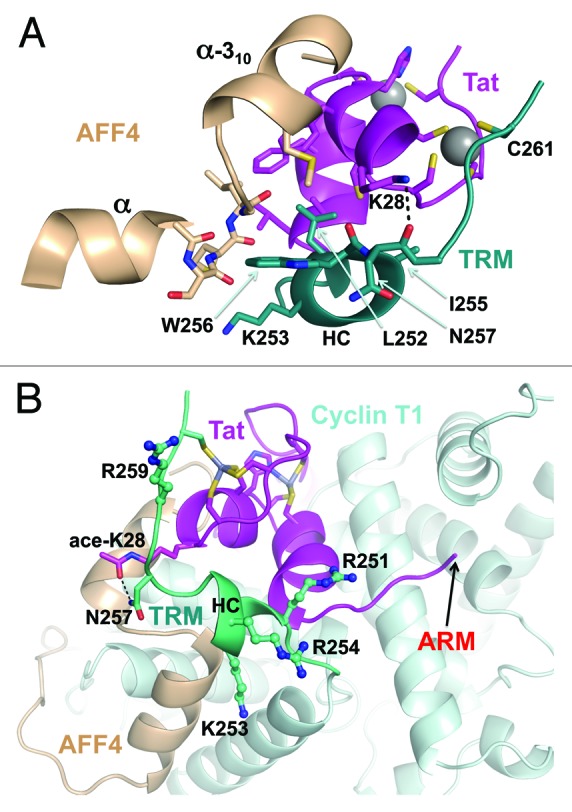
Figure 6. Cyclin T1 TRM and implications for TAR binding. (A) In addition to zinc coordination by Cys261, the residues from TRM helix HC interact with both Tat and AFF4. (B) Modeling of Tat Lys28 acetylation shows the formation of potential H-bond with the side chain of Asn257. The side chains of TRM arginines and lysine are shown as balls-and-sticks. The arrow indicates the location of Tat residue leading to its TAR-binding ARM.
The structure of the TRM shows that all 3 arginines within the TRM protrude toward the same surface as an RNA-binding arginine rich motif (ARM) of Tat (Fig. 6B), providing an additional surface for docking to TAR RNA. This is consistent with involvement of the TRM in the recruitment of TAR RNA.40 However, temperature factors of the TRM residues are relatively high pointing to the flexibility of the TRM (Fig. S4). This explains why the stabilization of the TRM by acetylation of Lys28 enhances the binding of TAR RNA.33 Indeed, modeling of Tat Lys28 acetylation places the acetyl group in a position that is favorable for H-bond formation with Asn257 in the TRM (Fig. 6B). This provides a structural basis for the modulation of TAR RNA binding by acetylation of Lys28 in Tat and for involvement of Asn257 in Cyclin T1.33
Discussion
The structure of the Tat•AFF4•P-TEFb complex described here defines the contacts that lead to simultaneous inclusion of both P-TEFb and Tat in the SEC. Both Tat and AFF4 interact primarily with Cyclin T1. As seen in both the Tat•P-TEFb26 and the Tat•AFF4•P-TEFb structures, Tat also interacts with the T-loop of Cdk9. Modeling of Tat from the Tat•P-TEFb structure into the AFF4•P-TEFb structure suggested that AFF4 might interact with Tat28 and our results here reveal a number of direct contacts that are orchestrated by the extensive interactions of both proteins with the surface of Cyclin T1. These contacts provide additional constraints that explain the limited range of Tat mutants that are tolerated in the AFF4•Tat interface. These constraints along with those created by the Tat•P-TEFb interface26 now explain almost all of the tolerated Tat activation domain mutants and this suggests that there are no other significant interactions made with the activation domain of Tat. In the presence of both Tat and AFF4 the structure of the TRM of Cyclin T1 was finally revealed with certainty and C261 is seen to interact with 1 of the 2 zinc atoms that are coordinated primarily with Tat.
Modeling studies starting with the Tat•AFF4•P-TEFb complex allows us to make additional predictions concerning modifications of Tat and selection of the AF4 family members in SECs. Acetylation of Lys28 of Tat has a positive impact on the association of the Tat•P-TEFb complex with TAR RNA.33 Modeling suggests that acetylation of Lys28 would provide additional stability to the TRM which would enhance the interaction of the TRM with the main loop in TAR, thereby explaining the positive effect of Lys 28 acetylation on TAR binding. Tat displays specificity for SECs that contain AFF1 and AFF4 rather than AFF2 or AFF3.17,25 The Met55 residue AFF4 is conserved in AFF1, but AFF2 and AFF3 have a threonine at the analogous position. Met55 in AFF4 determines the position of AFF4 α-helix relative to Cyclin T1 helix H5′ and modeling of a Met55Thr mutation in AFF4 shows structural changes leading to a significant clash with Tat. This is likely the explanation of why AFF2 and AFF3 are not bound by the Tat•P-TEFb complex.
Determination of structure of the Tat•AFF4•P-TEFb complex significantly advances our understanding of Tat function in recruiting P-TEFb and the SEC to the HIV-1 promoter, however, a number of issues remain to be determined. Tat can extract P-TEFb from the 7SK snRNP and this is important for efficient HIV replication.15,16,41 The 7SK snRNP is not tightly associated with chromatin and is rapidly extracted from nuclei even at very low salt.41 Expression of Tat in cells leads to disruption of the 7SK snRNP and the formation of a Tat•P-TEFb complex in the absence of any other viral proteins or the viral genome.15,16 However, components of the 7SK snRNP have been cross-linked to the HIV promoter and it has been suggested that the extraction of P-TEFb occurs at the promoter.42,43 Because Tat has low affinity to TAR in the absence of P-TEFb,13 whether the extraction takes place in the nucleoplasm or in close proximity to the HIV promoter is not clear. The order of assembly of the Tat•AFF4•P-TEFb complex is also not resolved. The structure described here does not indicate whether Tat binds to P-TEFb in the SEC or Tat•P-TEFb binds to the SEC. Although Tat and AFF4 have a significant interaction with each other when bound to P-TEFb, it is unlikely that they would interact with each other in the absence of P-TEFb. We favor the idea that Tat first extracts P-TEFb from the 7SK snRNP and then becomes associated with the SEC. Insight into the function of Tat will be aided by structurally defining the interactions of other proteins such as HEXIM1 and Brd4 with P-TEFb. In turn, this will aid the development of compounds with anti-viral activity that disrupts the Tat• P-TEFb and or the Tat•AFF4 interaction without affecting normal P-TEFb partners.
Materials and Methods
Expression of Tat•AFF4•P-TEFb
Sf21 insect cells were co-infected with 4 baculoviruses that individually directed the expression of N-terminally His-tagged human Cyclin T1 (1–266), human Cdk9 (7–332), HIV-1 Tat (1–48), and human AFF4 (27–69). To remove the His-tag from CyclinT1, TEV cleavage site (ENLYFQ) was inserted between His-tag and Cyclin T1. The 4 coding sequences were individually cloned into the pFastBac1 vector (Invitrogen) after PCR amplification. The baculoviruses were generated by Bac-to-Bac baculovirus expression system kit (Invitrogen, 10359-016), and these viruses were transfected into Sf21 cells following the manufacturer’s instructions. The viruses were amplified several times to obtain viral stocks with 108 pfu/ml. The presence of the gene of interest was determined by conducting PCR on bacmid DNA isolated by phenol extraction from the baculoviral stocks. Large scale production of the Tat•AFF4•P-TEFb complex was performed in 1 L of Sf-900 II serum-free medium (GIBCO 10902–088) with 1.25 × 106 Sf21 cells/ml. Typically, 1 L of cells was infected at once by adding 2.9 ml of each of the 4 viral stocks. The flasks were then incubated for an additional 72 h.
Purification of Tat•AFF4•P-TEFb
To avoid protein degradation, all solutions and equipment were kept cold and the purification was performed swiftly. Cells were harvested from 1 L cultures by spinning for 7 min at 200 × g in a Sorvall Legend RT 6441 rotor. The pellets were frozen at −80 °C overnight. Pellets were resuspended in Lysis Buffer (500 mM NaCl, 20 mM Tris pH 7.5, 10 mM K2HPO4, 2 mM MgCl2, 5 mM β-ME, 3% glycerol, and 0.1% PMSF solution). Lysates were combined and kept on ice for 10 min and then centrifuged at 16 000 × g for 40 min in a Sorvall RC5C SS-34 rotor in Oak Ridge tubes. The high-speed supernatant was loaded into 6 ml of Qiagen Ni-IDA Agarose column that had been equilibrated in Lysis Buffer. After loading, the column was washed with 35 ml Wash Buffer (2 mM imidazole, 200 mM NaCl, 20 mM Tris pH 7.5, 5mM β-ME, 3% glycerol). The column was then eluted with 45 ml of Wash Buffer supplemented with 150 mM imidazole. The eluted material was pooled and digested by TEV protease (1/10 molar ratio) for 16 h at 4 °C to remove the His-tag from Cyclin T1. The digested material was then dialyzed for 3 h. against 1 L of dialysis buffer (20 mM Tris pH 7.5, 25 mM NaCl, 5 mM β-ME, 3% glycerol). The dialyzed material was centrifuged at 16 000 × g for 20 min in a Sorvall RC5C SS-34 rotor and then loaded onto another 6 ml Qiagen Ni-IDA Agarose column that had been equilibrated in binding buffer (20 mM Tris pH7.5, 30 mM NaCl, 5 mM β-ME, 3% glycerol). The flow-through from second Ni-IDA column was passed through a 5 ml SP column and loaded onto a 5 ml Q column that had been equilibrated in binding buffer (10 mM HEPES pH7.5, 30 mM KCl, 5 mM BME, 3% glycerol). After 30 ml wash, the Q column was eluted with a 30 ml gradient from 69 to 108 mM KCl in elution buffer. The Tat•AFF4•P-TEFb complex eluted at 85 mM KCl. The peak fractions were free of contaminating proteins. The eluted peak fractions were pooled and brought to 5 mM ammonium sulfate by adding 2 M ammonium sulfate. Then the sample buffer was exchanged with optimal buffer (10 mM HEPES pH7.5, 60 mM KCl, 5 mM ammonium sulfate, 2 mM DTT) by 10 kDa MWCO Vivaspin-20 centrifugal devices (GE Healthcare). After buffer exchange, the sample was concentrated into 5.2–5.6 mg/ml. The purification protocol was performed more than 6 times with minor modifications and usually resulted in 1.8–3 mg of the Tat•AFF4•P-TEFb complex per 1 L of cells. The obtained samples were kept in small aliquots at −80 °C.
Crystallization and diffraction data collection
The frozen Tat•AFF4•P-TEFb samples were thawed at room temperature and Dynamic Light Scattering was used to monitor the state of sample aggregation. Initial crystallization screening was performed at a temperature of 295°K in 96-well plates using 50% diluted Crystal Screen and Crystal Screen 2 solutions (Hampton Research) by the sitting-drop vapor-diffusion method by mixing 1 μl of complex solution with 1 μl of reservoir solution. Tiny needle-like crystals appeared in the 22nd condition of the Crystal Screen 2 containing 50 mM MES buffer pH 6.5 and 6% w/v PEG 20 000. Variations in concentration of the components, molecular weight of PEGs, buffers and pH only slightly improved the shape and size of crystals. Screening of additives from Hampton Research Additive Screen resulted in a novel crystal type of rectangular plate shape in presence of YCl3. The final well solution producing best diffracting crystals contained 50 mM MES pH 6.5, 3.7–3.75% w/v PEG 20 000, 5 mM YCl3, 200 mM NDSB 211, 2 mM TCEP pH7.5. SDS-PAGE of dissolved crystals revealed all 4 subunits of Tat•AFF4•P-TEFb in stoichiometric (1:1:1:1) proportions.
The crystals were soaked in cryoprotectant for a few seconds, scooped in nylon-fiber loop and flash cooled in a dry nitrogen stream at 100°K. The cryoprotectant solution contained all components of well solution, with PEG 20 000 concentration increased to 8%, plus 24% v/v ethylene glycol. Preliminary X-ray examinations of crystals were performed using a Rigaku R-AXIS IV imaging plate with Osmic VariMax™ HR mirror-focused CuKα radiation from a Rigaku FR-E rotating-anode generator operated at 45 kV and 45 mA. The final data set was collected on Argonne National Laboratory Advanced Photon Source NE-CAT beamline 24ID-E using an ADSC Q315 detector. All intensity data were indexed, integrated and scaled with DENZO and SCALEPACK from the HKL2000 program package. The crystal parameters and data-processing statistics are summarized in Table 1.
Structure determination
The structure was determined by molecular replacement starting with the coordinates of Tat•P-TEFb (PDB entry 3MI9). Two molecules of Tat•AFF4•P-TEFb were located in an asymmetric unit. In addition to initial model, the electron density was clearly identifiable for AFF4 residues 32–69 in both molecules. The traceable density also was found for the residues 248–261 of Cyclin T1 in 1 molecule; however, in the second molecule the density for the residues 255–259 was of pour quality and was contiguous only at lower 0.6–0.7σ cutoffs, indicating high flexibility. The building of newly identified fragments and adjustments in the initial model were performed manually with TURBO-FRODO software. Due to anisotropic diffraction, the maps with the inclusion of data at resolution 2.9–2.8 Ε were noisy, so final refinement was performed at resolution of 2.9 Ε to an Rcryst of 22.5% and an Rfree of 26.7%. CNS version 1.144 was used for all crystallographic computing. Application of zonal scaling45 and bulk solvent correction improved the quality of electron density maps. The figures containing molecular structures were rendered with PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank J Lovelace and GE Borgstahl for maintenance and management of the Eppley Institute’s X-ray Crystallography facility and DG Vassylyev for the zonal scaling instruction files. This work is supported by NIH grants AI074392 and GM35500 to D.H.P, NE DHHS LB506 grant and in part by NIH grant GM101167 to T.H.T. The Eppley Institute’s core facilities are supported by the Cancer Center Support Grant P30CA036727. This work is also based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by NIH grant GM103403. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US. DOE under Contract No. DE-AC02-06CH11357.
Accession Numbers
Atomic coordinates and structure factors of Tat•AFF4•P-TEFb have been deposited in the Protein Data Bank ID code 4OR5.
Glossary
Abbreviations:
- P-TEFb
positive transcription elongation factor
- SEC
super elongation complex
- AD
activation domain
- CBS
Cyclin T1 binding site
- TRM
Tat-TAR recognition motif
- ARM
arginine rich motif
References
- 1.Trono D, Van Lint C, Rouzioux C, Verdin E, Barré-Sinoussi F, Chun TW, Chomont N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science. 2010;329:174–80. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 2.Lou Z, Sun Y, Rao Z. Current progress in antiviral strategies. Trends Pharmacol Sci. 2014;35:86–102. doi: 10.1016/j.tips.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karn J. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr Opin HIV AIDS. 2011;6:4–11. doi: 10.1097/COH.0b013e328340ffbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewin SR, Evans VA, Elliott JH, Spire B, Chomont N. Finding a cure for HIV: will it ever be achievable? J Int AIDS Soc. 2011;14:4. doi: 10.1186/1758-2652-14-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuritzkes DR. Drug resistance in HIV-1. Curr Opin Virol. 2011;1:582–9. doi: 10.1016/j.coviro.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He N, Zhou Q. New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC) J Neuroimmune Pharmacol. 2011;6:260–8. doi: 10.1007/s11481-011-9267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayton AI, Sodroski JG, Rosen CA, Goh WC, Haseltine WA. The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell. 1986;44:941–7. doi: 10.1016/0092-8674(86)90017-6. [DOI] [PubMed] [Google Scholar]
- 8.Guo J, Price DH. RNA polymerase II transcription elongation control. Chem Rev. 2013;113:8583–603. doi: 10.1021/cr400105n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81:119–43. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nilson KA, Price DH. The Role of RNA Polymerase II Elongation Control in HIV-1 Gene Expression, Replication, and Latency. Genet Res Int. 2011;2011:726901. doi: 10.4061/2011/726901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–8. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–32. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–62. doi: 10.1016/S0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 14.Peterlin BM, Brogie JE, Price DH. 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip Rev RNA. 2012;3:92–103. doi: 10.1002/wrna.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sedore SC, Byers SA, Biglione S, Price JP, Maury WJ, Price DH. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007;35:4347–58. doi: 10.1093/nar/gkm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barboric M, Yik JH, Czudnochowski N, Yang Z, Chen R, Contreras X, Geyer M, Matija Peterlin B, Zhou Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35:2003–12. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–51. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13:543–7. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- 19.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37:429–37. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Estable MC, Naghavi MH, Kato H, Xiao H, Qin J, Vahlne A, Roeder RG. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci. 2002;9:234–45. doi: 10.1007/BF02256070. [DOI] [PubMed] [Google Scholar]
- 21.Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KS, Allis CD, Roeder RG. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A. 2011;108:15751–6. doi: 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byun JS, Fufa TD, Wakano C, Fernandez A, Haggerty CM, Sung MH, Gardner K. ELL facilitates RNA polymerase II pause site entry and release. Nat Commun. 2012;3:633. doi: 10.1038/ncomms1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou S, Upton H, Bao K, Schulze-Gahmen U, Samelson AJ, He N, Nowak A, Lu H, Krogan NJ, Zhou Q, et al. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc Natl Acad Sci U S A. 2013;110:E123–31. doi: 10.1073/pnas.1216971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108:E636–45. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol Cell. 2010;38:428–38. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–51. doi: 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu H, Li Z, Xue Y, Schulze-Gahmen U, Johnson JR, Krogan NJ, Alber T, Zhou Q. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc Natl Acad Sci U S A. 2014;111:E15–24. doi: 10.1073/pnas.1318503111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, Krogan NJ, Zhou Q, Alber T. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. 2013;eLife2:e00327. doi: 10.7554/eLife.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27:1907–18. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campbell GR, Loret EP. What does the structure-function relationship of the HIV-1 Tat protein teach us about developing an AIDS vaccine? Retrovirology. 2009;6:50. doi: 10.1186/1742-4690-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo Z, Lin C, Guest E, Garrett AS, Mohaghegh N, Swanson S, Marshall S, Florens L, Washburn MP, Shilatifard A. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;32:2608–17. doi: 10.1128/MCB.00182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho S, Schroeder S, Kaehlcke K, Kwon HS, Pedal A, Herker E, Schnoelzer M, Ott M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J. 2009;28:1407–17. doi: 10.1038/emboj.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Orso I, Frankel AD. Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex. Proc Natl Acad Sci U S A. 2009;106:3101–6. doi: 10.1073/pnas.0900012106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ott M, Dorr A, Hetzer-Egger C, Kaehlcke K, Schnolzer M, Henklein P, Cole P, Zhou MM, Verdin E. Tat acetylation: a regulatory switch between early and late phases in HIV transcription elongation. Novartis Found Symp. 2004;259:182–93, discussion 193-6, 223-5. doi: 10.1002/0470862637.ch13. [DOI] [PubMed] [Google Scholar]
- 35.Kaehlcke K, Dorr A, Hetzer-Egger C, Kiermer V, Henklein P, Schnoelzer M, Loret E, Cole PA, Verdin E, Ott M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol Cell. 2003;12:167–76. doi: 10.1016/S1097-2765(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 36.Deng L, de la Fuente C, Fu P, Wang L, Donnelly R, Wade JD, Lambert P, Li H, Lee CG, Kashanchi F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology. 2000;277:278–95. doi: 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- 37.Ott M, Schnölzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–92. doi: 10.1016/S0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- 38.Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–18. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brès V, Tagami H, Péloponèse JM, Loret E, Jeang KT, Nakatani Y, Emiliani S, Benkirane M, Kiernan RE. Differential acetylation of Tat coordinates its interaction with the co-activators cyclin T1 and PCAF. EMBO J. 2002;21:6811–9. doi: 10.1093/emboj/cdf669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richter S, Ping YH, Rana TM. TAR RNA loop: a scaffold for the assembly of a regulatory switch in HIV replication. Proc Natl Acad Sci U S A. 2002;99:7928–33. doi: 10.1073/pnas.122119999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biglione S, Byers SA, Price JP, Nguyen VT, Bensaude O, Price DH, Maury W. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology. 2007;4:47. doi: 10.1186/1742-4690-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNamara RP, McCann JL, Gudipaty SA, D’Orso I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013;5:1256–68. doi: 10.1016/j.celrep.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Orso I, Jang GM, Pastuszak AW, Faust TB, Quezada E, Booth DS, Frankel AD. Transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol Cell Biol. 2012;32:4780–93. doi: 10.1128/MCB.00206-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 45.Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007;448:157–62. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.