

Abstract
α6β4 integrin is an adhesion molecule for laminin receptors involved in tumor progression. We present a link between β4 integrin expression and miR-221/222 in the most prevalent human mammary tumor: luminal invasive carcinomas (Lum-ICs). Using human primary tumors that display different β4 integrin expression and grade, we show that miR-221/222 expression inversely correlates with tumor proliferating index, Ki67. Interestingly, most high-grade tumors express β4 integrin and low miR-221/222 levels. We ectopically transfected miR-221/222 into a human-derived mammary tumor cell line that recapitulates the luminal subtype to investigate whether miR-221/222 regulates β4 expression. We demonstrate that miR-221/222 overexpression results in β4 expression downregulation, breast cancer cell proliferation, and invasion inhibition. The role of miR-221/222 in driving β4 integrin expression is also confirmed via mutating the miR-221/222 seed sequence for β4 integrin 3′UTR. Furthermore, we show that these 2 miRNAs are also key breast cancer cell proliferation and invasion regulators, via the post-transcriptional regulation of signal transducer and activator of transcription 5A (STAT5A) and of a disintegrin and metalloprotease-17 (ADAM-17). We further confirm these data by silencing ADAM-17, using a dominant-negative or an activated STAT5A form. miR-221/222-driven β4 integrin, STAT5A, and ADAM-17 did not occur in MCF-10A cells, denoted “normal” breast epithelial cells, indicating that the mechanism is cancer cell-specific.
These results provide the first evidence of a post-transcriptional mechanism that regulates β4 integrin, STAT5A, and ADAM-17 expression, thus controlling breast cancer cell proliferation and invasion. Pre-miR-221/222 use in the aggressive luminal subtype may be a powerful therapeutic anti-cancer strategy.
Keywords: ADAM-17, STAT5A, breast cancers, miR-221/222, β4 integrin
Introduction
Integrins are a family of heterodimeric transmembrane receptors that mediate cell–extracellular matrix (ECM) and cell–cell interactions.1 The β4 integrin subunit was initially identified as a tumor-related antigen in cancer and one that is associated with tumor progression.2 Human studies have demonstrated that β4 expression correlates with breast cancer size and grade as well as with a poor patient prognosis in a variety of tumors of epithelial origin.3,4 β4 integrin and its heterodimeric subunit, α6, are the receptors for laminins.5-7 In physiological conditions, β4 anchors the cytoskeleton to laminins in the basement membrane via hemidesmosomes located on the basal surface of epithelial cells.8 β4 integrin becomes diffusely distributed over the entire cell surface, in several tumors of epithelial origin that lack hemidesmosome anchorage, resulting in increased expression and signaling and, as a consequence, tumor invasion.9,10 Indeed, it has recently been demonstrated that β4 integrin depletion inhibits tumor cell expansion and invasion, mainly by impairing phosphatidylinositol 3-kinase (PI3K) pathway activity.11
Alternatively, cancer dissemination occurs via extracellular matrix (ECM) degradation. Members of the a disintegrin and metalloproteinase family (ADAM) have attracted interest in recent years because of their adhesive and proteolytic properties.12 In addition, ADAMs regulate signaling events that are relevant to cell proliferation.12 ADAM-17, a multi-domain protein,13,14 is the most extensively investigated member of this family.15 ADAM-17 is widely distributed, and its expression is modified not only during embryonic development, but also during an adult's life.16 Moreover, the ADAM-17 protein is upregulated in several diseases and tumors.17 In particular, increased ADAM-17 expression correlates with mammary cancer development and adverse outcomes in breast cancer patients.18
MicroRNAs (miRNAs), small endogenous non-coding single-stranded RNAs, are key players in gene regulation.19 Their pairing with the 3′untranslated region (3′UTR) of target mRNAs generally results in mRNA degradation or translational inhibition.19 In fact, the deregulation of miRNA expression is considered a hallmark of cancer.20,21 Although altered miRNA expression has been reported in breast cancers,22,23 the functional consequences of specific miRNA aberrant expression in tumor progression and invasion is still to be elucidated. miR-221/222 overexpression has recently been shown in several advanced malignancies, including mammary tumors.24,25 The signal transducers and activator of transcription (STAT) 5A is included in miR-221/222 target genes.26 Active STAT5 can promote breast cancer tumorigenesis in rodents,27 whereas it has been shown to positively correlate with the differentiation status of breast cancer tumors,28 with mammary epithelial cell differentiation and with endocrine therapy responsiveness in humans.29,30 Collectively, these studies point to the existence of a dual role for STAT5 in the mammary gland, both as a tumor formation initiator and as a promoter of established tumor differentiation. However, observations that STAT5 inhibition in breast cancers impairs the proliferative rate suggest that the role of STAT5 in breast cancer is still open to debate.31,32
Self-governing molecular mechanisms in breast cancer development and progression have been widely explored. Likewise, the role of β4 integrin as a signaling receptor in mammary tumors has been extensively documented.3,4 However, a new scenario in which β4 integrin expression modulates miRNA patterns has more recently been proposed for basal-like breast cancers.33 In this present study, we have performed in vitro and ex vivo investigations into the functional relationship between miR-221/222 and β4 integrin, ADAM-17, and STAT5 in the progression of luminal subtype breast cancers, which could benefit from further prognostic stratification.
Results
β4 integrin and miR-221/222 expression in human luminal invasive carcinomas (Lum-ICs)
It has been shown that β4 expression modulates miR-221/222 expression in basal-like carcinomas.33 We therefore decided to evaluate the expression of miR-221/222 and β4 integrin in estrogen receptor (ER)-positive (luminal) Lum-ICs, using the gene expression profiling data set generated by the TCGA consortium (The Cancer Genome Atlas Network),34 and it was found that both miR-221/222 and β4 are expressed in a significant fraction of luminal breast cancers (Fig. 1A). Further to this observation, we demonstrate that β4 and miR-221/222 are expressed in the human-derived luminal breast cancer cell lines MCF-7, MDA-MB361, and T47D (Fig. 1B and C).35 However, when β4 protein expression was evaluated on 15 primary human breast carcinoma samples (luminal subtypes), β4 was only found in 50% of preferentially high-grade and highly proliferating tumors (Fig. 1D). By contrast, all tumor samples expressed miR-221/222 (Fig. 1E) even though at different levels. Significantly, low levels of miR-221/222 were observed in samples expressing β4 integrin (Fig. 1D and E), while a high level of miR-221/222 was observed in low-grade tumors. Furthermore, miR221/222 expression inversely correlated (miR-221 P = 0.0013; miR-222 P = 0.037) with the proliferating index, evaluated by Ki67 nuclear expression (Fig. 1F). All primary samples features are reported in Table 1.

Figure 1. β4 integrin and miR-221/222 expression in Lum-IC samples. (A) β4 integrin and miR-221/222 distribution in basal- and luminal-like carcinomas from the TCGA consortium data set. (B) Cell extracts from luminal-derived MCF-7, MDA-MB361, and T47D cell lines were analyzed by western blot for β4 integrin content by densitometry (relative amount). Protein levels were normalized to β actin content. The results are representative of 4 different experiments performed in triplicate (n = 4). (C) MCF-7, MDA-MB361, and T47D cell lines were analyzed by qRT-PCR to evaluate miR-221 and miR-222 expression. The reported data were normalized to RNU6B and are representative of 5 different experiments performed in triplicate (n = 5). (D) Representative immunohistochemical staining for β4 integrin expression on human Lum-IC sections, including grade 1 and grade 3 samples. Scale bar: 80 μm (×40 magnification). (E) To evaluate miR-221 and miR-222 expression, qRT-PCR was performed in human Lum-IC samples (n = 6, grade 1; n = 9, grade 3). The reported data are normalized to RNU6B and are representative of all samples, performed in triplicate. The MCF-7 cell line was used as a control for miRNA expression. (***P < 0.001 grade 3 vs. grade 1 for miR-221; *P < 0.05 grade 3 vs. grade 1 for miR-222). (F) Correlation between the proliferating index Ki67 and miR-221 or miR-222 expression in the luminal carcinoma samples. r correlation coefficient and P values are reported.
Table 1. Histopathological and immunophenotypical features of primary human tumor samples of the luminal subtype (Lum-ICs).
| Sample | Histological type | Grade | %ER | %Ki-67 | %PR |
|---|---|---|---|---|---|
| 1 | IC-NST+ILC | 1 | 98 | 12 | 90 |
| 2 | IC-NST | 1 | 100 | 5 | 95 |
| 3 | IC-NST | 1 | 100 | 20 | 70 |
| 4 | IC-NST+ TUB | 1 | 95 | 16 | 90 |
| 5 | IC-NST | 1 | 95 | 6 | 95 |
| 6 | IC-NST | 1 | 85 | 15 | 80 |
| 7 | IC-NST | 3 | 98 | 47 | 25 |
| 8 | IC-NST + MPC | 3 | 95 | 35 | 20 |
| 9 | IC-NST | 3 | 98 | 23 | 95 |
| 10 | IC-NST | 3 | 95 | 60 | 75 |
| 11 | IC-NST + MPC | 3 | 75 | 39 | 30 |
| 12 | IC-NST | 3 | 98 | 46 | 0 |
| 13 | IC-NST | 3 | 100 | 31 | 30 |
| 14 | IC-NST | 3 | 100 | 39 | 70 |
| 15 | IC-NST | 3 | 90 | 30 | <1 |
All cases showed high estrogen receptor (ER) expression (range: 75–100%) with variable progesterone receptor (PR) expression and a proliferation index (Ki-67) ranging between 5% and 60%. ER, PR, and Ki-67 are expressed as percentage of positive cells. Histological type is categorized according to the latest WHO classification (IC-NST, invasive carcinoma of no special type; ILC, invasive lobular carcinoma; MPC, micropapillary carcinoma; TUB, tubular carcinomas).
β4 integrin expression is post-transcriptionally regulated by miR-221/222 in breast cancer cells
The above results gave us reason to hypothesize that miR-221/222 may control β4 integrin expression in the luminal breast cancer subtype, and gain-of-function experiments were performed in a MCF-7 wild-type (wt) cell line to validate this possibility (Fig. 2A). Data reported in Figure 2B show that miR-221/222 overexpression led to a downregulation of β4 integrin, suggesting that miR-221/222 may post-transcriptionally regulate β4 integrin expression. It has recently been reported that miRNAs interact in a non-canonical fashion with their putative target genes.36 Thus, the full-length 3′UTR nucleotide sequence of β4 integrin was analyzed for a miR-221/222 blasting sequence, and several base pairings were found (259–281bp 3′UTR β4 integrin) (Fig. 2C). The luciferase reporter vector, containing the full-length 3′UTR of β4 integrin, was then transfected into MCF-7 cells that overexpress miR-221/222. MCF-7 cells transfected with the luciferase reporter empty vector were used as a control. As expected, luciferase activity was not detectable in MCF-7 cells that overexpress miR-221/222 (Fig. 2D). These results were further confirmed using a luciferase reporter vector containing a point mutation in the seed sequence for miR-221/222 in the β4 integrin 3′UTR (Fig. 2E). The finding that miR-221/222 control β4 integrin expression, and not vice versa, is sustained by the results reported in Figure 2F and G. Indeed, β4 integrin transient silencing did not modify miR-221/222 expression in MCF-7 cells.

Figure 2. miR-221/222 post-transcriptionally regulate β4 integrin expression. (A) qRT-PCR was performed to evaluate miR-221 and miR-222 expression in MCF-7 wt transfected with pre-miR neg c or pre-miR-221 or pre-miR-222. The reported data are normalized to RNU6B and are representative of 5 experiments (n = 5) (***P < 0.001 miR-221 and miR-222 expression in MCF-7 wt cells transfected with pre-miR-221/222 vs. cells transfected with pre-miR neg c). (B) MCF-7 wild-type (wt) cells were transfected with pre-miR negative control (neg c) or with pre-miR-221 or pre-miR-222. Forty-eight hours later, cell extracts were subjected to western blot analysis to evaluate β4 integrin and β actin content (***P < 0.001 MCF-7 transfected with pre-miR-221 and pre-miR-222 vs. pre-miR neg c). The results are representative of 3 different experiments performed in triplicate (n = 3). (C) Blast analysis of the human miR-221 sequence and 3′UTR full-length of β4 integrin shows several base pairings from bp 259 to 281 of the β4 integrin 3′UTR. (D) pGL3 empty vector or pGL3-3′UTR β4 integrin luciferase constructs were transfected into MCF-7 wild-type cells, previously transfected with pre-miR neg c or with pre-miR-221 or pre-miR-222. Relative luciferase activity is reported (***P < 0.001 pre-miR neg c in presence of pGL3–3′UTR β4 vs. pGL3; ***P < 0.001 pre-miR-221 or pre-miR-222 vs. pre-miR neg c in pGL3–3′UTR β4-transfected cells) (n = 4). (E) Luciferase activity was evaluated in MCF-7 cells that had been co-transfected with pre-miR neg c or pre-miR-221 or pre-miR-222 precursors and pGL3 or pGL3–3′UTR β4 integrin that had been mutated into the seed sequence (mut). Relative luciferase activity is reported (***P < 0.001 pre-miR neg c in presence of pGL3-3′UTRmut β4 vs. pGL3) (n = 3). (F) MCF-7 wild-type cells were transfected for 48 h with scramble or short hairpin (sh)-β4 integrin and evaluated for β4 integrin and β actin content. (***P < 0.001 sh-β4 integrin vs. scramble for β4 integrin content). The results are representative of 4 different experiments performed in triplicate (n = 4). (G) qRT-PCR was performed to evaluate miR-221 and miR-222 expression in MCF-7 wild-type cells transfected with scramble or sh-β4 integrin. The reported data were normalized to RNU6B and are representative of 5 different experiments performed in triplicate (n = 5).
Finally, both β4 integrin and miR-221/222 expression was evaluated in MCF-10A cells to evaluate whether miR-221/222-driven β4 expression specifically regulates cancer cell fate. As shown in Figure 3A and B, these cells express low levels of β4 integrin and high levels of miR-221/222. Moreover, no changes in β4 expression were detected (Fig. 3A), when loss-of-function experiments were performed (Fig. 3C). This suggests that miR-221/222-driven β4 integrin expression is specific to tumor cells.

Figure 3. miR-221/222 expression in MCF-10A cells does not affect β4 integrin, STAT5A, or ADAM-17 expression. (A) MCF-10A cells transfected with anti-miR neg c or anti-miR-221 or anti-miR-222 were analyzed for β4 integrin, STAT5A, and ADAM-17 content. Protein levels were normalized to β actin content. The results are representative of 3 different experiments, performed in triplicate (n = 3). (B) qRT-PCR was performed to evaluate miR-221 and miR-222 expression in MCF-7 wt and MCF-10A cells. The reported data are normalized to RNU6B and are representative of 5 experiments (n = 5) (***P < 0.001 miR-221 and miR-222 expression in MCF-10A vs. MCF-7 wt cells). (C) MCF-10A cells transfected with anti-miR neg c or anti-miR-221 or anti-miR-222 were evaluated by qRT-PCR for miR-221 and miR-222 expression. The reported data are normalized to RNU6B and are representative of 5 experiments (n = 5) (*P < 0.05 miR-221 and miR-222 expression in MCF-10A cells transfected with anti-miR-221/222 vs. cells transfected with anti-miR neg c).
miR-221/222-driven β4 integrin downregulation is crucial for MCF-7 proliferation and invasion
Functional studies were performed in MCF-7 wt cells that overexpress miR-221/222 in order to investigate the biological relevance of the miR-221/222-induced post-transcriptional regulation of β4 integrin. miR-221/222 overexpression, resulting in β4 integrin downregulation, was associated with the impaired ability of cells to pass across the extracellular matrix and with impaired cell proliferation, as sustained by cell counting, by DNA-polymerase-δ auxiliary protein expression (e.g., proliferating-cell nuclear antigen, PCNA), by cyclin D1 expression, and by phosphorylated Akt content (Fig. 4A–D). FACS analysis demonstrates that miR-221/222 overexpression led to G0/G1 cell cycle arrest (Fig. 4E). The biological relevance of miR-221/222 overexpression in MCF-7 cells was also confirmed by a clonogenic assay (Fig. 4F). As expected, we were unable to detect a significant increase in MCF-10A cell proliferation when miR-221/222 antagomir were used (data not shown).

Figure 4. miR-221/222 overexpression impairs MCF-7 cell proliferation and invasion. (A) A cell proliferation assay was performed for the indicated time intervals on MCF-7 wild-type cells transfected with pre-miR neg c, pre-miR-221 or pre-miR-222 (***P < 0.001 pre-miR neg c vs. pre-miR-221 and pre-miR-222 transfected MCF-7 cells) (n = 4). (B) Representative FACS analysis to evaluate PCNA expression in MCF-7 cells, transfected as above. PCNA staining (48 h after transfection) is reported. Percentage ± SEM of reduced proliferation: pre-miR-221, 60 ± 5.6; pre-miR-222, 40 ± 4.4, ***P < 0.001. (C) Cyclin D1 and phospho (p)Akt content in pre-miR neg c-, pre-miR-221-, and pre-miR-222-transfected MCF-7 cells was evaluated by western blot analysis. Protein levels were normalized to β actin or Akt content. The results are representative of 4 different experiments performed in triplicate (n = 4) (***P < 0.001 pre-miR-221 and pre-miR-222 vs. pre-miR neg c transfected MCF-7 cells). (D) An invasion assay was performed on MCF-7, transfected as above. The percentage of invading cells is reported (***P < 0.001 pre-miR-221 and pre-miR-222 vs. pre-miR neg c transfected MCF-7 cells) (n = 4). (E) A cell proliferation assay was performed by FACS analysis to evaluate the percentage of cells in each cell cycle phase. The results are representative of 3 different experiments performed in triplicate (data are expressed as mean, n = 3). (F) Clonogenic assay was performed on MCF-7 cells, treated as above. The number of formed clones per field (20× magnification) is reported in the histogram (***P < 0.001 pre-miR-221 and pre-miR-222 vs. pre-miR neg c transfected MCF-7 cells) (n = 3).
These data were further confirmed using a stable clone that lacked the β4 integrin subunit (MCF-7 β4i), generated from the MCF-7 cell line.11 As shown in Figure 5A, the level of β4 integrin subunit in the MCF-7 scr-shRNA clone (MCF-7c) was similar to that of the parental cells, and, as such, these cells were used as an internal control. As expected, functional studies performed in these 2 clones showed a 50% reduction in the proliferation rate and invasiveness of β4 integrin-depleted cells (Fig. 5B–D). Unexpectedly, MCF-7 β4i cells displayed a 10-fold increase in miR-221/222 expression (Fig. 5E) compared with the MCF-7c clone. This suggests that, as happens in vivo, the miR-221/222 level might control the proliferative and invasive capability of this tumor subtype. Loss-of-function experiments were thus performed in MCF-7 β4i cells to evaluate this possibility (Fig. 6A). As shown in Figure 6, antagomir expression promoted MCF-7 β4i cell proliferation (Fig. 6B and C), cyclin D1 expression, Akt phosphorylation (Fig. 6D), and invasion (Fig. 6E).

Figure 5. β4 integrin depletion led to cell proliferation and cell invasion inhibition in breast cancer cells. (A) Cell extracts from MCF-7 wt cells, MCF-7 scr-shRNA clone (MCF7c), or MCF-7 β4-shRNA clone (MCF-7 β4i) were subjected to SDS-PAGE to evaluate β4 integrin and β actin content (***P < 0.001 MCF-7 β4i vs. MCF-7 wt and MCF-7c cells). The results are representative of 3 different experiments, performed in triplicate (n = 3). (B) A cell proliferation assay was performed for indicated time intervals on MCF-7c and MCF-7 β4i clones (***P < 0.001 MCF-7 β4i vs. MCF-7c) (n = 3). (C) PCNA staining in MCF-7 cell clones analyzed by FACS. Percentage ± SEM of reduced proliferation: MCF-7 β4i 50 ± 5.7, ***P < 0.001. (D) An invasion assay was performed on MCF-7c and MCF-7 β4i cells. Percentage of invading cells is reported (***P < 0.001 MCF-7 β4i vs. MCF-7c) (n = 4). (E) miR-221 and miR-222 expression was analyzed by qRT-PCR on MCF-7c and MCF-7 β4i cells. The reported data were normalized to RNU6B and are representative of 4 different experiments, performed in triplicate (n = 4) (***P < 0.001 miR-221 and miR-222 expression in MCF-7 β4i vs. MCF-7c).

Figure 6. miR-221/222 loss-of-function in the MCF-7 β4i clone rescues proliferation and invasiveness. (A) qRT-PCR was performed to evaluate miR-221 and miR-222 expression in MCF-7 β4i cells transfected with anti-miR neg c or anti-miR-221 or anti-miR-222. The reported data are normalized to RNU6B and are representative of 5 experiments (n = 5) (**P < 0.01 miR-221 and miR-222 expression in MCF-7 β4i cells transfected with anti-miR-221/222 vs. cells transfected with anti-miR neg c). (B) A cell proliferation assay was performed for indicated time intervals on MCF-7 β4i cells transfected with anti-miR neg c, anti-miR-221, or anti-miR-222 (***P < 0.001, anti-miR-221 or anti-miR-222 vs. anti-miR neg c transfected MCF-7 β4i cells) (n = 3). (C) PCNA staining in MCF-7 β4i cells transfected as above. Percentage ± SEM of increased proliferation: anti-miR-221, 60 ± 3.7; anti-miR-222, 50 ± 4.2 ***P < 0.001. (D) MCF-7 β4i cells, treated as above, were analyzed for cyclin D1, pAkt, and β4 integrin content. Protein levels were normalized to β actin or Akt content. The results are representative of 3 different experiments, performed in triplicate (n = 3) (***P < 0.001 anti-miR-221 and anti-miR-222 vs. anti-miR neg c for cyclin D1 and pAkt content). (E) An invasion assay was performed on MCF-7 β4i cells treated as indicated. Percentage of invading cells is reported (***P < 0.001 anti-miR-221 and anti-miR-222 vs. anti-miR neg c) (n = 4).
miR-221/222 target STAT5A to regulate breast cancer cell proliferation
The above results suggest that miR-221/222 still control breast cancer cell behavior by regulating additional targets, even when β4 integrin is silenced. It was decided to explore other known target genes as a means to investigate this hypothesis. As shown in Figure 7A, p27Kip1and p57Kip2 expression levels were unchanged in MCF-7 β4i and in MCF-7c cells. Similar results were obtained in MCF-7 wt that overexpress miR-221/222 (data not shown). By contrast, STAT5A expression inversely correlated with miR-221/222 levels in the 2 different clones (Fig. 7A). Similar results were obtained when gain- and loss-of-function experiments were performed (Fig. 7A and B). The post-transcriptional regulation of STAT5A by miR-221/222 in our experimental models was further confirmed by the luciferase reporter assay performed in both clones, using the full-length 3′UTR of the STAT5A gene or the point-mutated 3′UTR seed sequence of STAT5A (Fig. 8A and B). Functional studies were then performed in both MCF-7 clones by overexpressing a dominant-negative STAT5A (ΔSTAT5A) or a constitutively activated STAT5 construct (1*6 STAT5A).37 As reported in Figure 8C, STAT5A inactivation in MCF-7c and STAT5A induction in MCF-7 β4i cells led to inhibition and cell proliferation induction, respectively. However, neither the activation nor inactivation of STAT5A leads to detectable changes in cell invasiveness (Fig. 8D). These data, along with the finding that high levels of STAT5 expression were found in highly proliferative MDA-MB361 and T47D cell lines (Fig. 9A and B), suggest that miR-221/222-driven STAT5A expression strictly controls breast cancer cell proliferation. Furthermore, STAT5A expression was not modulated by antagomir expression in MCF-10A cells (Fig. 3A).
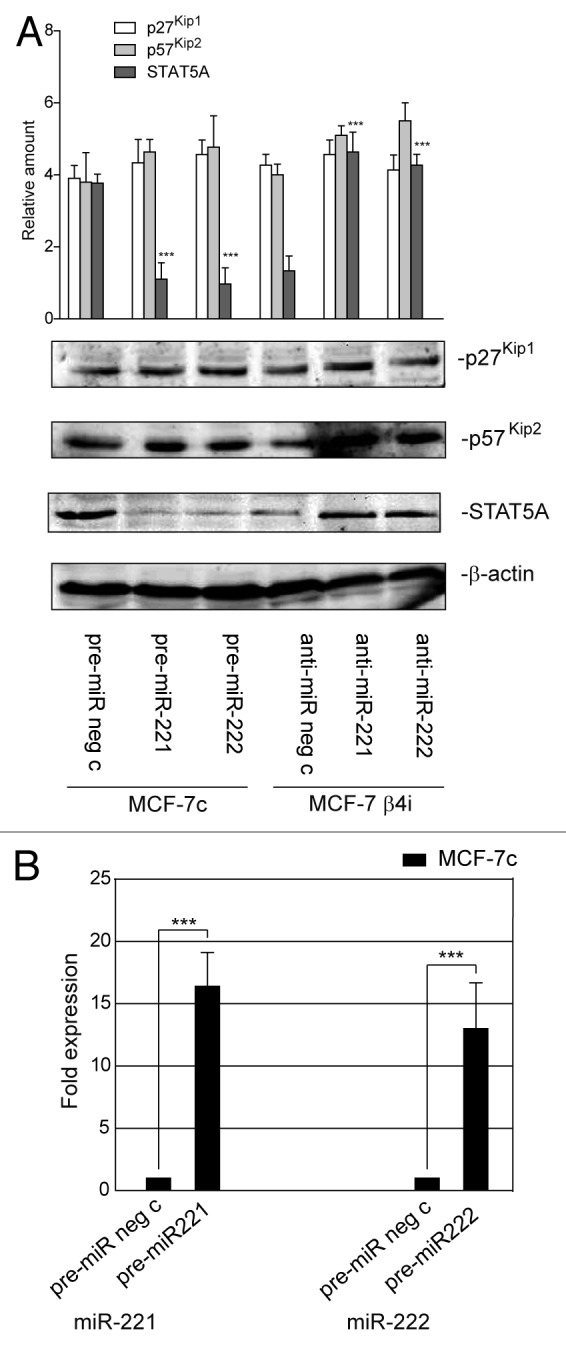
Figure 7. STAT5A expression is under control of miR-221/222 in mammary tumor cells. (A) p27Kip1, p57Kip2, STAT5A, and β actin content were analyzed by western blot in MCF7-7c cells transfected with pre-miR neg c, pre-miR-221, or pre-miR-222 precursors or in MCF-7 β4i cells transfected with anti-miR neg c, anti-miR-221, or anti-miR-222 oligonucleotides. The results are representative of 3 different experiments performed in triplicate (n = 3) (***P < 0.001 gain- and loss-of-function values vs. control values for STAT5A content). (B) qRT-PCR was performed to evaluate miR-221 and miR-222 expression in MCF-7c cells transfected with pre-miR neg c or pre-miR-221 or pre-miR-222. The reported data are normalized to RNU6B and are representative of 5 experiments (n = 5) (***P < 0.001 miR-221 and miR-222 expression in MCF-7c cells transfected with pre-miR-221/222 vs. cells transfected with pre-miR neg c).

Figure 8. miR-221/222 drive mammary tumor cell proliferation by regulating STAT5A. (A) Luciferase activity was evaluated in MCF-7 clones treated as indicated. Relative luciferase activity is reported (***P < 0.001 pre-miR neg c-transfected MCF-7c in presence of pGL3–3′UTR STAT5A vs. pGL3; ***P < 0.001 pre-miR-221 or pre-miR-222 vs. pre-miR neg c transfected MCF-7c; ***P < 0.001 anti-miR-221 or anti-miR-222 vs. anti-miR neg c transfected MCF-7 β4i) (n = 4). (B) pGL3 empty vector or the pGL3–3′UTR STAT5A mutated seed sequence (mut) were co-transfected with pre-miR neg c or pre-miR-221 or pre-miR-222 precursors in MCF-7c. Relative luciferase activity is reported (***P < 0.001 pre-miR neg c transfected MCF-7c in presence of pGL3–3′UTRmut STAT5A vs. pGL3) (n = 3). (C) A cell proliferation assay was performed for indicated times in MCF-7c cells, transfected with pCNeo empty vector or pCNeo-ΔSTAT5A construct, and in MCF-7 β4i cells, transfected with pCNeo empty vector or pCNeo-1–6*STAT5A construct (***P < 0.001 pCNeo-ΔSTAT5A and pCNeo-1–6 *STAT5A constructs vs. pCNeo empty vector) (n = 3). (D) An invasion assay was performed on MCF-7 clones treated as above. Percentage of invading cells is reported (n = 4).
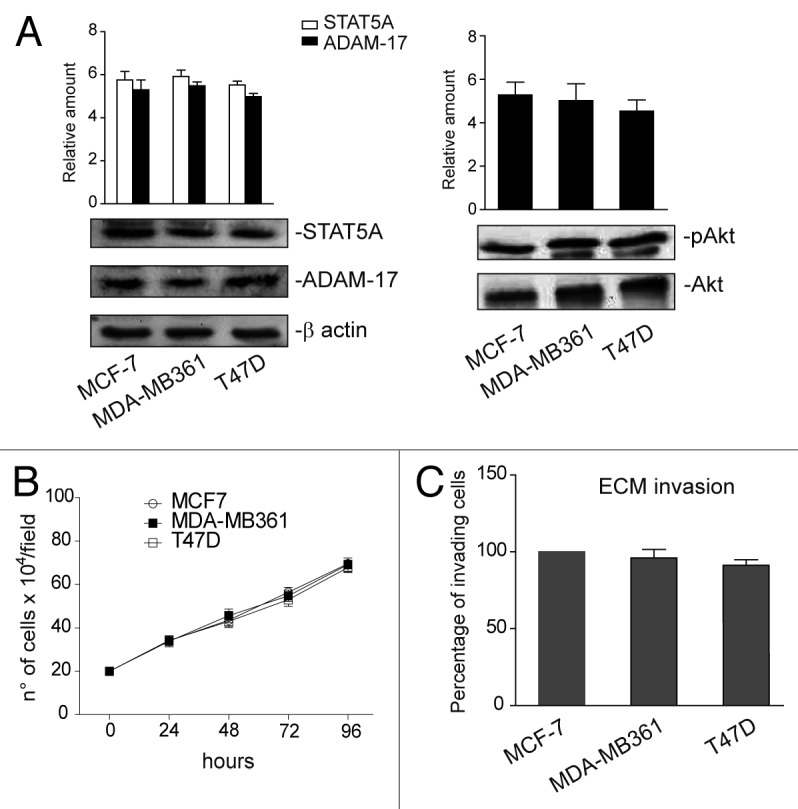
Figure 9. STAT5A, ADAM-17, and pAkt content in MCF-7, MDA-MB361, and T47D cell lines. (A) Cell extracts from MCF-7, MDA-MB361, and T47D cell lines were analyzed by western blot for STAT5A, ADAM-17, and pAkt content by densitometry (relative amount). Protein levels were normalized to β actin (STAT5A and ADAM-17) or to Akt (pAkt) content. The results are representative of 4 different experiments, performed in triplicate (n = 4). (B) A cell proliferation assay was performed for the indicated time intervals on luminal cell lines (n = 4). (C) An invasion assay was performed on MCF-7, MDA-MB361, and T47D cells. The percentage of invading cells is reported (n = 4).
miR-221/222 target ADAM-17 to drive cell invasion
Finally, the possibility that proteins involved in cell invasion could also be post-transcriptionally regulated by miR-221/222 was evaluated. A number of different prediction software programs (miRBase-MicroCosm version v5, miRanda, TargetScan, and RNAhybrid) were used to this end. Particular attention was paid to ADAM-17, which was found to be expressed in cells highly invasive obtained from the MCF7c, MDA-MB361, and T47D cell lines, but not from the MCF-7 β4i clone (Figs. 9A and C and 10B). Indeed, we found that miR-222 matched the 3′UTR of the ADAM-17 gene (Fig. 10A). The inverse correlation between ADAM-17 and miR-221/222 in the 2 clones (MCF-7c and MCF-7 β4i) (Figs. 10B and 5E) was further confirmed by loss- and gain-of-function experiments (Fig. 10C). Thus, the luciferase reporter vector, containing the full-length 3′UTR of ADAM-17 was transfected into MCF-7c and MCF-7 β4i cells, and luciferase activity was evaluated. As expected, luciferase activity was only detected in MCF-7c cells (Fig. 10D). These results were confirmed by the overexpression or downregulation of miR-221/222 in MCF7c and MCF7b4i cells, respectively (Fig. 10D). The specificity of the miR-221/222-mediated post-transcriptional regulation of ADAM-17 was further confirmed using a luciferase reporter vector that contained a point mutation in the seed sequence of ADAM-17 3′UTR (Fig. 10E).

Figure 10. miR-221/222 post-transcriptionally regulate ADAM-17 expression. (A) Alignment of miR-222 with its potential binding site in the 3′UTR of ADAM-17 mRNA (miRANDA website). (B) ADAM-17 and β actin content in MCF-7c and MCF-7 β4i clones (***P < 0.001 MCF-7 β4i vs. MCF-7c). The results are representative of 3 different experiments, performed in triplicate (n = 3). (C) Cell extracts from MCF-7c and MCF-7 β4i cells subjected to gain- and loss-of-function experiments, respectively, were analyzed by western blot for ADAM-17 content. Protein levels were normalized to β actin content (***P < 0.001 gain- and loss-of-function values vs. control values for ADAM-17 content). The results are representative of 4 different experiments performed in triplicate (n = 4). (D) Luciferase activity was evaluated in MCF-7 clones, treated as indicated. Relative luciferase activity is reported (***P < 0.001 pre-miR neg c-transfected MCF-7c in presence of pGL3–3′UTR ADAM-17 vs. pGL3; ***P < 0.001 pre-miR-221 or pre-miR-222 vs. pre-miR neg c transfected MCF-7c; ***P < 0.001 anti-miR-221 or anti-miR-222 vs. anti-miR neg c transfected MCF-7 β4i) (n = 4). (E) pGL3 empty vector or pGL3–3′UTR ADAM-17 mutated seed sequence (mut) were co-transfected with pre-miR neg c or pre-miR-221 or pre-miR-222 precursors in MCF-7c. Relative luciferase activity is reported (***P < 0.001 pre-miR neg c transfected MCF-7c in presence of pGL3–3′UTRmut ADAM-17 vs. pGL3) (n = 3).
The biological relevance of ADAM-17 in our models was evaluated using siRNA technology (Fig. 11A). Figure 11A–C shows that ADAM-17 depleted MCF-7c cells were still able to proliferate as sustained by cell counting, cyclin D1 expression, and phosphorylated Akt content. On the other hand, ADAM-17 depletion impairs cell invasive capability (Fig. 11E) without affecting β4 integrin expression (Fig. 11D). Antagomir expression in MCF-10A cells had no effect on ADAM-17 expression once again (Fig. 3A).

Figure 11. miR-221/222-regulated ADAM-17 expression drives mammary tumor cell invasion. (A) ADAM-17, cyclin D1 and β actin content were analyzed by western blot in MCF-7c cells transfected for 48 h with scramble or ADAM-17 siRNA (***P < 0.001 siRNA ADAM-17 vs. scramble for ADAM-17 content). The results are representative of 4 different experiments, performed in triplicate (n = 4). (B) Western blot analysis for pAkt and Akt content in MCF-7c, whether it was silenced for ADAM-17 or not. The results are representative of 3 different experiments performed in triplicate (n = 3). (C) A cell proliferation assay for indicated time intervals on MCF-7c cells, treated as above (n = 4). (D) Cell extracts from MCF-7c cells, silenced or not, for ADAM-17 were analyzed by western blot to evaluate β4 integrin and β actin content. The results are representative of 3 different experiments performed in triplicate (n = 3). (E) An invasion assay was performed on MCF-7c cells, treated as above. The percentage of invading cells is reported (***P < 0.001 siRNA ADAM-17 vs. scramble) (n = 4). (F) Co-immunoprecipitation experiments were performed in MCF-7 wt cells using ADAM-17 and β4 integrin antibodies. β4 integrin and ADAM-17 content was evaluated. The results are representative of 3 different experiments performed in triplicate (n = 3).
Finally, the presence of a disintegrin domain in the ADAM-17 sequence led us to investigate whether β4 integrin and ADAM-17 act in cooperation to mediate cell invasion. Indeed, we found that β4 integrin is physically associated to ADAM-17 in our experimental conditions (Fig. 11F).
Discussion
In this study, we demonstrate that miR-221/222 plays a role in regulating the tumor growth and invasion of luminal subtype breast carcinomas by regulating a number of genes. miRNAs are abnormally expressed in a variety of cancer types, including breast cancers, and they can act as oncomiRs or oncosuppressor-miRs depending on cellular contest.38 This is particularly true for miR-221/222 that may act as oncomiRs in tumors of epithelial origin39 and as oncosuppressors or oncomiRs in hemopoietic malignancies.40,41 A correlation between miR-221/222 and the response of breast cancer cells to tamoxifen has been reported.42 Moreover, the miRNA microarray screening of luminal- and basal-like subtypes revealed that basal-like tumors express high miR-221/222 levels.43,44 Likewise, Pincini et al.45 demonstrated that in an ErbB2-transformed mammary cell model Crk-associated substrate (p130Cas) overexpression turns on a specific invasive signature resulting from deregulation of genes and miRs, including miR-221/222. More recently, these miRs33 have been found to be inversely correlated with β4 integrin expression, while we have found, by analyzing the gene expression profiling data set generated by the TCGA consortium,34 that miR-221/222 and β4 integrin are expressed in luminal carcinomas as well. However, no inverse correlation between miR-221/222 and β4 integrin expression was found. Since TCGA is a genomic database that does not consider tumor grade, the possibility of a correlation between miR-221/222 and β4 integrin protein expression was investigated in human carcinoma samples of luminal subtype, which display distinct differentiation grade and proliferation activity. It was found that β4 expression was associated with poor primary luminal Lum-IC differentiation (G3 tumors) and with low miR-221/222 expression, which is consistent with β4 integrin's role in tumor progression. On the other hand, an inverse correlation between the expression of both miRs and the tumor proliferating index, Ki67, was discovered.
The expression, localization, and cytoskeletal interactions of β4 integrin are crucial drivers of cancer cell proliferation and invasion.46 The increase in β4 expression in cancers of epithelial origin has suggested that β4 expression might be regulated, at least in part, at the transcriptional level.47 In the present study, we demonstrate, for the first time, that β4 integrin expression is under the control of miR-221/222 in the MCF-7 cell line, a known breast carcinoma cell line of the luminal subtype.48 The concept of miR-221/222 dictating tumor aggressiveness by regulating β4 expression was then further validated by functional studies. Cells that overexpress miR-221/222 displayed low proliferation rate and invasiveness as well as almost undetectable PI3K/Akt activation.49 The finding that miR-221/222 downregulation in MCF-10A cells did not affect β4 integrin expression further sustains that such a mechanism specifically controls cancer cell fate.
The role of the PI3K signaling cascade in mediating breast tumor progression50 has spurred the development of numerous classes of PI3K, Akt, and mTOR inhibitors that are currently used in clinics.50,51 However, the appearance of resistance to such approaches highlighted the need for novel anti-cancer strategies. The combined targeting of the PI3K/mTOR and the JAK2/STAT5 pathways in breast cancers has provided a new therapeutic opportunity.52 Indeed, the STAT pathway is activated in response to different stimuli,53-55 and cross-talk between STAT5 and integrins has been also reported.56-58 We herein demonstrate, for the first time, that miR-221/222 strictly control STAT5A expression in breast cancer cells that recapitulate luminal subtype. Moreover, loss-of-function experiments in β4 integrin-depleted cells indicate that such a post-transcriptional event is independent of β4 integrin expression, but depends on miR-221/222 deregulation. Although the role of STAT5 in breast cancers is still being debated,28 agents that downregulate STAT5 reduce the growth of breast cancer cell lines derived from different breast cancer sub-types.31,32 In fact, we demonstrated that low STAT5 expression was associated with a low proliferation rate. The biological relevance of STAT5 in mediating breast cancer cell proliferation is further sustained by the ectopic expression of the constitutively active form or the dominant-negative STAT5 construct. STAT5 is crucial for biological functions such as cell proliferation and survival, but also for cell migration.56,59 We failed to detect any relevant role for STAT5 in cell invasion in our models. This suggests that STAT5 primarily controls cell proliferation in carcinomas of the luminal subtype.
Cell invasion in tumors lacking hemidesmosome anchorage is facilitated by β4 integrin switching into a more active signaling receptor.9,10 However, extracellular proteases, such as ADAMs, also actively participate in invasion by acting as a control device for cell–ECM interactions.60 ADAMs have a complex multi-domain structure with proteolytic potential and adhesive and signaling properties.59 Data from pre-clinical cancer models point to the “shedding” activities of ADAMs; cleaving or solubilizing the ectodomain of cytokines, growth factors, receptors, and adhesion molecules may regulate activities that include cell migration and proliferation.60 Indeed, a correlation between ADAM-17 expression and high-grade invasive breast tumors has been reported in humans.18 In keeping with clinical data, we demonstrate that human-derived cell lines, which display high proliferative rate and invasive capability, express ADAM-17. Possibly due to the presence of a disintegrin domain, it is able to bind and to regulate the activity of integrins.12,16 Furthermore, ADAM-17 physically interacts with β4 integrin and controls β4 integrin-mediated cell invasion, but not cell proliferation. This is particularly true, as ADAM-17 knockdown was associated with impaired β4 integrin-mediated invasion, while it had no effect on cell proliferation. Finally, we herein demonstrate, for the first time, that ADAM-17 expression is strictly controlled by post-transcriptional mechanisms involving miR-221/222, specifically in tumor cells.
β4 overexpression in cancer cells supports many of the cellular events that are involved in tumor progression.9,10 Although the precise mechanisms involved are still unclear, we identify a post-transcriptional mechanism driven by miR-221/222 that modulates β4 integrin, STAT5A and ADAM-17 expression in luminal carcinomas that display more aggressive behavior. This suggests that miR-221/222 expression levels in combination with those of β4 integrin, ADAM17, and STAT5 may confer additional benefits to the prognostic stratification of luminal breast cancer subtypes. Moreover, the pleiotropic effects of miR-221/222, which only occur in cancer cells, indicate that efforts should now be made to develop successful in vivo delivery systems in order to investigate the therapeutic potential of pre-miR-221/222 in reverting cancer cell behavior.
Materials and Methods
Reagents and antibodies
RPMI medium, bovine serum albumin (BSA), fetal calf serum (FCS), low gelling temperature agarose, RNase, propidium iodide, SDS, PIPES, Triton X-100, Nonidet P-40, NaCl, NaF, NaOV4, Na4P2O7, MgCl2, KCl, HCl, Na-azide, Hepes, Tris, EDTA, EGTA, ethanol, aprotinin, PMSF, DTT, leupeptin, penicillin-streptomycin, and HEPES were purchased from Sigma-Aldrich. Protein molecular weight markers, Acrylammide, polyvinylidene difluoride (PVDF) membranes, chemiluminescence reagents (ECL), and HRP-conjugated anti-rabbit or anti-mouse IgG were purchased from Bio-Rad. Gene Ruler™ DNA ladder mix and Gene Ruler™ DNA ladder plus were purchased from Fermentas International Inc. Lipofectin® Reagent and TRIzol were purchased from Invitrogen™ (Life Technologies). Monoclonal anti-PCNA antibody was purchased from Abcam. Anti-p57Kip2, anti-p27Kip1, anti-β-actin, anti-STAT5A, anti-ADAM-17, and anti-cyclin D1 antibodies were purchased from S. Cruz Biotechnology. Anti-phospho Akt (Ser-473/Thr-308) and anti-Akt antibody were purchased from Cell Signaling. Anti-β4 antibodies (439-9B and 450-11A) were prepared as described by Gambaletta et al.9
Laminin 5 preparation
A laminin 5-enriched matrix was prepared from 804G cells as previously described.61 Briefly, confluent 804G cells, in either 100-mm dishes or 96-well plates, were washed in sterile PBS and detached from the underlying laminin 5-enriched matrix via treatment in 20 mmol/L NH4OH at 4 °C for 10 min and were subsequently washed twice with sterile PBS. Poly-L-lysine (Calbiochem) was used as a control matrix at a concentration of 10 mg/mL.
Cell lines
The human breast carcinoma cell lines MCF-7, MDA-MB361, and T47D were obtained from the American Type Culture Collection, maintained in RPMI medium supplemented with 10% FCS (Invitrogen), and cultured on laminin. The generation of MCF-7 scrambled short hairpin RNA (scr-shRNA), defined MCF7c and the MCF-7 β4 shRNA cell subclones, defined MCF7 β4i, whether they expressed α6β4 integrin or not, were obtained as previously described.22 The rat bladder epithelial cell line 804G used to produce matrigel was kindly provided by Dr G Meneguzzi (Faculty of Medicine, Institut National de la Sante´ et de la Recherche Médicale U634). The human mammary cell line MCF-10A, used as a control,62 was kindly provided by Prof D Taverna (Department of Molecular Biotechnology and Health Sciences, University of Torino).
Cell proliferation
Proliferative activity was assayed via direct cell count by 3 individual operators in triplicate, as previously described (number ± SEM of cells per field, 10× magnification).55 Proliferation was also evaluated as the expression of the DNA-polymerase-δ auxiliary protein (e.g., proliferating-cell nuclear antigen, PCNA) or by flow cytometer (FACScan, Becton Dickinson Immunocytometry Systems) analysis. Cell cycle phases were also analyzed; breast cancer cells, treated as indicated, were fixed with 70% ethanol. DNA was stained with propidium iodide and analyzed with FACScan after digestion with RNase.56
Cell invasion
Cell invasion was assessed using a 48-well modified Boyden chamber (NeuroProbe) and 8-μm pore polyvinyl pyrrolidone-free polycarbonate Nucleopore filters (Costar). The filters were coated with 3 mg/mL Cultrex (Trevigen). The lower compartment of the chamber was filled with conditioned serum-free medium, produced from NIH3T3 fibroblasts, for 24 h. MCF-7 wt, MCF-7c, and MCF-7 β4i cells (5 × 104cells/ml), treated as indicated, were harvested and placed in the upper compartment of the Boyden chamber. After 8 h of incubation at 37 °C, the cells migrated onto the lower surface of the filters and were fixed and stained with DiffQuick (Merz-Dade). The migrated cells in 12 high-power fields were counted. Each assay was performed in quadruplicate and repeated at least 3 times. The ability of the cells to adhere to the filters was verified by staining the upper side of the filter for each cell line.
Clonogenic assay
To investigate the ability of tumor cells to form colonies, 1 × 105 cells were re-suspended in 0.3% agarose (low gelling temperature agarose), whether they overexpressed miR-221/222 or not, and seeded on 6-well plates pre-plated with 0.6% regular agarose. Colonies were counted after 3 wk by 3 individual operators in triplicate (number ± SEM of cells per field, 10× magnification)
Analysis of TCGA data
Gene and microRNA expression profiling data of primary breast tumors were obtained from the website (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/),34 associated to TCGA consortium (PMID 2300897), and selected based on the clinical and molecular parameters reported on the same website.
Human carcinoma samples (luminal subtype, Lum-ICs)
A series of 15 ER-positive invasive carcinomas of no special type63 were retrieved from the archives of the Pathology Unit at our institution. The study was approved by the ethic institutional review board for “Biobanking and use of human tissue for experimental studies” at the Pathology Services of the Azienda Ospedaliera Città della Salute e della Scienza di Torino. Written informed consent was obtained from all patients to authorize their tissue for use in research.
The cohort included 6 low-grade (G1) carcinomas and 9 high-grade (G3) carcinomas.64,65 ER positivity was scored according to the ASCO/CAP guidelines,65 and proliferation index was evaluated as a continuous variable (percentage of stained cells).66
Immunohistochemistry (IHC)
IHC was performed on 3-μm thick sections of formalin-fixed paraffin-embedded tissues (FFPE) using the Ventana BenchMark® XT automated immunostainer (Ventana Medical Systems). Slides were incubated with anti-β4-integrin antibody (diluition 1:200) for 32 min at room temperature, after protease 1 (Ventana Medical Systems) pre-treatment (4 min). Positive and negative controls (omission of the primary antibody and IgG-matched serum) were included for each immunohistochemical run.
RNA isolation and quantitative real-time PCR (qRT-PCR) for miRNAs
Total RNA was isolated, using the mirVana extraction kit (Ambion), from MCF-7 wt and cell clones, from MDA-MB361, T47D, and MCF-10A cells. RNA was also extracted from fresh frozen blocks. The number of sections needed to obtain a nucleic acid yield adequate for molecular analysis depended on the type of tissue samples (fibrosis, cellularity) and sample dimension. Sections were collected in a 1.5-ml sterile Eppendorf tube. RNA extraction from fresh frozen sections was performed with 1 ml of Trizol reagent (Invitrogen) according to manufacturer’s instructions. Isolated RNA was then reverse-transcribed using a TaqMan microRNA RT kit specific for miR-221 and miR-222.
Reverse miRNAs were subjected to quantitative real-time PCR using the TaqMan microRNA assay kit and the ABI PRISM 7700 sequence detection system (Applied Biosystems). miRNA expression was normalized to small nuclear RNA, RNU6B. Gain- and loss-of-function experiments were performed in MCF-7 wt, MCF-7c, MCF-7 β4i, or MCF-10A cells as previously described.26
ADAM-17 silencing by small interfering RNAs (siRNA)
To obtain ADAM-17 inactivation, MCF-7c cells were transiently transfected with MISSION siRNA for ADAM-17 or with duplex siRNAs purchased from Sigma-Aldrich. Transfection was performed according to vendor’s instructions. Forty-eight hours later whole-cell extracts were processed. Cell viability was evaluated at the end of each experiment.
Dominant-negative STAT5A (ΔSTAT5A) and STAT1*6 vector transfection
MCF-7c and MCF-7 β4i cells were transiently transfected with the ΔSTAT5A construct or with STAT1*6 plasmid vector, as previously described.37 The empty vector pCNeo was used as control.
Luciferase miRNA target reporter assay
The luciferase reporter assay was performed using a construct generated by sub-cloning into the restriction site XbaI of the luciferase reporter vector pGL3 Basic Vector (Promega). The PCR products were amplified from full-length 3′UTR of ADAM-17, STAT5A and β4 integrin DNA. The PCR products were obtained using the following primers:
ADAM-17: sense, 5′TCTAGATTTA GTTCTCAGCT CTTCTGAC3′
antisense, 5′TCTAGAGTCT CACTCTGTCA CCCA3′:
STAT5A: sense, 5′AAGAGCTCAT GTTTGAATCC CACGCT3′
antisense,5′TTGAGCTCAC ACAAATGTGT GGTCTT3′:
β4 integrin: sense, 5′TCTAGATGAC CGCACCCTGC CCCACC3′
antisense, 5′TCTAGAAGCA GTAGCAAAAC CATTAT3′
The site-directed mutagenesis of 3′UTR ADAM-17 and 3′UTR β4 integrin amplified PCR product was performed to obtain the mutated miR-222 or miR-221 binding site, respectively. The sequence was generated using the Quik-Change SiteDirect Mutagenesis kit (Stratagene). The oligonucleotides used were: sense, 5′CTAGTTATTA CCTATATTTT TTATGTAGC3′ for ADAM-17 and sense, 5′TGTAACCAAA GATATGTAAG CAGCACAAG3′ for β4 integrin, which contained the desired mutation, was designed according to the manufacturer’s instructions (the mutated nucleotides are underlined and italicized). The mutated 3′UTR STAT5A luciferase vector was obtained as previously described.26
The insert identities were verified by sequencing. The pGL3, pGL3–3′UTR ADAM-17, pGL3–3′UTR STAT5A, and pGL3–3′UTR β4 full-length or mutated reporter vectors were transiently co-transfected in MCF-7c or MCF-7 β4i cells, treated as indicated, at a 30:1 molar ratio with the pRL vector coding for the Renilla luciferase used as an internal control for the luciferase assay as previously described.67 Luciferase activities were analyzed 48 h after transfection by Dual-Luciferase Report Assay System (Promega) as previously described.26,67
Statistical analysis
Comparison and significance of differences tests between 2 groups were performed using a t test. Comparisons between 3 or more groups were performed using one-way ANOVA, and significance of difference tests were evaluated with Newman–Keuls multicomparison post-test. P values * < 0.05, ** < 0.01, and *** < 0.001 were considered significant and were indicated with different symbols, as detailed in each figure legend. All statistical analyses were performed with Graph Pad Prism version 5.04 software (Graph Pad Software, Inc). Densitometric analysis was used to calculate the differences in the fold induction of protein levels and normalized to β actin or to Akt content and reported as “relative amount” in figures.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Dr Brizzi is the guarantor of this work, and as such had full access to all the data and takes full responsibility for the integrity of data and the accuracy of data analysis. We are grateful to Prof D Taverna for providing us with the MCF-10A cell line. This work has been supported by grants from the Italian Association for Cancer Research (AIRC) to M.F.B. (IG 5649). AIRC 5 × 1000 (SPMCO 9979) and Filas Lazio to R.F.
Glossary
Abbreviations:
- STAT
signal transducer and activator of transcription
- ADAM
a disintegrin and metalloprotease
- ECM
extracellular matrix
- 3′UTR
3′untranslated region
- PCNA
Proliferating-Cell Nuclear Antigen
- scr-shRNA
scramble-short hairpin ribonucleic acid
- wt
wild-type
- PI3K
phosphatidylinositol 3-kinase
References
- 1.Hynes RO. The dynamic dialogue between cells and matrices: implications of fibronectin’s elasticity. Proc Natl Acad Sci U S A. 1999;96:2588–90. doi: 10.1073/pnas.96.6.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falcioni R, Perrotti N, Piaggio G, Kennel SK, Sacchi A. Insulin-induced phosphorylation of the beta-4 integrin subunit expressed on murine metastatic carcinoma cells. Mol Carcinog. 1989;2:361–8. doi: 10.1002/mc.2940020611. [DOI] [PubMed] [Google Scholar]
- 3.Raymond K, Kreft M, Song JY, Janssen H, Sonnenberg A. Dual Role of alpha6beta4 integrin in epidermal tumor growth: tumor-suppressive versus tumor-promoting function. Mol Biol Cell. 2007;18:4210–21. doi: 10.1091/mbc.E06-08-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu S, Simin K, Khan A, Mercurio AM. Analysis of integrin beta4 expression in human breast cancer: association with basal-like tumors and prognostic significance. Clin Cancer Res. 2008;14:1050–8. doi: 10.1158/1078-0432.CCR-07-4116. [DOI] [PubMed] [Google Scholar]
- 5.Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J Cell Biol. 1997;139:1873–84. doi: 10.1083/jcb.139.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spinardi L, Einheber S, Cullen T, Milner TA, Giancotti FG. A recombinant tail-less integrin beta 4 subunit disrupts hemidesmosomes, but does not suppress alpha 6 beta 4-mediated cell adhesion to laminins. J Cell Biol. 1995;129:473–87. doi: 10.1083/jcb.129.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee EC, Lotz MM, Steele GD, Jr., Mercurio AM. The integrin alpha 6 beta 4 is a laminin receptor. J Cell Biol. 1992;117:671–8. doi: 10.1083/jcb.117.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaapveld RQ, Borradori L, Geerts D, van Leusden MR, Kuikman I, Nievers MG, Niessen CM, Steenbergen RD, Snijders PJ, Sonnenberg A. Hemidesmosome formation is initiated by the beta4 integrin subunit, requires complex formation of beta4 and HD1/plectin, and involves a direct interaction between beta4 and the bullous pemphigoid antigen 180. J Cell Biol. 1998;142:271–84. doi: 10.1083/jcb.142.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gambaletta D, Marchetti A, Benedetti L, Mercurio AM, Sacchi A, Falcioni R. Cooperative signaling between alpha(6)beta(4) integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J Biol Chem. 2000;275:10604–10. doi: 10.1074/jbc.275.14.10604. [DOI] [PubMed] [Google Scholar]
- 10.Mercurio AM, Bachelder RE, Rabinovitz I, O’Connor KL, Tani T, Shaw LM. The metastatic odyssey: the integrin connection. Surg Oncol Clin N Am. 2001;10:313–28, viii-ix. [viii-ix.] [PubMed] [Google Scholar]
- 11.Bon G, Folgiero V, Bossi G, Felicioni L, Marchetti A, Sacchi A, Falcioni R. Loss of beta4 integrin subunit reduces the tumorigenicity of MCF7 mammary cells and causes apoptosis upon hormone deprivation. Clin Cancer Res. 2006;12:3280–7. doi: 10.1158/1078-0432.CCR-05-2223. [DOI] [PubMed] [Google Scholar]
- 12.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–89. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan H, Turck CW, Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J Biol Chem. 2003;278:18617–27. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- 14.Díaz-Rodríguez E, Montero JC, Esparís-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13:2031–44. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Black RA. Tumor necrosis factor-alpha converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/S1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 16.Reiss K, Saftig P. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–37. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Duffy MJ, McKiernan E, O’Donovan N, McGowan PM. Role of ADAMs in cancer formation and progression. Clin Cancer Res. 2009;15:1140–4. doi: 10.1158/1078-0432.CCR-08-1585. [DOI] [PubMed] [Google Scholar]
- 18.McGowan PM, McKiernan E, Bolster F, Ryan BM, Hill AD, McDermott EW, Evoy D, O’Higgins N, Crown J, Duffy MJ. ADAM-17 predicts adverse outcome in patients with breast cancer. Ann Oncol. 2008;19:1075–81. doi: 10.1093/annonc/mdm609. [DOI] [PubMed] [Google Scholar]
- 19.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 21.Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009;60:167–79. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 22.Iorio MV, Casalini P, Piovan C, Braccioli L, Tagliabue E. Breast cancer and microRNAs: therapeutic impact. Breast. 2011;20(Suppl 3):S63–70. doi: 10.1016/S0960-9776(11)70297-1. [DOI] [PubMed] [Google Scholar]
- 23.Heneghan HM, Miller N, Lowery AJ, Sweeney KJ, Kerin MJ. MicroRNAs as Novel Biomarkers for Breast Cancer. J Oncol. 2009;2009:950201. doi: 10.1155/2010/950201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah MY, Calin GA. MicroRNAs miR-221 and miR-222: a new level of regulation in aggressive breast cancer. Genome Med. 2011;3:56. doi: 10.1186/gm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen WX, Hu Q, Qiu MT, Zhong SL, Xu JJ, Tang JH, Zhao JH. miR-221/222: promising biomarkers for breast cancer. Tumour Biol. 2013;34:1361–70. doi: 10.1007/s13277-013-0750-y. [DOI] [PubMed] [Google Scholar]
- 26.Dentelli P, Rosso A, Orso F, Olgasi C, Taverna D, Brizzi MF. microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler Thromb Vasc Biol. 2010;30:1562–8. doi: 10.1161/ATVBAHA.110.206201. [DOI] [PubMed] [Google Scholar]
- 27.Shan L, Yu M, Clark BD, Snyderwine EG. Possible role of Stat5a in rat mammary gland carcinogenesis. Breast Cancer Res Treat. 2004;88:263–72. doi: 10.1007/s10549-004-0805-2. [DOI] [PubMed] [Google Scholar]
- 28.Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int J Cancer. 2004;108:665–71. doi: 10.1002/ijc.11619. [DOI] [PubMed] [Google Scholar]
- 29.Furth PA, Nakles RE, Millman S, Diaz-Cruz ES, Cabrera MC. Signal transducer and activator of transcription 5 as a key signaling pathway in normal mammary gland developmental biology and breast cancer. Breast Cancer Res. 2011;13:220. doi: 10.1186/bcr2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita H, Iwase H, Toyama T, Fujii Y. Naturally occurring dominant-negative Stat5 suppresses transcriptional activity of estrogen receptors and induces apoptosis in T47D breast cancer cells. Oncogene. 2003;22:1638–52. doi: 10.1038/sj.onc.1206277. [DOI] [PubMed] [Google Scholar]
- 31.Lim EJ, Hong DY, Park JH, Joung YH, Darvin P, Kim SY, Na YM, Hwang TS, Ye SK, Moon ES, et al. Methylsulfonylmethane suppresses breast cancer growth by down-regulating STAT3 and STAT5b pathways. PLoS One. 2012;7:e33361. doi: 10.1371/journal.pone.0033361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JH, Darvin P, Lim EJ, Joung YH, Hong DY, Park EU, Park SH, Choi SK, Moon ES, Cho BW, et al. Hwanggeumchal sorghum induces cell cycle arrest, and suppresses tumor growth and metastasis through Jak2/STAT pathways in breast cancer xenografts. PLoS One. 2012;7:e40531. doi: 10.1371/journal.pone.0040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerson KD, Maddula VS, Seligmann BE, Shearstone JR, Khan A, Mercurio AM. Effects of β4 integrin expression on microRNA patterns in breast cancer. Biol Open. 2012;1:658–66. doi: 10.1242/bio.20121628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–65. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brizzi MF, Dentelli P, Rosso A, Calvi C, Gambino R, Cassader M, Salvidio G, Deferrari G, Camussi G, Pegoraro L, et al. RAGE- and TGF-beta receptor-mediated signals converge on STAT5 and p21waf to control cell-cycle progression of mesangial cells: a possible role in the development and progression of diabetic nephropathy. FASEB J. 2004;18:1249–51. doi: 10.1096/fj.03-1053fje. [DOI] [PubMed] [Google Scholar]
- 38.Hummel R, Hussey DJ, Haier J. MicroRNAs: predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur J Cancer. 2010;46:298–311. doi: 10.1016/j.ejca.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 39.Volinia S, Calin GA, Liu C-G, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Vecchiarelli-Federico LM, Li YJ, Egan SE, Spaner D, Hough MR, Ben-David Y. The miR-17-92 cluster expands multipotent hematopoietic progenitors whereas imbalanced expression of its individual oncogenic miRNAs promotes leukemia in mice. Blood. 2012;119:4486–98. doi: 10.1182/blood-2011-09-378687. [DOI] [PubMed] [Google Scholar]
- 41.Di Martino MT, Gullà A, Cantafio ME, Lionetti M, Leone E, Amodio N, Guzzi PH, Foresta U, Conforti F, Cannataro M, et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget. 2013;4:242–55. doi: 10.18632/oncotarget.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao JJ, Lin J, Yang H, Kong W, He L, Ma X, Coppola D, Cheng JQ. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem. 2008;283:31079–86. doi: 10.1074/jbc.M806041200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, et al. miR-221/222 targeting of trichorhinophalangeal 1 (TRPS1) promotes epithelial-to-mesenchymal transition in breast cancer. Sci Signal. 2011;4:pt5. doi: 10.1126/scisignal.2002258. [DOI] [PubMed] [Google Scholar]
- 44.Radojicic J, Zaravinos A, Vrekoussis T, Kafousi M, Spandidos DA, Stathopoulos EN. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle. 2011;10:507–17. doi: 10.4161/cc.10.3.14754. [DOI] [PubMed] [Google Scholar]
- 45.Pincini A, Tornillo G, Orso F, Sciortino M, Bisaro B, Leal MdelP, Lembo A, Brizzi MF, Turco E, De Pittà C, et al. Identification of p130Cas/ErbB2-dependent invasive signatures in transformed mammary epithelial cells. Cell Cycle. 2013;12:2409–22. doi: 10.4161/cc.25415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C, Yang X, Pursell B, Mercurio AM. Id2 complexes with the SNAG domain of Snai1 inhibiting Snai1-mediated repression of integrin β4. Mol Cell Biol. 2013;33:3795–804. doi: 10.1128/MCB.00434-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bon G, Di Carlo SE, Folgiero V, Avetrani P, Lazzari C, D’Orazi G, Brizzi MF, Sacchi A, Soddu S, Blandino G, et al. Negative regulation of beta4 integrin transcription by homeodomain-interacting protein kinase 2 and p53 impairs tumor progression. Cancer Res. 2009;69:5978–86. doi: 10.1158/0008-5472.CAN-09-0244. [DOI] [PubMed] [Google Scholar]
- 48.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bon G, Folgiero V, Di Carlo S, Sacchi A, Falcioni R. Involvement of alpha6beta4 integrin in the mechanisms that regulate breast cancer progression. Breast Cancer Res. 2007;9:203. doi: 10.1186/bcr1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 51.Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog. 2012;17:69–95. doi: 10.1615/CritRevOncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- 52.Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Müller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 53.Dentelli P, Rosso A, Calvi C, Ghiringhello B, Garbarino G, Camussi G, Pegoraro L, Brizzi MF. IL-3 affects endothelial cell-mediated smooth muscle cell recruitment by increasing TGF beta activity: potential role in tumor vessel stabilization. Oncogene. 2004;23:1681–92. doi: 10.1038/sj.onc.1207290. [DOI] [PubMed] [Google Scholar]
- 54.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dentelli P, Rosso A, Olgasi C, Camussi G, Brizzi MF. IL-3 is a novel target to interfere with tumor vasculature. Oncogene. 2011;30:4930–40. doi: 10.1038/onc.2011.204. [DOI] [PubMed] [Google Scholar]
- 56.Defilippi P, Rosso A, Dentelli P, Calvi C, Garbarino G, Tarone G, Pegoraro L, Brizzi MF. beta1 Integrin and IL-3R coordinately regulate STAT5 activation and anchorage-dependent proliferation. J Cell Biol. 2005;168:1099–108. doi: 10.1083/jcb.200405116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uberti B, Dentelli P, Rosso A, Defilippi P, Brizzi MF. Inhibition of β1 integrin and IL-3Rβ common subunit interaction hinders tumour angiogenesis. Oncogene. 2010;29:6581–90. doi: 10.1038/onc.2010.384. [DOI] [PubMed] [Google Scholar]
- 58.Brizzi MF, Tarone G, Defilippi P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr Opin Cell Biol. 2012;24:645–51. doi: 10.1016/j.ceb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Bernaciak TM, Zareno J, Parsons JT, Silva CM. A novel role for signal transducer and activator of transcription 5b (STAT5b) in beta1-integrin-mediated human breast cancer cell migration. Breast Cancer Res. 2009;11:R52. doi: 10.1186/bcr2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8:929–41. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 61.Kennel SJ, Epler RG, Lankford TK, Foote LJ, Dickas V, Canamucio M, Cavalierie R, Cosimelli M, Venturo I, Falcioni R, et al. Second generation monoclonal antibodies to the human integrin alpha 6 beta 4. Hybridoma. 1990;9:243–55. doi: 10.1089/hyb.1990.9.243. [DOI] [PubMed] [Google Scholar]
- 62.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–86. [PubMed] [Google Scholar]
- 63.Lakhani S, Ellis IO, Schnitt SJ, Hoon Tan P, van de Vijer M. WHO Cassification of Tumors of the Breast. Lyon: International Agency for Research on Cancer; 2012. [Google Scholar]
- 64.Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991;19:403–10. doi: 10.1111/j.1365-2559.1991.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 65.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, et al. American Society of Clinical Oncology. College of American Pathologists American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version) Arch Pathol Lab Med. 2010;134:e48–72. doi: 10.5858/134.7.e48. [DOI] [PubMed] [Google Scholar]
- 66.Dowsett M, Nielsen TO, A’Hern R, Bartlett J, Coombes RC, Cuzick J, Ellis M, Henry NL, Hugh JC, Lively T, et al. International Ki-67 in Breast Cancer Working Group Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103:1656–64. doi: 10.1093/jnci/djr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Togliatto G, Trombetta A, Dentelli P, Rosso A, Brizzi MF. MIR221/MIR222-driven post-transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high-glucose- and AGE-mediated vascular cell damage. Diabetologia. 2011;54:1930–40. doi: 10.1007/s00125-011-2125-5. [DOI] [PubMed] [Google Scholar]