

Abstract
We established new methods for cloning cDNA ends that start with reverse transcription (RT) and soon proceed with the synthesis of the second cDNA strand, avoiding manipulations of fragile RNA. Our 3′-end cloning method does not involve poly-dT primers and polymerase chain reactions (PCR), is low in efficiency but high in fidelity and can clone those RNAs without a poly-A tail. We also established a cDNA protection assay to supersede RNA protection assay. The protected cDNA can be amplified, cloned and sequenced, enhancing sensitivity and fidelity. We report that RT product using gene-specific primer (GSP) cannot be gene- or strand-specific because RNA sample contains endogenous random primers (ERP). The gene-specificity may be improved by adding a linker sequence at the 5′-end of the GSP to prime RT and using the linker as a primer in the ensuing PCR. The strand-specificity may be improved by using strand-specific DNA oligos in our protection assay. The CDK4 mRNA and TSPAN31 mRNA are transcribed from the opposite DNA strands and overlap at their 3′ ends. Using this relationship as a model, we found that the overlapped sequence might serve as a primer with its antisense as the template to create a wrong-template extension in RT or PCR. We infer that two unrelated RNAs or cDNAs overlapping at the 5′- or 3′-end might create a spurious chimera in this way, and many chimeras with a homologous sequence may be such artifacts. The ERP and overlapping antisense together set complex pitfalls, which one should be aware of.
Keywords: antisense, chiermic RNA, cDNA protection assay, reverse transcription, polymerase chain reaction, cDNA cloning, RNA protection assay
Introduction
A recent advance in RNA research suggests that virtually the entire non-repeat part of the human genome is transcribed, at least at some times or in some cell types,7,17 with a tally of 161,000 transcripts so far.25 Moreover, it is estimated that over 63% of RNA transcripts are accompanied by antisense counterparts,24 and the Unigene database of the National Center for Biotechnology Information (NCBI) contains over 123,000 human antisense entries.26 One meaning of these figures is that, for most genomic loci, both strands of the DNA double helix are transcribed.6,19 Most antisense transcripts may be non-coding, but there are still many that do encode proteins. For example, the DNA strand opposite to the one encoding the THRA (17q11.2), CDK4 (12q14.1), CCND1 (11q13) and GAPDH (12p13) genes harbors the NR1D1, TSPAN31, LOC100996515 and LOC100996356 protein-coding genes, respectively, as shown in the NCBI. Cloning cDNA often involves reverse-transcription (RT) and polymerase chain reactions (PCR), but the situation wherein antisense is also expressed often sets pitfalls and hurdles, which are widely neglected, in our way of cloning the 5′- or 3′-end of cDNA or determining from which DNA strand an RNA is transcribed. For instance, it may not be easy to clone the 5′ and 3′ ends of the so-called ncRNACCND143 and to determine whether it is transcribed from the same strand as the CCND1 or as the LOC100996515.
Another ribonomic advance suggests that transcripts from about 65% of the human genes form chimeric RNA with a transcript from another gene. This other gene in most cases is nearby on the same chromosome but can also be located on another chromosome.7,17 Actually, modern RNA-sequencing technologies have provided us with thousands of RNA chimeras.14 A tiny number of them are known to be transcribed from fusion genes that are formed due to genetic alterations, such as chromosomal translocation and genomic DNA deletion or amplification.9 Unfortunately, the vast remaining majority, i.e., those not associated with a known genomic alteration, remain putative and are not very meaningful to us so far, because their existence has not been verified with a vigorous method and because their full-length sequence has not been cloned and, thus, their open reading frame is unclear.16 These weaknesses are due mainly to the lack of reliable and efficient approaches of cloning and verification. Current RNA sequencing technologies are reliant on RT or on the principles similar to RT,13 provide only short sequences, and have poor strand-specificity,35 thus only suitable for screening, but not for verification, of long RNA. Cloning methods involving RT-PCR may result in artificial chimeric cDNA8,11,23,30,35,36,38in part because template switching may occur during RT11,23,33and mis-priming can occur in PCR. RNA protection assay is the most commonly used method to verify the true existence of an RNA, in which an in vitro synthesized complementary RNA (cRNA) is used to hybridize with the parental RNA in solution, followed by RNase digestion of the non-hybridized RNA. This method does not involve RT or PCR, which on one hand increases its reliability, but on the other hand makes the method very inefficient.13 Also problematically, the protected RNA cannot be directly sequenced to confirm its identity. For these reasons, most attempts to verify chimeric RNAs still, unfortunately, use problematic RT-PCR. Moreover, it needs to distinguish a true chimeric cDNA end cloned with a routine 5′ or 3′ RACE (rapid amplification of cDNA end) method from the cDNA end of the parent mRNA, as depicted in Figure 1A. Actually, because in most cases the mRNA of each parent gene is more abundant, the cloned cDNA end is more likely to belong to the parent mRNA.

Figure 1. Cloning RNA 3′ end. (A) Two hurdles for cloning the 5′ or 3′ end, and for PCR amplification, of chimeric cDNA: (1) Gene-specific primer (GSP) used in RACE amplifies the 5′ or 3′ end of not only the chimeric cDNA but also the cDNA of a parent gene (black or red line). (2) Forward (F) or reverse (R) primer primes not only the chimeric cDNA but also the cDNA of a parent gene, making the first several cycles of PCR less efficient. (B) Our strategy for cloning RNA 3′ end: After RT with random hexamers, a forward primer of the gene of interest and Taq are used to synthesize the 3′ part of the second cDNA strand. S1 is added to cut the 3′-overhang of the first strand. The cDNA blunt ends are then appended with a dA by Taq, followed by T-A cloning. (C) Illustration of the locations of primers and the S1 cutting site on the CDK4 mRNA. (D) Part of the 3′ sequence obtained, in which the lowercase “a” is added by Taq and the underlined sequence belongs to the T-A vector. The sequence matches completely to the CDK4 mRNA. Note that there is an internal poly-A sequence 21 nt upstream of the authentic poly-A tail. (E–G) Both pairs of primers (F665+R1086 and F136+R1086) designed to amplify the 5′ and the middle regions of the mRNA, respectively, can amplify the RT product (cDNA) of RNA from HeLa cells without S1 digestion (arrowheads). F655+R1086 can still yield a band from the cDNA digested with 10 or 15 units of S1, while the F136+R1086 yielded only a very faint band (arrow) when 10 units were used and no band when 15 units were used.
We attempt to develop methods that are devoid of the above-described weaknesses in cloning and verifying chimeric or antisense-accompanied RNA. Although not yet reaching this aim, we have established new methods for cloning cDNA ends and have established a cDNA protection assay to supersede RNA protection assay, as described in this report. Sometimes “RNA,” but not “mRNA,” is termed herein because our methods are also suitable for cloning those long RNAs and chimeric RNAs that are non-coding. Some pitfalls and artifacts of RT and PCR that are widely neglected in the literature are also described to alert the peers.
Results
Cloning RNA 3′ end
The 3′ end of long RNAs is usually cloned by using a poly-dT oligo to prime the poly-A tail of the RNA or by ligating a linker sequence to the 3′ end since about 50% of the long RNAs lack a poly-A tail,37 although they likely have a poly-A signal. In our strategy, RNA can be primed with random hexamers in RT. Taq DNA polymerase (Taq for brevity) and a forward primer of the interested gene are used to synthesize the 3′ part of the second cDNA strand, which is also the 3′ part of the parental RNA. S1 nuclease (S1 for brevity) is added to cut off the 3′-overhang of the 1st cDNA strand (Fig. 1B), since S1 digests single-stranded, but not double-stranded, DNA or RNA. The blunt ends of the double-stranded cDNA are then appended with a dA by Taq to allow cloning the fragment into a T-A vector (Fig. 1B).
As an example, unDNased RNA from HeLa cells was converted in RT to the first cDNA strand by random hexamers. The CDK4F665 forward primer (all primers listed in Table 1) and PCR Mastermix were mixed with the RT product to synthesize the second strand of CDK4 cDNA by incubation at 72°C for 10 min. S1 was added to cut off the single-stranded part of the 1st cDNA strand upstream of the F665 primer (Fig. 1C). After inactivation of S1 and purification of the double-stranded cDNA, PCR of the cDNA with F136+R1086 primers did not yield signal, which confirmed that the region upstream of F665 (including the F136 sequence) had been digested by S1, while PCR with F665+R1086 yielded a band (Fig. 1E–G), indicating that the double-stranded part could withstand the S1. The amount of S1 might need to be optimized for different target genes, due to the difference in the residuals of RNAs and single stranded cDNAs to be digested, since herein 10 units of S1 was not sufficient (Fig. 1F). A portion of the purified double-stranded cDNA was then cloned into a T-A vector. Sequencing a resultant plasmid and aligning the sequence with the CDK4 mRNA sequence (NM_000075.3) revealed that the canonical 3′ end, including the whole poly-A tail (Fig. 1D), was fully cloned.
Table 1. Primers used.
| Primer | Sequence | Primer | Sequence |
|---|---|---|---|
| NewA | 5′-GTGGAGTCTACGCGAACTTGTCCT17–3′ | CDK4R822 | 5′-TCCACATGTCCACAGGTGTTGC-3′ |
| NewC | 5′-GTGGAGTCTACGCGAACTTGTCC-3′ | CDK4F933 | 5′-GATGACTGGCCTCGAGATGT-3′ |
| NewB mixture | 5′-TCAGGATTGATGGTGCCTACAGC13V-3′ (V = A,G,T) | CDK4R1086 | 5′-AGGCAGAGATTCGCTTGTGT-3′ |
| NewD | 5′-TCAGGATTGATGGTGCCTACAGC-3′ | CDK4F1096 | 5′-TGCAGCACTCTTATCTACATAAGGAT-3′ |
| HPRT1F123 | 5′-CTTCCTCCTCCTGAGCAGTC-3′ | TSPAN31F73 | 5′-AAGCTGTCGGGGTCCTGGAA-3′ |
| HPRT1R683 | 5′-AACACTTCGTGGGGTCCTTT-3′ | TSPAN31F647 | 5′-CTTAAGCATTCAGACGAAGC-3′ |
| CCND1F70 | 5′-TAGCAGCGAGCAGCAGAGTC-3′ | TSPAN31R860 | 5′-ACCCTAGATATTCCCTAAGG-3′ |
| CCND1F183 | 5′-CCCAGCTGCCCAGGAAGAGC-3′ | TSPAN31R1668 | 5′-CTTGGAAGAAGGGACTTTCC-3′ |
| CCND1R981 | 5′-TTGACTCCAGCAGGGCTTCG-3′ | MYCF125 | 5′-GCGCTGAGTATATAAAAGCCGGTT-3′ |
| CCND1R1067 | 5′-TGTGCAAGCCAGGTCCACCT-3′ | MYCR838 | 5′-CCACCGCCGTCGTTGTCTCC-3′ |
| CDK4F136 | 5′-GTATGGGGCCGTAGGAACCG-3′ | BCAS4E1F | 5′-TCCTGATGCTGCTCGTGGAC-3′ |
| CDK4F665 | 5′ TCTGGTGACAAGTGGTGGAA 3′ | BCAS3E25R | 5′-CATACACAGGGACCGAGCTT-3′ |
| NewDCF933 | 5′-TCAGGATTGATGGTGCCTACAGCGATGACTGGCCTCGAGATGT-3′ | ||
| NewDTF647 | 5′-TCAGGATTGATGGTGCCTACAGCCTTAAGCATTCAGACGAAGC-3′ | ||
Note: The number in the primer indicates the first (for forward) or the last (for reverse) nucleotide of that primer in the position of the mRNA. Thus, the range between the F and R numbers should normally be the size of the RT-PCR amplified DNA fragment in agarose gel.
Cloning RNA 5′ end by G-tailing
Tailing the 3′ end of a DNA with terminal deoxynucleotidyl transferase (TdT) is a traditional method for several different purposes, including cloning of the 3′ end of a cDNA. We prime the RNA of interest with a reverse primer in RT to convert the RNA to the first cDNA strand (Fig. 2). After removal of dNTP and short oligos, TdT and dGTP are used to append a poly-dG tail to the 3′ end of the cDNA, which is the RNA 5′ end. A poly-dC mixture is used to prime the poly-dG for synthesis of the second cDNA strand. This poly-dC mixture, referred to as NewB, contains four oligos with a linker sequence (dubbed as NewD) at the 5′ end and with one of the four bases at the 3′ end, so that one of the four oligos can be anchored on the last nucleotide (nt) of the cDNA. A reverse primer and NewD will then be used in PCR to amplify the double-stranded 3′ part of the cDNA (Fig. 2).

Figure 2. Cloning RNA 5′ end. In our strategy, RNA is converted in RT to the first strand of cDNA with a reverse primer of the interested gene. TdT and dGTP are used to append a poly-dG to the cDNA 3′ end, which is the 5′ end of the RNA. NewB is used to prime the synthesis of the second cDNA strand with PCR Mastermix. NewB is a mixture of four poly-dC oligos with a linker sequence (NewD) at the 5′ end and one of the four bases at the 3′ end (Table 1) and, thus, can be anchored on the last nt of the cDNA. The NewD and a reverse primer of the desired gene are then used in PCR to amplify the double-stranded cDNA, followed by T-A cloning. As an example, RNA from HeLa cells was RT with the HPRT1R683 primer. The HPRT1 cDNA was tailed with a poly-dG, followed by PCR with NewD+R683 that yielded a fuzzy band. Excision and purification of this band (boxed) as the template for a second round of PCR with the same primers resulted in a dominant band and several smaller bands. Cloning and sequencing the dominant band confirm that it is G-tailed 5′ end of HPRT1 mRNA. In the sequence obtained, the lowercase “t” before the NewB (underlined) and the “a” (added by Taq) after the R683 (underlined) were the cloning sites. The sequences before the “t” and after the “a” belong to the T-A vector. The lowercase “g” after NewB is the first nt of HPRT1 mRNA anchored by the NewB.
As an example, unDNased RNA from HeLa cells was converted in RT with a HPRT1 reverse primer (R683) to the first cDNA strand, followed by removal of dNTP, primers and other short oliogs by running the reaction through a RapidTip2, followed by washing and precipitation with ethanol. The cDNA was then tailed with a poly-dG using TdT and dGTP. NewB was used to prime the poly-dG tail for synthesis of the second cDNA strand. NewD and the R683 were used as the primers in PCR to amplify the double-stranded part of the HPRT1, which resulted in a fuzzy band. Purification of this fuzzy band from agarose gel followed by second PCR resulted in a clear band of the correct size, which was cloned into a T-A vector. Sequencing one resultant plasmid followed by alignment of the sequence with the HPRT1 mRNA sequence (NM_000194.2) confirms that a NewB primer has indeed anchored on the last nt of the HPRT1 (Fig. 2).
cDNA protection assay
We established a new strategy using cDNA to supersede cRNA and accordingly using S1 to replace RNase in RNA protection assay. In this strategy, an RNA aliquot is primed by random hexamers in RT and converted to cDNA. An aliquot of the cDNA is used to hybridize with a commensurate amount of the RNA (Fig. 3A). S1 is added to digest the non-hybridized cDNA and RNA and then is inactivated. PCR with gene-specific primers (GSP) ensues to amplify a fragment of the RNA-protected cDNA. As a negative control, an equal aliquot of the cDNA is directly digested with the same amount of S1 to ensure that without hybridization, no cDNA is left to be amplified in PCR. The amount of S1 may need to be optimized for each target gene because too-low enzyme activity may not be sufficient to remove all single-stranded cDNAs and may thus cause false positivity, whereas too much enzyme is known to have weak activity toward double-stranded DNA.
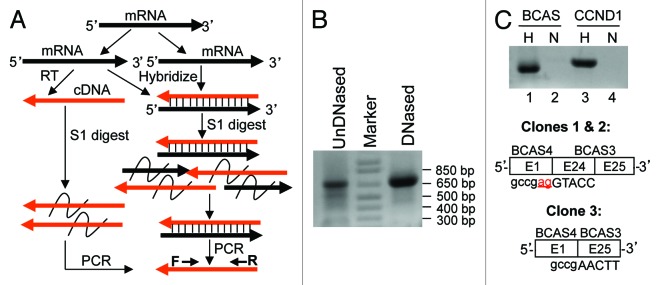
Figure 3. cDNA protection assay. (A) In this strategy, an RNA aliquot is converted to cDNA in RT with random hexamers. An aliquot of the cDNA is hybridized with an equivalent amount of RNA, followed by digestion of the non-hybridized cDNA and RNA with S1. S1 is inactivated and PCR with gene-specific primers (F and R) ensues to amplify the RNA-protected cDNA. As a negative control, an equal aliquot of cDNA is digested with S1 to ensure that without hybridization, no cDNA is left for amplification by PCR. (B) RT with random hexamers and with RNA sample from MCF7 cells that was not treated or was treated with DNase I followed by inactivation of the enzyme. PCR with BCS4F1+BCAS3R25 primers detects a dominant band at about 650 bp and several minor and smaller bands. (C) An equal amount of RT product (cDNA) was hybridized (H) or non-hybridized (N) with a commensurate amount of RNA, followed by S1 digestion. PCR with BCAS4F1+BCAS3R25 primers detected the dominant BCAS4-BCAS3 cDNA (BCAS) in hybridized, but not in non-hybridized, RNA aliquot. As a control, a CCND1 PCR fragment was added into the hybridization reaction as an indicator that double-stranded DNA could withstand the hybridization and the S1 digestion as it can be amplified by PCR, whereas single-stranded CCND1 cDNA in non-hybridized RT product was digested by S1 and thus could not be amplified by PCR. Cloning the BCAS band and sequencing three randomly selected plasmid clones reveal that in clones 1 and 2, the 3′ end of exon 1 of BCAS4 was fused to the 5′ end of exon 24 of BCAS3, whereas clone 3 lacks the last two nt (underlined “ag” shown in clones 1 and 2) of exon 1 of BCAS4 and the whole exon 24 of BCAS4.
In the MCF7 human breast cancer cell line, the BCAS4 (breast cancer amplified sequence 4) at the 20q13 of the human genome forms a fusion gene with the BCAS3 at the 17q23, which is transcribed and alternatively spliced to different chimeric RNAs, most of which contain exon 1 of BCAS4 and exons 24 and 25 of BCAS3.5,21,29 We performed RT with random hexamers and then PCR with primers at exon 1 of BCAS4 and exon 25 of BCAS3, which resulted in a dominant band at about 650 bp and several minor and smaller bands (Fig. 3B). Pretreatment of the RNA sample with DNase I followed by inactivation of the enzyme (as discussed later) caused partial losses of the minor bands, which redistributed the primers and thus increased the abundance of the dominant band (Fig. 3B). An aliquot of the cDNA was hybridized with an equivalent amount of the RNA sample. Moreover, the RT product (1/20) was also used in PCR to amply a fragment of CCND1, which was purified from agarose gel. Half of the purified CCND1 cDNA was added into the hybridization reaction as an indicator of whether double-stranded DNA could withstand the hybridization and the S1 digestion. After hybridization, S1 was added to digest the non-hybridized RNAs and cDNAs, while the same amount of the cDNA as used in hybridization was also S1-treated as a negative control (Fig. 3C). After inactivation of S1, PCR with the BCAS primers yielded a band from the S1-treated cDNA/RNA hybrids, but not from the non-hybridized cDNA, as expected (Fig. 3C). Similarly, PCR amplification of CCND1 as done in the above also yielded the anticipated band from the hybridized cDNA but not from the non-hybridized counterpart (Fig. 3C). Cloning the BCAS4-BCAS3 band and sequencing three resultant plasmid clones reveal that in clones 1 and 2, the canonical 3′ end of exon 1 of BCAS4 was fused to the canonical 5′ end of exon 24 of BCAS3, whereas the clone 3 lacked the last 2 nt (i.e., AG) of exon 1 of BCAS4 and the whole exon 24 of BCAS3 (Fig. 3C).
RT product primed by endogenous random primers in RNA samples
When performing RT, we often set up a reaction without adding primers as a negative control, but this reaction still and always yielded cDNAs. As examples, PCR with 1/20 (1 µl) of such non-primer RT product as the template could amplify CCND1, CDK4 or HPRT1 cDNA (Fig. 4A). The same CDK4 primers did not produce any band when the template was replaced by water (lanes 1 vs 3 in Fig. 4A), confirming that the PCR reagents were not contaminated by cDNA templates.
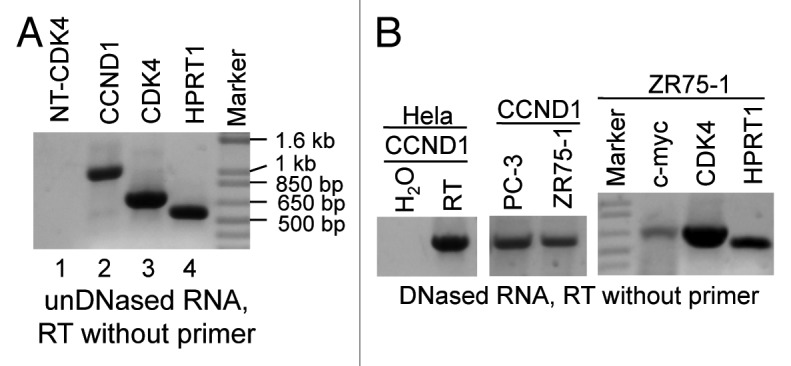
Figure 4. RT primed by endogenous random primers (ERP). (A) UnDNased RNA sample from HeLa cells was used in RT without adding primers. The RT product (1/20) was used as the template in PCR with the F70+R1067 for CCND1, F136+R822 for CDK4 and F123+R683 for HPRT1, respectively. As a negative control, the same CDK4 primers were used in a PCR with H2O to replace the RT product as the template (lanes 1 vs. 3). (B) RNA samples from Hela, PC-3 and ZR75-1 cell lines were treated with DNase I, followed by inactivation of the enzyme. RT was then performed without adding primers. The RT product was used as the template in PCR amplification of CCND1, CDK4 and HPRT1 as in (A) or of c-myc with the F125+R838 primers. In an addition, CCND1 PCR, the RT product, was superseded by H2O as the template.
In our routine practice, we often treated RNA samples with low concentration (several units) of DNase I, followed by inactivation of the enzyme with different methods including protein extraction with phenol/chloroform, so that genomic DNA residuals would not be mis-primed in the ensuing RT-PCR.41 RNA samples from HeLa, PC-3 and ZR75-1 cell lines that were pre-treated with DNase I followed by inactivation of the enzyme with 15 mM EDTA at 72°C for 15 min were used in RT without adding primer. PCR with the RT product as the template could still amplify several genes’ cDNA (Fig. 4B). Treatment of RNA samples with a much larger amount of DNase I could not eliminate the PCR-amplified bands, although the DNase activity could not be completely inactivated and the remaining activity decreased the detected level (data not shown). These results suggest that RNA specimens contain endogenous random primers (ERP) for RT that cannot be removed by DNase treatment.
Antisense-caused RT-PCR artifacts
We infer that when an antisense is expressed and overlaps with the sense RNA at their 5′ or 3′ ends, any cloning approaches that involve RT-PCR, including our method, may create artifacts, although many peers still use RT-PCR in cloning under this situation. We used the CDK4/TSPAN31 relationship to test this hypothesis, since the CDK4 mRNA and the TSPAN31 mRNA overlap at their last 517 nt (Fig. 5A). We designed a forward primer at the penultimate exon of CDK4 (CF933) or TSPAN31 (TF647); one set of these two primers also contained newD sequence at the 5′ end as a linker (NewDCF933 and NewDTF647). Other primers are illustrated in Figure 5A. Forward primer of one strand can also serve as reverse primer of the opposite strand. UnDNased RNA from HeLa cells was used in RT with the NewDTF647 as the GSP, which should specifically convert the CDK4 mRNA to cDNA (RT-B in Fig. 5B) if the mRNA reaches the TF647 region as we hypothesized. PCR using this RT-B product as the template and the CF136+R822 as the primer pair could indeed amplify a correct CDK4 band as expected (lane 4 in Fig. 5B). PCR with NewD as the reverse and the CF1096 as the forward primers resulted in a 1.5 kb band (Lane 6 in Fig. 5B). Cloning and sequencing this band confirmed that it was part of CDK4 mRNA containing the 84-bp intron 5 of TSPAN31. These results together indicate that some CDK4 mRNAs reach at least the TF647 region, making the overlapped region much longer than what is shown in the NCBI (thick arrow in Fig. 5A).
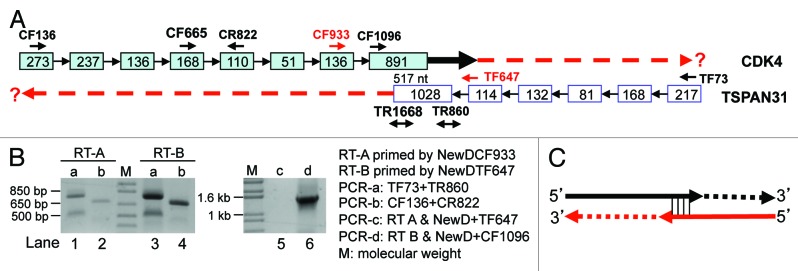
Figure 5. RT with linker-containing GSP to detect CDK4 (NM_000075.3) and TSPAN31 (NM_005981.3). (A) Illustration of the CDK4/TSPAN31 relationship according to the NCBI, with the locations of CDK4 forward (CF) or reverse (CR) primers and TSPAN31 forward (TF) or reverse (TR) primers indicated. Boxes represent exons with their length indicated as the number of nt. Note that the two mRNAs overlap at their last 517 nt. (B) RT of unDNased RNA from Hela cells primed by the NewDCF933 should specifically convert the TSPAN31 mRNA to cDNA (RT-A), whereas RT primed by the NewDTF647 should specifically convert the CDK4 mRNA to cDNA (RT-B), if the mRNAs reach the regions of these primers. These two RT products were used as the template in PCR with either the TF73+TR860 (PCR-a, lanes 1 and 3) or the CF136+CR822 (PCR-b, lanes 2 and 4) as the primer pair or in PCR with the primer pair of NewD+TF647 (PCR-c, lane 5) or NewD+CR1096 (PCR-d, lane 6). The results in lanes 4 and 6 together with sequence data suggest that some CDK4 transcripts reach the TF647 region (thick black arrow in A). (C) When sense or antisense RNA has an unprotected 3′ end overlapping with the other, the overlapping sequence may serve as a primer in RT to extend its 3′ end with its antisense as the template. The extension may also occur in the ensuing PCR, as depicted in Figure 6A. Because the mRNA does not really have this extended part (dashed lines), the corresponding PCR product, such as the bands in lanes 1, 2 and 3 in (B), is a wrong-template artifact.
Similarly, RT primed by the NewDCF933 should specifically convert the TSPAN31 mRNA to cDNA if the mRNA reaches the CF933 region (RT-A in Fig. 5B). PCR using this RT-A product as the template and the TF73+TR860 as the primer pair yielded the correct TSPAN31 band (the top band in lane 1 in Fig. 5B). However, a smaller band was also produced that was confirmed by T-A cloning and sequencing to be the LMTK2 mRNA from chromosome 7, but not the TSPAN31. Alignment of the LMTK2 and CDK4 sequences suggests that the TMTK2 cDNA is more likely to be primed by an endogenous primer in the RNA sample as described above, but not by the NewDCF933, indicating that RT using GSP is not so gene- and strand-specific as it is supposed to be.
PCR using the RT-A product as the template and the NewD+TF647 as the primer pair did not yield any band, which was discrepant to the results in lane 1 (lanes 1 vs 5 in Fig. 5B). More surprisingly, PCR using TF73+TR860 as the primers and RT-B as the template yielded the same two bands as when RT-A was used as the template (Lanes 1 vs 3 in Fig. 5B), although the RT-B primed by NewDTF647 was not supposed to convert the 5′ part of TSPAN31, or any RNA from this region, to cDNA. Similarly, PCR using CF136+R822 as the primers and RT-A as the template generated the same band as when RT-B was used as the template (Lanes 2 vs 4 in Fig. 5B), although the RT-A primed by NewDCF933 was not supposed to convert the 5′ part of CDK4, or any RNA from this region, to cDNA. A reasonable explanation for these inconsistent results is that some CDK4 and TSPAN31 mRNAs have an unprotected 3′-end at the overlapped region, serving as the primer to extend its cDNA as illustrated in Figure 5C. This extension may happen in RT or in the ensuring PCR as discussed later. In other words, the bands in lanes 2 and 3 in Figure 5B do not have the corresponding RNA as the original template and thus are wrong-template artifacts. The TSPAN31 band (the top one) in lane 1 of Figure 5B might be such wrong-template artifact as well, which explains why NewD+TF647 failed to yield a PCR product (lane 5 in Fig. 5B). In line with this conjecture, a PCR with the RT-A product as the template and the CF933+TR1668 as the primer pair did not produce any band (data not shown).
Discussion
Features of our cloning methods
Routine 5′ or 3′RACE usually can only clone short cDNA fragments, sometimes making it unclear whether the cloned cDNA end belongs to a chimeric RNA or to an mRNA of the parent gene (Fig. 1A), in part because the first and last exons are often very large. Moreover, 5′RACE is difficult as its first several steps are manipulations of fragile RNA. Our cloning methods start with RT and soon proceed with the synthesis of the second cDNA strand, with all later steps involving only double-stranded cDNA that is much more stable. Since almost the entire second strand can be synthesized, either one of our cloning methods can clone virtually the entire cDNA. A difference is that our 5′ cloning method involves PCR amplification and, thus, is more efficient, whereas our 3′ cloning method is a non-amplified approach with low efficiency but high fidelity. In addition, our 3′ method does not require a poly-dT primer, allowing cloning those RNAs without a poly-A tail and eliminating mis-priming to an internal poly-A sequence. Actually, we once used 3′RACE to clone the 3′ end of TSPAN31 with a poly-dT primer that contains a linker (coined as NewA; Table 1), followed by PCR with the linker sequence as a primer (coined as NewC; Table 1). The 3′ end cloned lacks the last 17-nt sequence (GACCATTAAAAAAAAAA) because there is a 14-adenine sequence in front of it (data not shown). If needed, however, our method can still use poly-dT primers in RT, alone or in combination with PCR, to enhance the cloning efficiency.
In our practice of molecular cloning, poly-dT primer is often used to prime poly-A tail in RT, whereas a poly-dG oligo longer than a hexamer (GGGGGG) is technically difficult to be synthesized, purified and verified and, thus, is much more expensive. Therefore, tailing a cDNA with poly-dG followed by priming it with a poly-dC oligo becomes the only practical choice for our 5′ end cloning method. The length of the poly-dG tail may be different among tailed targets, but one of the four oligos in the NewB mixture (Table 1) should be anchored on the last nt of the targeted cDNA, regardless of the length of the poly-dG tail and whether the 3′ end of the first cDNA strand has been added with several nt by the MMLV during the RT.4,18 However, it remains possible that the NewB mis-primes an internal poly-G sequence, which has actually happened in our practice.
Similar to routine 5′ and 3′ RACEs, our methods only use a single GSP and, thus, may still cause unspecific bands as shown in Figure 2. Moreover, cDNA may have breakages and, similar to routine RACEs, our methods cannot distinguish a genuine cDNA end from a spurious one. In addition, transcription may be initiated from or terminated at alternative sites. Therefore, cloning multiple bands and sequencing multiple plasmid clones are strongly recommended, not only to avoid artifacts but also to increase the chance of identifying alternative 5′ or 3′ ends.
Merits of cDNA protection assay
The strategy to protect a cDNA instead of the parental RNA has four major merits: (1) After being protected by the parental RNA, the cDNA can be PCR-amplified, which dramatically increases the sensitivity. If part of the cDNA is an RT artifact, it will not be protected because the single-stranded part of the cDNA or of the parental RNA will be digested by S1. Single-stranded DNA is about 5-fold more sensitive to S1 than RNA, as stated in the supplier’s datasheet of S1 nuclease. (2) The protected cDNA can be directly cloned and sequenced to confirm its identity, whereas in RNA protection assay, the protected RNA still needs to be converted to cDNA if a long fragment needs to be sequenced at a high quality. (3) It is still technically difficult to determine from which DNA strand an RNA is transcribed. Use of strand-specific DNA oligos to supersede cDNA in our protection assay may be the best way for this purpose, as discussed later. (4) DNA/RNA hybrid has its unique structure and compositions that are distinguishable from DNA/DNA or RNA/RNA hybrid,40 in part because DNA/DNA contains dA and dT, RNA/RNA contains rA and rU, while DNA/RNA contains all four. These differences should provide us with unique strategies to develop sensitive methods and instruments for the detection and quantification of those DNA/RNA hybrids that are at very low abundance. Such strategies should be applicable and, thus, intriguing, as endogenous DNA/RNA hybrids in eukaryotic cells are many fewer than the DNA/DNA and RNA/RNA hybrids, especially when a larger DNA/RNA fragment is designed for protection.
In most assays the probe is used at great excess compared with the target. We suggest that if our method is used mainly to verify the true existence of an RNA transcript, the RNA sample should be considered as the probe and, thus, used in great excess, relative to the cDNA. Conversely, if the aim is to quantify the RNA expression level, the cDNA should be regarded as the probe and used in great excess. A set of nested PCR, including those with one primer in the S1-digested region, should help in authenticating the RNA and, thus, is highly recommended, especially when T-A cloning and sequencing the resultant plasmids are omitted due to whatever considerations.
Unvanquished obstacles set by ERP in RT
Although retrovirus uses cellular tRNA to prime mRNA for reverse transcriptases to synthesize the 1st DNA strand,31,32 endogenous small RNAs such as mRNA fragments can efficiently prime cDNA synthesis by reverse transcriptases.12,20,22 RNA samples contain a huge number of short RNA fragments, such as degraded RNAs, excised introns and other processed mRNAs that are known to us recently,1 which can serve as ERP for RT. This is likely the reason why RT can occur without addition of primers, a phenomenon coined by others as “background priming.”2,15 This also explains why DNase treatment of RNA samples cannot eliminate cDNA generation in the ensuing RT. Actually, during DNase treatment and inactivation, some RNAs are likely degraded to be ERP. Besides, short genomic and mitochondrial DNA fragments resulting from degradation or incomplete DNase digestion can also serve as ERP.
The presence of ERP should not affect the RT results from random hexamers, and may not affect the results from poly-dT primer either if polyadenylation is not a specific concern. However, the results from GSP may no longer be gene- and strand-specific, not only because GSP may mis-prime, which is familiar to us, but also because ERP can prime other RNAs, including the antisense of the interested RNA if it exists. The gene-specificity may be improved by adding a linker sequence, herein NewD, to the 5′ end of the GSP and using it as one primer in the ensuing PCR. The strand-specificity may also be improved in this way if the antisense RNA level is relatively low and the amount of Linker-GSP is carefully managed, as shown in lanes 5 vs 6 in Figure 5B and as depicted in Figure 6A. However, this strategy may not always work well. When we tested this strategy by determining the existence of LOC100996515 RNA with primers at the region overlapped by CCND1, CCND1 as its antisense was often detected because of its much higher abundance. A better way to ensure the strand-specificity may be to use strand-specific, probably labeled, DNA oligos to replace cDNA probes in hybridization with the RNA of interest. Such strand-specific DNA oligos can be in vitro synthesized like a primer or made by other ways,10 including PCR with one biotinylated primer followed by capture with streptavidin-coated magnetic beads or PCR with one 5′phosphorylated primer10 followed by digestion of the useless, 5′phosphorylated strand using lambda exonuclease.10,34

Figure 6. Depiction of artifacts caused by ERP or by 5′ or 3′ overlapping. (A) Because the RNA sample contains ERP, RT with Linker-GSP will also generate the first cDNA strand of the antisense RNA, besides the cDNA of the desired sense RNA. When the RT product (usually only 1 µl) is added into the PCR mixture as the template, some Linker-GSP residual is transferred together, which primes the synthesis of a linker containing antisense fragment. The fragment is amplified in the later PCR cycles. However, because the PCR mixture contains many more copies of the GSP and the gene-specific reverse primer (GSRP) than the Linker-GSP residual, the first PCR cycle should generate many more copies of the desired sense cDNA, which titrates out the antisense in later PCR cycles, unless the antisense RNA is expressed at a much higher level than the sense. We use as small an amount of linker-GSP as possible in the RT to minimize its residual in the RT product. (B) Two cDNAs that overlap at their 5′ ends, no matter whether they are unrelated or are originated respectively from a sense and an antisense transcripts that overlap at their 3′ ends (like CDK4 and TSPAN31), can be converted to 3′-overlapped counterparts after one round of PCR by GSP or ERP, which, in turn, creates wrong-template extension in later PCR cycles as depicted in Figure 5C. (C) If a cDNA has an unprotected 3′ end that is reverse-complementary to an unrelated cDNA (in red color), this matched part (e.g., ATCGA/TAGCT) and this other cDNA may serve in PCR as the primer and the template, respectively, to create a spurious chimera.
Although GSP has been widely used in RT-PCR for decades, to our knowledge none of the published studies has addressed the possible spuriousness and provided a corrective measure as did we herein. Since routine GSP-primed RT is, likely, neither gene- nor strand-specific, whether those published data need to be reevaluated or reinterpreted becomes an uncomfortable but unavoidable question that peers need to bear in mind, in our humble opinion.
Unsolved, overlap-caused artifacts of cDNA end and chimeras
The NCBI has updated several times the sequences of CDK4 and TSPAN31 and keeps extending the overlapped region. One of the CDK4 sequences we obtained is longer than the latest NCBI version. We surmise that all old and new sequences may be correct, representing different variants with different lengths of the overlap. However, cloning the 3′ end of each of these mRNAs is technically difficult due to two major reasons: (1) ERP will result in cDNA of the antisense in RT (Fig. 6A). (2) One of the mRNA molecules, either CDK4 or TSPAN31, may have an unprotected 3′ end at the overlapped region, due to reasons such as degradation (breakage), premature transcription, deadenylation, early termination of RT, etc., occurring either as a physiological event or as an artifact. This unprotected 3′ end will serve as a primer to extend its cDNA with an antisense RNA as the template in RT, creating a wrong-template artifact (Fig. 5C). In this situation, the 3′ end cloned by any RT-PCR involved approach, including our method, could be an artifact. This artifact may also occur in PCR, if not already in RT, because one round of PCR, primed by either GSP or ERP, will convert two 5′-overlapped cDNAs to two 3′-overlapped ones (Fig. 6B). This pitfall should be particularly alerted to the biomedical society because so often RT-PCR is used to clone RNA without preclusion of the existence of overlapped antisense RNA. So far we are still unable to get out from this trap and, thus, unable to clone the genuine 3′-end of the extended CDK4 mRNA shown in lane 6 of Figure 5B, and to determine whether TSPAN31 also has mRNA variant(s) extended beyond the latest NCBI version.
Our results also alert us to anther pitfall that if routine or quantitative RT-PCR is used to determine the expression level of an RNA that is accompanied by an overlapped antisense transcript, PCR with primers at the overlapped region starts with two templates and, thus, will falsely double the expression level. Therefore, the locations of the primers matter, and primers at different regions of the RNA should be used. Since over 63% of RNA transcripts may be accompanied by antisense counterparts,24 peers should be alerted to the pitfalls described above.
A huge number of putative chimeric RNAs encompass a short homologous sequence shared by the two partners.27 The reason is unknown but it has led to discussions on how such chimeras are formed.3,27,39 Our observation of wrong-template extension created by overlap at the 5′ or 3′ end enlightens us in that some, likely many, of this type of chimeras may simply be RT-PCR spuriousness: If, as described above, an RNA or a cDNA has an unprotected 3′ end that is reversely complementary to an unrelated (i.e., not its antisense) RNA or cDNA, a chimeric sequence may be generated in RT or PCR (Fig. 6C). Since a pentamer can prime RT or PCR efficiently, the homologous part can be a 5-nt sequence, although whether a shorter oligo still has some priming ability is not so clear. Because the RNA repository in any human cell contains numerous such short homologous sequences, we tend to believe that many of those chimeras containing a short homologous sequence and obtained by approaches that involve RT, PCR or similar methods are such technical artifacts.
Summary
We describe two new methods for cloning cDNA ends and a cDNA protection assay to supersede RNA protection assay. We also report that GSP-primed RT product is neither gene- nor strand-specific because the RNA sample contains ERP. The gene-specificity may be improved by adding a linker sequence to the GSP and then using the linker as a primer in the ensuing PCR, whereas the strand-specificity may be improved by using strand-specific DNA oligos as the probe in our protection assay. Using the CDK4/TSPAN31 relationship as a model, we find that when sense and antisense RNAs overlap at their 3′ ends, the overlapped sequence might serve as a primer with its antisense as the template to create a wrong-template extension in RT or PCR, resulting in a spurious 3′ end. This result edifies us that two unrelated RNAs or cDNAs that overlap at the 5′- or 3′-end may also create a chimeric sequence in this way. Therefore, many chimeric RNAs containing a short homologous sequence and obtained by approaches involving RT or PCR may be such artifacts and, thus, need to be vigorously verified with, such as, our protection assay. The ERP and the 5′- or 3′-overlapping antisense together set more complex pitfalls in our way of RNA cloning, which should be highly alerted to the peers. Our methods cannot fully circumvent these traps but should be good alternative or corrective measures to the available ones for cloning chimeric or antisense-accompanied RNA, both together constituting the majority of the cellular RNA repository.
Materials and Methods
RNA preparation, DNase I treatment and RT
Total RNA was extracted from indicated cell lines using TRIzol (Invitrogen, Cat. 15596-026). In some experiments, RNA was treated with DNase I (1–3 units) to remove genomic DNA residuals, followed by inactivation of the DNase with 15 mM EDTA at 72°C for 15 min. An aliquot (4–5 µg) of total RNA was reverse-transcribed to the first strand of cDNA with indicated primers and M-MLV Reverse Transcriptase (Promega, Cat #. M1705; www.promega.com), following the manufacturer's instruction, but in a 20–25 µl volume.
Primer nomenclature
We used “F” and “R” to indicate a forward and a reverse primer, respectively. Each primer’s name ends with a number that indicates the first (for F) or the last (for R) nt of that primer in the position, i.e., the distance from the first nt, of the mRNA. Thus, the F-to-R range is the size of an RT-PCR amplified DNA fragment in agarose gel. All primers are listed in Table 1. More details of the primer design principle were described before.41
Purification of DNA and T-A cloning
PCR-amplified cDNA fragment was fractioned in 1% agarose gel and visualized by ethidium bromide staining. The desired band was then excised out and purified with UltraClean Gel DNA Extraction Kit (ISC BioExpress; www.bioexpress.com) following the manual, or with a simple method we described before.42 The purified DNA was ligated into a pGEM-T Easy Vector (Promega; www.promega.com).
RNA 3′ end cloning
RT was performed using RNA from Hela cells and primed by random hexamers, with other conditions as described above. The 3′ part of the second strand of CDK4 cDNA was synthesized using 1/3 to 1/2 of the RT products,100 nM CDK4 F665 primer and 1x PCR Mastermix, with one cycle of 95°C for 5 min, 60°C for 2 min and 72°C for 15 min in a thermocycler. Ten or 15 units of S1 nuclease (Cat # 18001-016; www.invitrogen.com) was added, followed by incubation at room temperature for 60 min to digest the 3′ overhang of the first cDNA strand and all single stranded cDNAs or mRNAs. EDTA was added to a final concentration of 10–15 mM with incubation at 72°C for 15 min to inactivate S1. To remove EDTA, the reaction was transferred to an Eppendorf tube with additions of 0.35 ml H2O and 1.2 ml 95% ethanol, followed by precipitation at -20°C for 20 min and then centrifugation at 13,000 rpm at 4°C for 15 min. The ethanol was discarded and the cDNA pellet was suspended with 14 µl H2O. To ensure that the 3′ overhang of the first cDNA strand had been removed by S1 but the double-stranded fragment was protected, 2 µl of the recovered double-stranded cDNA was used as the template to run 40 cycles of PCR with the F136+R1086 or the F665+R1086 primer pair (Fig. 1). The remaining (10 µl) double-stranded cDNA was then added with 10 µl PCR Mastermix, followed by incubation at 72°C for 10 min to append a dA at the cDNA blunt ends. A portion (herein 6 µl) of the dA-appended cDNA was cloned into a T-A vector. The resultant plasmid clones were first confirmed by PCR with the F665+R1086 primers and then sequenced with a vector primer.
RNA 5′ cloning with G-tailing
RT was performed using RNA from Hela cells as above-described, but with HPRT1R683 as a gene specific reverse primer. After being run through RapidTip2 (cat # RT050-096; www.midsci.com) to remove primers, enzymes, dNTP and debris, an aliquot (1/4) of the RT product was transferred into a 500-µl tube with additions of 30 units of TdT (www.promega.com; Cat# M828C), two units of RNase H, 4 mM dGTP, and 2 mM MnCl2 in a final volume of 25 µl, followed by incubation at 37°C for 30–60 min to synthesize a poly-dG tail. The TdT was inactivated by heating to 72°C for 10 min. About half of the dG-tailed product was primed by a NewB mixture (Table 1), with 1x PCR Mastermix in a 20-µl volume, to synthesize the second cDNA strand by one cycle of 95°C for 5 min, 60°C for 2 min and 72°C for 15 min in a thermocycler. About 1/4 of the double-stranded HPRT1 cDNA was then used as the template to run PCR with the NewD+HPRT1R683 primer pair for 40 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 60 sec. The PCR product appeared as a fuzzy band in agarose gel and, thus, was excised out and purified as the template for a second round of PCR, followed by excision and purification of the dominant band for T-A cloning (Fig. 2).
cDNA protection assay
RT was performed using RNA from MCF7 cells in a 25-µl volume and primed with random hexamers. The RT product was incubated at 72°C for 15 min with about 10–15 mM EDTA to inactivate RNase H and DNA polymerase activities of the MMLV. To remove EDTA, the RT product was transferred to an Eppendorf tube with additions of 0.35 ml H2O and 1.2 ml 95% ethanol, followed by precipitation of the cDNA as described above. The cDNA was suspended in 20 µl of H2O. In a 500-µl tube, the hybridization was set up with 1/10–1/5 (2–4 µl) of the cDNA and an equivalent amount of the RNA sample in a 50-µl solution containing 25% formamide (v/v), 600 mM NaCl, 30 mM Tris-HC (pH 7.5), 0.1% SDS, 10 mM DTT and 4 mM EDTA, as described before.28 Moreover, 1 µl (1/20) of the cDNA was also used in PCR to amplify the CCND1 cDNA with the F70+R1067 primer pair, and the PCR product was purified from agarose gel. About half of the purified CCND1 PCR product was added into the hybridization reaction as an indicator of whether the hybridization and the ensuing S1 digestion degrade double-stranded DNA. After topping with 35 µl mineral oil (purchased from a Walmart store; product #831432DB1) to prevent evaporation, the hybridization reaction was performed at 68°C for 8 h or longer. After transfer to an Eppendorf tube, the reaction was diluted and precipitated with additions of 0.35 ml H2O and 1.2 ml 95% ethanol as described above. The hybrids were suspended in 18 µl H2O and divided to three aliquots for digestion with 0, 10 or 15 units of S1 in a final volume of 20 µl at room temperature for 60 min. As a negative control (Fig. 2), a separate S1 digestion was set up with equal amounts of cDNA (the RT product) and S1. The S1 was then inactivated with 10–15 mM EDTA at 72°C for 15 min. The EDTA was removed by dilution and precipitation with additions of 0.35 ml H2O and 1.2 ml 95% ethanol at -20°C as described above. The cDNA/RNA hybrids were suspended in 16 µl H2O, 3 µl of which was used to run PCR with the BCAS4E1F+hBCAS3E25R primers and the CCND1F183+R1067 primers to ensure that the BCAS4-BCAS3 and the CCND1 cDNAs had been protected. The BCAS4-BCAS3 band was then purified from agarose gel and cloned into a T-A vector for sequencing verification.
Acknowledgments
We want to thank Fred Bogott, MD, Ph.D., at Austin Medical Center, for his excellent English editing of this manuscript. This work was supported by a grant from the Department of Defense of United States (DOD Award W81XWH-11-1-0119) to D.J.L. The funding agency had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Affymetrix ENCODE Transcriptome Project. Cold Spring Harbor Laboratory ENCODE Transcriptome Project Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature. 2009;457:1028–32. doi: 10.1038/nature07759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adrover MF, Muñoz MJ, Baez MV, Thomas J, Kornblihtt AR, Epstein AL, et al. Characterization of specific cDNA background synthesis introduced by reverse transcription in RT-PCR assays. Biochimie. 2010;92:1839–46. doi: 10.1016/j.biochi.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 3.Al-Balool HH, Weber D, Liu Y, Wade M, Guleria K, Nam PL, et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011;21:1788–99. doi: 10.1101/gr.116442.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods. 2009;177:381–5. doi: 10.1016/j.jneumeth.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bärlund M, Monni O, Weaver JD, Kauraniemi P, Sauter G, Heiskanen M, et al. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–7. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- 6.Beiter T, Reich E, Williams RW, Simon P. Antisense transcription: a critical look in both directions. Cell Mol Life Sci. 2009;66:94–112. doi: 10.1007/s00018-008-8381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. ENCODE Project Consortium. NISC Comparative Sequencing Program. Baylor College of Medicine Human Genome Sequencing Center. Washington University Genome Sequencing Center. Broad Institute. Children’s Hospital Oakland Research Institute Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brakenhoff RH, Schoenmakers JG, Lubsen NH. Chimeric cDNA clones: a novel PCR artifact. Nucleic Acids Res. 1991;19:1949. doi: 10.1093/nar/19.8.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celestino R, Sigstad E, Løvf M, Thomassen GO, Grøholt KK, Jørgensen LH, et al. Survey of 548 oncogenic fusion transcripts in thyroid tumors supports the importance of the already established thyroid fusions genes. Genes Chromosomes Cancer. 2012;51:1154–64. doi: 10.1002/gcc.22003. [DOI] [PubMed] [Google Scholar]
- 10.Civit L, Fragoso A, O’Sullivan CK. Evaluation of techniques for generation of single-stranded DNA for quantitative detection. Anal Biochem. 2012;431:132–8. doi: 10.1016/j.ab.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Cocquet J, Chong A, Zhang G, Veitia RA. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006;88:127–31. doi: 10.1016/j.ygeno.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 12.Colett MS, Larson R, Gold C, Strick D, Anderson DK, Purchio AF. Molecular cloning and nucleotide sequence of the pestivirus bovine viral diarrhea virus. Virology. 1988;165:191–9. doi: 10.1016/0042-6822(88)90672-1. [DOI] [PubMed] [Google Scholar]
- 13.Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge JM, et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012;7:e28213. doi: 10.1371/journal.pone.0028213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frech B, Peterhans ERT-PCR. RT-PCR: ‘background priming’ during reverse transcription. Nucleic Acids Res. 1994;22:4342–3. doi: 10.1093/nar/22.20.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frenkel-Morgenstern M, Lacroix V, Ezkurdia I, Levin Y, Gabashvili A, Prilusky J, et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–42. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gingeras TR. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–11. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginsberg SD, Che S. RNA amplification in brain tissues. Neurochem Res. 2002;27:981–92. doi: 10.1023/A:1020944502581. [DOI] [PubMed] [Google Scholar]
- 19.Grinchuk OV, Jenjaroenpun P, Orlov YL, Zhou J, Kuznetsov VA. Integrative analysis of the human cis-antisense gene pairs, miRNAs and their transcription regulation patterns. Nucleic Acids Res. 2010;38:534–47. doi: 10.1093/nar/gkp954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gubler U. Second-strand cDNA synthesis: mRNA fragments as primers. Methods Enzymol. 1987;152:330–5. doi: 10.1016/0076-6879(87)52038-9. [DOI] [PubMed] [Google Scholar]
- 21.Hahn Y, Bera TK, Gehlhaus K, Kirsch IR, Pastan IH, Lee B. Finding fusion genes resulting from chromosome rearrangement by analyzing the expressed sequence databases. Proc Natl Acad Sci USA. 2004;101:13257–61. doi: 10.1073/pnas.0405490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzig E, Voronin N, Hizi A. The removal of RNA primers from DNA synthesized by the reverse transcriptase of the retrotransposon Tf1 is stimulated by Tf1 integrase. J Virol. 2012;86:6222–30. doi: 10.1128/JVI.00009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houseley J, Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, et al. RIKEN Genome Exploration Research Group. Genome Science Group (Genome Network Project Core Group) FANTOM Consortium Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–6. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 25.Kowalczyk MS, Higgs DR, Gingeras TR. Molecular biology: RNA discrimination. Nature. 2012;482:310–1. doi: 10.1038/482310a. [DOI] [PubMed] [Google Scholar]
- 26.Li K, Ramchandran R. Natural antisense transcript: a concomitant engagement with protein-coding transcript. Oncotarget. 2010;1:447–52. doi: 10.18632/oncotarget.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Zhao L, Jiang H, Wang W. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J Mol Evol. 2009;68:56–65. doi: 10.1007/s00239-008-9187-0. [DOI] [PubMed] [Google Scholar]
- 28.Liao D, Porsch-Hällström I, Gustafsson JA, Blanck A. Sex differences at the initiation stage of rat liver carcinogenesis--influence of growth hormone. Carcinogenesis. 1993;14:2045–9. doi: 10.1093/carcin/14.10.2045. [DOI] [PubMed] [Google Scholar]
- 29.Løvf M, Thomassen GO, Bakken AC, Celestino R, Fioretos T, Lind GE, et al. Fusion gene microarray reveals cancer type-specificity among fusion genes. Genes Chromosomes Cancer. 2011;50:348–57. doi: 10.1002/gcc.20860. [DOI] [PubMed] [Google Scholar]
- 30.Mader RM, Schmidt WM, Sedivy R, Rizovski B, Braun J, Kalipciyan M, et al. Reverse transcriptase template switching during reverse transcriptase-polymerase chain reaction: artificial generation of deletions in ribonucleotide reductase mRNA. J Lab Clin Med. 2001;137:422–8. doi: 10.1067/mlc.2001.115452. [DOI] [PubMed] [Google Scholar]
- 31.Mak J, Kleiman L. Primer tRNAs for reverse transcription. J Virol. 1997;71:8087–95. doi: 10.1128/jvi.71.11.8087-8095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marquet R, Isel C, Ehresmann C, Ehresmann B. tRNAs as primer of reverse transcriptases. Biochimie. 1995;77:113–24. doi: 10.1016/0300-9084(96)88114-4. [DOI] [PubMed] [Google Scholar]
- 33.McManus CJ, Duff MO, Eipper-Mains J, Graveley BR. Global analysis of trans-splicing in Drosophila. Proc Natl Acad Sci USA. 2010;107:12975–9. doi: 10.1073/pnas.1007586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Null AP, Hannis JC, Muddiman DC. Preparation of single-stranded PCR products for electrospray ionization mass spectrometry using the DNA repair enzyme lambda exonuclease. Analyst. 2000;125:619–26. doi: 10.1039/a908022h. [DOI] [PubMed] [Google Scholar]
- 35.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pääbo S, Irwin DM, Wilson AC. DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem. 1990;265:4718–21. [PubMed] [Google Scholar]
- 37.Ponting CP, Belgard TG. Transcribed dark matter: meaning or myth? Hum Mol Genet. 2010;19(R2):R162–8. doi: 10.1093/hmg/ddq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiu X, Wu L, Huang H, McDonel PE, Palumbo AV, Tiedje JM, et al. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol. 2001;67:880–7. doi: 10.1128/AEM.67.2.880-887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritz K, van Schaik BD, Jakobs ME, Aronica E, Tijssen MA, van Kampen AH, et al. Looking ultra deep: short identical sequences and transcriptional slippage. Genomics. 2011;98:90–5. doi: 10.1016/j.ygeno.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Shaw NN, Arya DP. Recognition of the unique structure of DNA:RNA hybrids. Biochimie. 2008;90:1026–39. doi: 10.1016/j.biochi.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Sun Y, Li Y, Luo D, Liao DJ. Pseudogenes as weaknesses of ACTB (Actb) and GAPDH (Gapdh) used as reference genes in reverse transcription and polymerase chain reactions. PLoS One. 2012;7:e41659. doi: 10.1371/journal.pone.0041659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Y, Sriramajayam K, Luo D, Liao DJ. A quick, cost-free method of purification of DNA fragments from agarose gel. J Cancer. 2012;3:93–5. doi: 10.7150/jca.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]