

Abstract
Autophagy and the ubiquitin–proteasome pathway (UPP) are the major protein degradation systems in eukaryotic cells. Whereas the former mediate a bulk nonspecific degradation, the UPP allows a rapid degradation of specific proteins. Both systems have been shown to play a role in tumorigenesis, and the interest in developing therapeutic agents inhibiting protein degradation is steadily growing. However, emerging data point to a critical role for autophagy in cellular senescence, an established tumor suppressor mechanism. Recently, a selective protein degradation process mediated by the UPP was also shown to contribute to the senescence phenotype. This process is tightly regulated by E3 ubiquitin ligases, deubiquitinases, and several post-translational modifications of target proteins. Illustrating the complexity of UPP, more than 600 human genes have been shown to encode E3 ubiquitin ligases, a number which exceeds that of the protein kinases. Nevertheless, our knowledge of proteasome-dependent protein degradation as a regulated process in cellular contexts such as cancer and senescence remains very limited. Here we discuss the implications of protein degradation in senescence and attempt to relate this function to the protein degradation pattern observed in cancer cells.
Keywords: E3 ligases, ERK kinases, Ras oncogene, ubiquitin, SASP
Introduction to Cellular Senescence
The long lifespan and constant cell turnover of complex organisms pose the challenge of dealing with the inevitable accumulation of DNA damage and gene mutations that drive carcinogenesis. Fortunately, multiple mechanisms have evolved to detect DNA aberrations and oncogenic stress and protect against the initiation and progression of neoplastic growth. Among these, cellular senescence is a stable cell cycle arrest triggered by a variety of insults including short telomeres, activated oncogenes, DNA damage, and reactive oxygen species.1 However, how these stresses converge to regulate a common cellular state is not currently well understood. Senescence is a complex multifaceted cellular phenotype, without an exclusive hallmark, with a broad range of proposed effector mechanisms, and, still, with an ambiguous definition. Indeed, different senescent cells are characterized by a wide range of biomarkers (reviewed in refs. 1 and 2), many of which are neither exclusive to senescence nor universally present in senescent cells. Because of this phenotypic heterogeneity and often imprecise definition, the assessment of senescence should be carefully addressed and should attempt to rigorously define a combination of senescence-associated features. Moreover, it needs to be recognized that this diversity in the phenotypic traits could reflect a concomitant heterogeneity at the level of the effector programs.
At the molecular level, senescence triggers important changes in gene expression patterns, but there is little overlap between different cell types.3 For example, a comparison between young and senescent human fibroblasts and mammary epithelial cells (HMEC) revealed a transcriptional fingerprint unique to senescence, but limited similarity between the 2 cell lineages.4 Other gene expression analyses have revealed a proinflammatory gene profile in senescent cells under the regulation of the NF-κB transcription factor5-7 or a downregulation of E2F target genes under the regulation of the retinoblastoma tumor suppressor (RB).8,9 However, cells with inactivation in NF-κB or RB can senesce in response to multiple stressors,5-9 indicating that the programs they control are not essential for the initiation of the process. Several target genes of the tumor suppressor p53 (TP53) were also reported to mediate senescence, such as p21 (CDKN1A),10-12 the tumor suppressor promyelocytic leukemia (PML),13,14 the plasminogen activator inhibitor-1 (PAI-1),15 DEC1, and E2F7.16 Again, a p53-dependent transcriptional pattern is not a prerequisite for senescence, and its relative contribution to the process depends on the cell type and the status of the p16INK4A–RB pathway.17 Our current knowledge thus suggests that senescence is consistent with distinct gene expression profiles and a variety of effector mechanisms, depending on the triggers, cell types, and cellular context.
Beyond transcriptional regulatory networks that characterize senescence, direct control of protein levels also appears strikingly affected. This involves the regulation of mRNA translation and protein stability of specific senescence mediators, such as p5318-20 and PML.21-23 In addition, it is thought that a global upregulation of translation may contribute to senescence, since the key regulator of protein synthesis, mTOR, has been shown to favor senescence in different contexts1 and total protein synthesis is increased in Ras-induced senescent cells.24 Similarly, a more general function of protein degradation now emerges as critical to reorganize the proteome of cells undergoing senescence. Here, we will discuss the impact of protein degradation on the senescence-associated proteome and how this mechanism could contribute to the onset of cellular senescence. Thus, we will effectively address the question: how does a pre-neoplastic cell destroy the machinery required for its subsequent progression to a cancer?
Protein Degradation and Senescence
The lysosomal degradation pathway is the principal system used by eukaryotic cells to degrade and recycle cytosolic components and organelles. A cytoplasmic cargo is engulfed into vesicles and delivered to the lysosome by the process of autophagy, which can be divided into 3 classes: (1) chaperone-mediated autophagy; (2) microautophagy; and (3) macroautophagy.25 The latter is mainly a nonspecific cytoplasmic degradation mechanism that has been shown to support tumorigenesis in Ras-expressing cancer cells,26 pancreatic tumors,27 lymphomas,28 and breast cancer.29 Macroautophagy is required to eliminate abnormal mitochondria, reduce the production of reactive oxygen species and replenish tricarboxylic acid (TCA) cycle metabolites.26,27 Given its catabolic capacity, macroautophagy improves the survival of both normal and cancer cells under metabolic stress by maintaining the availability of building blocks in order to preserve essential cellular functions.30
It is now appreciated that in addition to supporting cell viability in established tumors, macroautophagy has context-specific tumor-suppressor functions. The first evidence of such a function came from the discovery that the haploinsufficiency of the autophagy-related gene Beclin1 (BECN1) leads to cancer predisposition in mice.31 Moreover, many effectors of macroautophagy, including Atg5,32 Atg7,32,33 Atg4C,34 Bif-135, and UVRAG,36,37 have been linked to tumor suppression, further supporting its importance in anticancer mechanisms. Mechanistically, macroautophagy may circumvent malignant transformation by inducing autophagic cell death38 or cellular senescence24 in the context of oncogenic stress. Despite the demonstration that chaperone-mediated autophagy is downregulated in senescent cells,39 and that macroautophagy may prevent senescence in some contexts,40 a growing number of observations show a correlation between markers of autophagy and the senescence phenotype.41-43 Also, numerous studies have now demonstrated the critical role of macroautophagy during the establishment of senescence triggered by various stresses.24,44-49 Interestingly, some recent work suggests an intimate relationship between macroautophagy and the senescence-associated secretory phenotype (SASP).50,51 These studies propose that macroautophagy is required to attenuate the proteotoxic stress induced by the high protein synthesis rate involved in the SASP and to supply the process with building blocks and energy.25,52 The SASP has been linked to the deleterious effects of senescence,7,53 but also to the auto/paracrine reinforcement of the phenotype5,54-56 and to the immune clearance of senescent cells,57-60 thereby suggesting that autophagy might play a central function to explain the pathophysiological relevance of senescence.
During the molecular characterization of the role of the ERK kinases in Ras-induced senescence in human fibroblasts, our group discovered that senescence depends on high-strength ERK signals. In this context, we serendipitously found that some ERK targets were degraded. This initial observation lead to the identification of multiple actively degraded phosphoproteins during Ras-induced senescence.61 Consistent with increased macroautophagy during senescence, we observed an increase in overall protein degradation in oncogenic Ras-expressing senescent cells (Fig. 1A and B) but no increase in the total amount of polyubiquitinated conjugates (Fig. 1C) or upregulation of the proteasome activity as measured with the proteasome activity probe Me4BodipyFL-Ahx3Leu3VS (Fig. 1D).62 However, by in-depth characterization of an array of proteins that we found to be degraded, we discovered that the degradation process involved ubiquitination and the proteasome. This senescence-associated degradation program was conserved in multiple contexts, including mouse fibroblasts and human mammary epithelial cells expressing oncogenic Ras and in human fibroblasts with short telomeres. Thus, the second major degradation system used by eukaryotic cells, the selective degradation by the ubiquitin–proteasome pathway (UPP), is also engaged in senescent cells and allows the degradation of specific proteins. We called the process senescence-associated protein degradation or SAPD.61 Although its exact contribution to senescence needs further study, depletion of some individual SAPD targets was sufficient to trigger senescence, thereby illustrating the relevance of this mechanism for the onset and/or maintenance of senescence. We hypothesized that under mitogenic stress, such as conferred by hyperactivation of the ERK/MAPK pathway, the downstream effectors of mitogenic signaling undergo proteasome-dependent degradation, and that their depletion accounts for different characteristics of senescent cells.63 Consistent with this model, a phosphoproteomic analysis of Ras-expressing senescent cells treated with the proteasome inhibitor MG132 revealed many proteasome targets whose downregulation can contribute to senescence (Fig. 2; Table 1). We will thus discuss the features of senescence that are most likely to be induced or affected by the SAPD.
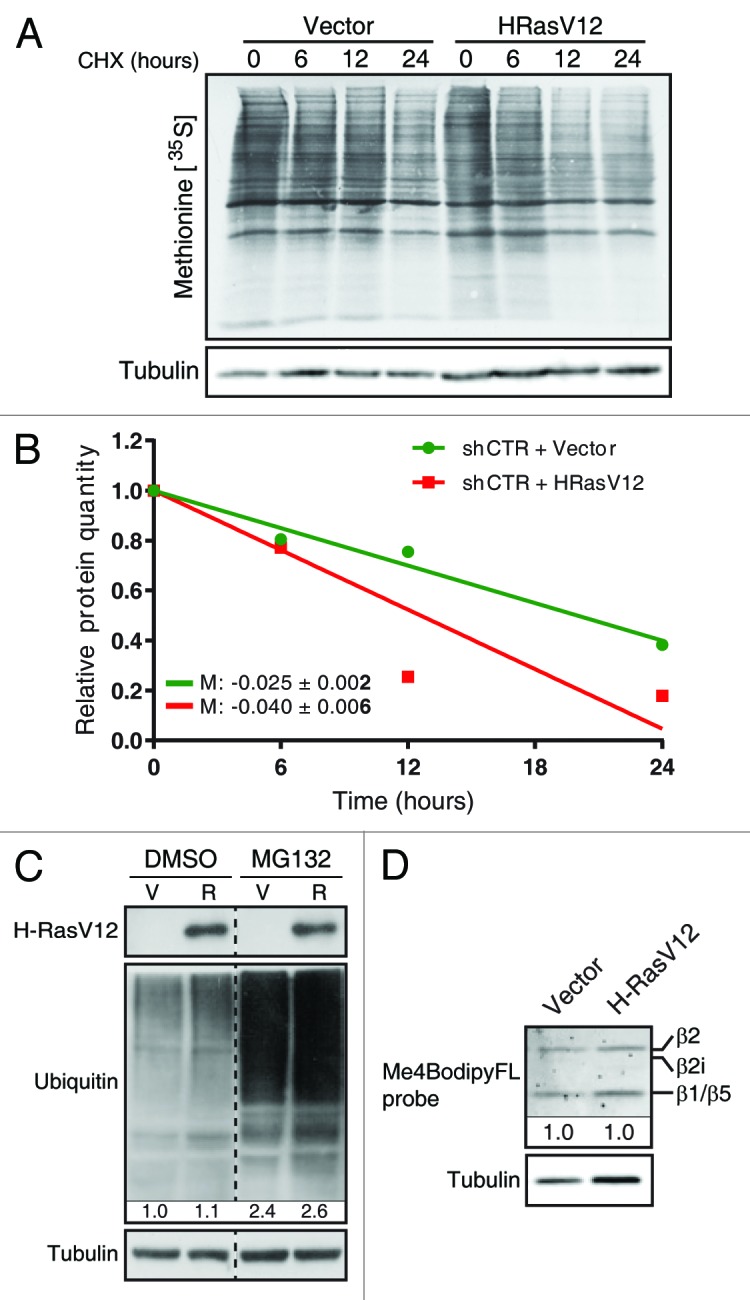
Figure 1. Oncogenic Ras increases overall protein degradation, but does not increase proteasome activity. (A) Normal human fibroblasts (IMR90; from ATCC) cultured in Dulbecco modified Eagle medium (DMEM; Wisent) and expressing oncogenic Ras (R) or an empty pWZL vector (V), 10 d after retroviral infection. Total protein extracts after a pulse with 0.5 µCi [35S]-methionine for 2 h, followed by a treatment with 75 µg/mL cycloheximide (CHX; Sigma-Aldrich) for the indicated times. (B) Bands were quantitated using Image Lab 4.0 (M = slope). An immunoblot for α-tubulin (1:5000; clone B-5–1-2, T6074, Sigma-Aldrich) was used for normalization. (C) Immunoblots for HRas (1:250; clone F235, Sc-29, Santa Cruz), α-tubulin and mono-polyubiquitylated conjugates (1:1000; clone FK2, BML-PW8810, Enzo Life sciences). Protein extracts from IMR90 cells as in (A), but treated with DMSO or 20 µM MG132 (Sigma-Aldrich) for 18 h. (D) Fibroblasts as in (A) were treated with 500 nM of the proteasome activity probe Me4BodipyFL-Ahx3Leu3VS (Boston Biochem, I-190) for 1 h. Total protein extracts were subjected to SDS-PAGE, and fluorescence was analyzed on a ChemiDoc™ MP System (Bio-Rad). Multiple catalytic subunits are visible (β1, 2, and 5).
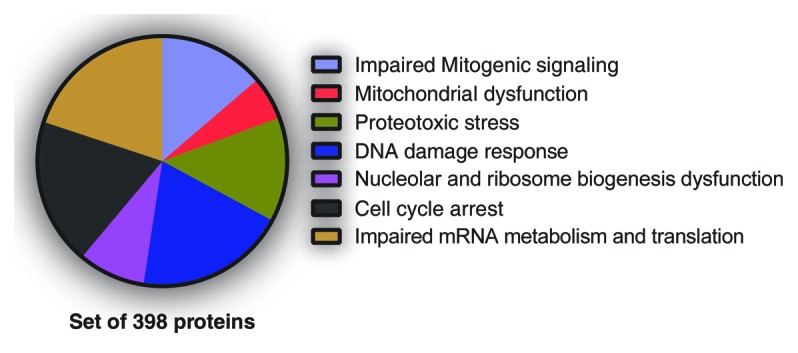
Figure 2. Senescence-associated phenotypes likely regulated by SAPD targets. Normal human fibroblasts, 10 d after infection with H-RasV12, were treated 18 h with DMSO (control) or 20 µM MG132 (Sigma-Aldrich). Then, cells were harvested, and protein extracts were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for phosphoproteomics. Almost 3000 phosphopeptides from 1018 proteins were enriched. A FatiGO single enrichment analysis with the Babelomics 4.3 platform was perform in order to identify Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome terms significantly enriched. The terms related to a senescence phenotype and their associated peptides (398 proteins) were grouped into the indicated phenotypes.
Table 1. Potential SAPD targets and the senescence-associated phenotypes they could regulate.
| Phenotypes | Proteins | Functions |
|---|---|---|
| Mitochondrial dysfunction | ATP5B | β-Subunit of the ATP synthase catalytic core (F1); ATP synthesis66 |
| STAT3 | Modulates respiration via complex I and II69,70 | |
| HSP1A1 (HSP70) | Component of the TOM complex; import of matrix mitochondrial proteins74 | |
| TOMM70A | Component of the TOM complex; import of matrix mitochondrial proteins74 | |
| TOMM34 | Component of the TOM complex; import of matrix mitochondrial proteins74 | |
| Proteotoxic stress | HSPA1A (HSP70) | Protein refolding under stress conditions; supports oncogenesis82,83,89 |
| HSPA5,7,8 and 9 | Protein refolding under stress conditions; supports oncogenesis82,83,89 | |
| HSPB1 (HSP27) | Protein refolding under stress conditions; suppresses cellular senescence82,83,89,95 | |
| HSPD1 (HSP60) | Protein refolding under stress conditions82,83,89 | |
| HSP90AB1 | Protein refolding under stress conditions; supports oncogenesis82,83,89 | |
| DNA damage response | CCDC6 | DNA damage checkpoints and DNA repair109,110,116 |
| SOD1 | Superoxide detoxification113,114,120 | |
| TOP2 | Relaxes topological constraints during DNA replication; chromosomes segregation117 | |
| TERF2IP | Component of the telosome; tethering telomeres to the nuclear envelope; protect telomere ends from NHEJ and HDR124-126,128,267,268 | |
| Nucleolar and ribosome biogenesis dysfunction | NOLC1 | Polymerase I coactivator; scaffold protein for nucleolar assembly137,138 |
| NOP56 and 58 | Components of the box C/D snoRNPs139 | |
| DDX51 | RNA helicase; processing of the 3′ end of the 28S rRNA140 | |
| NOL6 | Processing of the 18S rRNA141 | |
| NOC2L | Processing of the 18S, 28S and 5.8S rRNAs142 | |
| NCL | Polymerase I transcription; rRNA processing; ribosome assembly and transport143 | |
| RPLP1 | Translational elongation; overexpression bypasses replicative senescence144 | |
| RSL1D1 | Regulates the nucleolar localization of nucleostemin; rRNA processing146,149 | |
| NPM1 | Processing of the 32S pre-rRNA to the mature 28S rRNA153,154 | |
| Cell cycle arrest | YAP1 | Regulates apoptosis; regulates organ size; liver oncogene167,269 |
| MCM2 | Initiation and progression of DNA replication172 | |
| LRWD1 (ORCA) | stabilizes the origin recognition complex (ORC) to chromatin173 | |
| MYC | Promotes DNA replication177 | |
| JUN | Promotes G1-to-S-phase progression189 | |
| KAP1 (TRIM28) | Destabilized the tumor suppressor p53192-194 | |
| TBX2 | Repression of P19ARF; repression of the CDK inhibitors p21 and p27199-201 | |
| Impaired mRNA metabolism and translation | YB-1 (YBX1) | mRNA stability, mRNA packaging, splicing and translational initiation; oncogene204,207 |
| SRm160 (SRRM1) | Coactivator for exonic splicing enhancers and for 3′-end processing of specific pre-mRNAs210-213 | |
| SRm300 (SRRM2) | Coactivator for exonic splicing enhancers and for 3′-end processing of specific pre-mRNAs210,211,213 |
SAPD and the Senescent Phenotype
Mitochondrial dysfunction
Mitochondria are dysfunctional in senescent cells,64,65 but the mechanism explaining their alterations is unknown. The ATP synthase enzyme uses the proton gradient generated by the electron transport chain in inner mitochondrial membrane to catalyze ATP production.66 The ATP synthase subunit ATP5B is degraded by the proteasome in Ras-induced senescence (Table 1), and an increase of its turnover might explain the drop in ATP levels in senescent cells reported in some studies.65,67,68 This might contribute to senescence, since inhibition of ATP synthase with oligomycin has been shown to induce a partial senescence phenotype.65
The signal transducer and activator of transcription 3 (STAT3) is a transcription factor activated by the JAK kinases in response to cytokines. However, a pool of this protein has been shown to be imported into mitochondria and incorporated to complex I via GRIM-19.69 Mitochondrial STAT3 modulates respiration, mainly by promoting the activity of complex I and II of the electron transport chain.70 This function of STAT3 appears to support Ras-driven transformation and ensures the proper functioning of mitochondria.71 Indeed, impaired levels or regulation of STAT3 have been shown to induce mitochondrial dysfunction and ROS production.72,73 Interestingly, STAT3 is a confirmed SAPD target, and its degradation may thus link senescence to mitochondrial dysfunction (Table 1).61
In addition, 3 components of the TOM complex were found to be unstable during Ras-induced senescence: HSP70 (HSP1A1), TOMM70A, and TOMM34 (Table 1).61 The TOM complex is responsible for the import of matrix mitochondrial proteins involved in the TCA cycle and β-oxidation.74 This complex is assisted by the chaperone ATPase HSP70, which is very unstable in senescent cells.61 It is thus possible that defects in mitochondrial protein import due to degradation of TOM complex components contribute to the mitochondrial dysfunction observed in senescent cells. It is known that the TOM complex is regulated by phosphorylation,75 and we found phosphorylation of serine 91 of TOMM70A and serine 186 of TOMM34 in Ras-induced senescence. It will be of considerable interest to address whether these sites are phosphorylated by the ERKs or other kinases and mediate recognition of E3 ligases. Of note, HSP70 regulates oncogene-induced senescence (OIS), and knockdown of this protein is associated to an increase in ERK activity,76 perhaps creating a positive feedback loop that plays a role in maintaining OIS.
Proteotoxic stress
Accumulation of damaged and misfolded proteins leads to chronic proteotoxic stress, which is intimately linked to organismal aging and associated pathologies.77 The oxidative stress resulting from either mitochondrial dysfunction65,78 or upregulation of oxidative metabolism51,79 can promote protein oxidation,80,81 thereby leading to protein misfolding.82,83 Also, it is proposed that the high production of secreted cytokines in the SASP overcomes the cellular capacity for accurate protein synthesis and thus produces improper proteins and proteotoxic stress.51 Interestingly, we found that the main housekeeping system to maintain protein homeostasis, the heat-shock proteins (HSPs), is also downregulated in senescence.61 Indeed, the proteasomal degradation of HSP70 has been confirmed, and an impressive number of HSP proteins are unstable in Ras-induced senescence (Table 1). This is consistent with the demonstration that chaperone-mediated autophagy, but not macroautophagy, is downregulated in senescent cells.39 Further supporting our observations, several reports have shown either a decrease in HSPs during senescence or a direct function of these proteins in opposing the induction of senescence.76,84-89 Conversely, high levels of HSPs support tumorigenesis by circumventing a toxic accumulation of misfolded proteins in cancer cells that frequently experience proteotoxic stress, suggesting a widespread vulnerability that can be targeted therapeutically.82,90-93 Taken together, the observations discussed above strongly suggest that a breakdown of protein homeostasis is an important feature of cellular senescence and therefore of tumor suppression.
Beyond these correlative findings, we are tempted to speculate that downregulation of HSP activity might have a primary and critical role during the establishment of a senescent program. First, the reduction of protein refolding might stimulate abnormal protein clearance by degradation, either by macroautophagy or proteasomal-dependent degradation.25,94-96 Notably, a decrease in HSP levels correlates with an elevated activity of the CHIP ligase during senescence, suggesting that this E3 ligase could play a pivotal role in targeting misfolded proteins to the UPP.97 Somehow, the directed degradation of HSPs could reinforce the main cellular protein degradation mechanisms in order to eliminate dysfunctional proteins instead of investing energy in protein repair. Senescent cells use energy to support production of signaling molecules and secretion products. Protein degradation produces amino acids used as building blocks and substrates to feed the TCA cycle, thereby supporting metabolite synthesis and energy production.25 Perhaps protein degradation is a better investment than protein repair to support the SASP. This could be particularly true if low-cost degradation processes are favored during senescence. In this regard, it has been reported that ubiquitin and ATP-independent proteasomal degradation, accomplished by the 20S proteasomes, is the predominant mechanism to remove damaged proteins in oxidative contexts,98 as is the case in senescent cells. If this speculation proves true, this may be the designated route to optimize production of building blocks and energy saving.
DNA damage response
Cellular senescence induced by various stresses is characterized by an inability to properly repair DNA breaks and thus by a permanent DNA damage response (DDR).99-102 The latter is thought to contribute to both the induction and maintenance of senescence.103-108 The coiled-coil domain containing protein 6 (CCDC6) is a component of the DNA damage checkpoint machinery, and its corresponding gene is rearranged in 20% of papillary thyroid carcinomas.109 During DDR, CCDC6 is stabilized by ATM and contributes to proper DNA repair.110 Interestingly, we found that CCDC6 is unstable in Ras-induced senescence (Table 1), and its degradation may thus contribute to the persistent DDR observed in senescent cells.100-102,104-108 Moreover, this protein is a target for the tumor suppressor E3 ligase SCF-FBW7, suggesting a role of this E3 ligase in SAPD.110
In addition to limiting proper DNA repair, the SAPD could itself promote DNA damage. The DNA breaks that underlie senescence can be triggered by different stresses. One of these is the increase in reactive oxygen species (ROS),111-113 resulting from abnormal mitochondrial activities during senescence as discussed previously. Surprisingly, we found that the copper zinc superoxide dismutase 1 (SOD1) is unstable in Ras-dependent senescent cells (Table 1). Since this enzyme metabolizes superoxide radicals to molecular oxygen and hydrogen peroxide, and therefore is a major component of the antioxidant defenses within the cell,113,114 SOD1 depletion could cooperate with mitochondrial generation of ROS to increase the total amount of these reactive ions and concomitant DNA damage. Further supporting this conjecture, SOD1 deficiency has been shown to induce persistent DNA damage in mice115 and senescence in human fibroblasts.116 In addition to increased ROS levels, oncogenic activation drives an initial phase of DNA hyper-replication, leading to premature termination of replication forks, thereby producing DNA damage that triggers senescence.106,107 Intriguingly, another candidate target of SAPD identified by proteomics is TOP2 (Table 1), which is known to relax topological constraints during DNA replication and to allow chromosome segregation.117 Accordingly, a deficiency in this topoisomerase could increase fork collapses by preventing their progression, causing aberrant replication intermediates and the activation of DDR.118 Also, a lack in TOP2 can impair completion of DNA replication by interfering with the proper resolution of replication forks at chromosomal termination regions (TERs),119,120 thus generating DNA damage at TERs and even more when cells undergo mitosis.120-122 Furthermore, TOP2 has been shown to play an architectural function at intergenic regions adjacent to transcribed genes during S phase, and this seems to protect against collisions between replication forks and transcription sites. This role appears critical to avoid replication-induced DNA damage, since cells deficient in TOP2 experience DNA breaks at normally TOP2-bound regions.123 Taken together, the functions of TOP2 suggest that it plays a critical role to maintain genome integrity in cycling cells, and that its depletion in cells experiencing hyper-replication is likely to trigger DNA damage-promoted senescence. Finally, another proposed source of DNA damage leading to senescence is telomere dysfunction, which leads to telomere dysfunction-induced foci (TIF).99,105 The shelterin complex (telosome) associates with telomeres and protects chromosome ends.124 The human ortholog of the yeast telomere binding protein Rap1, TERF2IP,125 is part of the complex and has been shown to play a role in tethering telomeres to the nuclear envelope126 and to protect telomere ends from non-homologous end joining (NHEJ).127,128 TERF2IP downregulation may trigger telomere dysfunction-induced DNA damage (Table 1), which contributes to DDR in OIS.100,129,130 Interestingly, TERF2IP interacts with PML,131 a protein forming PML nuclear bodies (PML-NBs) during senescence and that has been implicated in protein degradation.132-135 This suggests that senescence-associated PML-NBs could be a specialized compartment where nuclear proteins are degraded during SAPD.
Dysfunction in nucleolar and ribosome biogenesis
The nucleolus is the principal site of ribosome synthesis, where RNA polymerase I (PolI) transcribes rRNA genes (rDNA) to produce the 47S rRNA (rRNA) precursor. The 47S precursor is cleaved and modified by 2’-O-methylation and pseudouridylation of specific nucleotides to form the mature 18S and 28S rRNAs. These processes are guided by small nucleolar RNAs (snoRNAs) assembled into RNA/protein complexes called small nucleolar ribonucleoproteins (snoRNPs). Mature rRNAs are assembled with ribosomal proteins (RPs), inside the nucleolus, to produce the 40S and 60S ribosomal subunits, which then migrate toward the cytoplasm.136 Approximately 50% of the energy of proliferating eukaryotic cells is dedicated to ribosome biogenesis, and the process requires approximately 200 snoRNAs, more than 80 RPs, and hundreds of accessory proteins.136 We found that many proteins implicated in rRNA transcription and maturation are unstable in Ras-induced senescence. They include NOLC1,137,138 NOP58, NOP56,139 DDX51,140 NOL6,141 and NOC2L.61,142 We also found unstable proteins which are implicated in late steps of ribosome synthesis, such as nucleolin (NCL),143 the ribosomal protein P1 (RPLP1),144 and the ribosomal protein L23 (RPL23) (Table 1). Although we do not know yet whether the instability of the proteins discussed above causes a decrease in their levels in senescent cells, such a reduction may lead to defects in ribosome biogenesis or may simply be part of a compensatory mechanism that degrades these proteins when ribosome biogenesis is reduced.
We confirmed the proteasome-dependent degradation of the ribosomal L1 domain-containing 1 protein (RSL1D1 or CSIG) and its decrease in Ras-induced senescence (Table 1).61 Interestingly, this protein was previously found downregulated in senescent cells.145 RSL1D1 regulates the nucleolar localization of nucleostemin (NS), which, in turn, regulates the nucleolar localization of DDX21.146 Nucleostemin and DDX21 have been shown to be important for the processing of pre-rRNA.147,148 Using RNAi screening, a role in rRNA processing was also shown for RSL1D1 together with NOP56, DDX51, NOL6/NRAP, NOC2L/NIR, and nucleolin (NCL).149 We knocked-down the expression of this protein in normal human fibroblasts, and this resulted in the induction of the senescent phenotype.61 Hence, a reduction in RSL1D1 can be causative for senescence, and its role in ribosomal biogenesis suggests that defects in this process may be another effector mechanism of senescence.
It has been shown that the alternative reading frame protein (ARF, also known as p19ARF), a well-known inducer of senescence, stabilizes p53 by inhibiting the E3 ubiquitin ligase MDM2 (HDM2).150-152 However, ARF also inhibits cell proliferation by targeting nucleophosmin (NPM1/B23) for sumoylation and degradation and in this way regulates the processing of the 32S pre-rRNA to the mature 28S rRNA.153,154 Furthermore, ARF and NPM1 control the sub-nuclear localization of the transcription termination factor I (TTF-1), which has been shown to regulate PolI transcription initiation/termination and rRNA processing.155 Therefore, in addition to inducing the senescence phenotype through the MDM2–p53 axis, ARF affects ribosome biogenesis, and we can hypothesize that this function may also reinforce the senescence program. Supporting this, we found that NPM1 is unstable in Ras-induced senescence (Table 1).
In light of the results presented above, it is tempting to suggest that defects in ribosome biogenesis can be an important mediator of senescence. This is in agreement with recent reports showing that CX-5461, an inhibitor of rRNA synthesis, induces cellular senescence in solid tumor cell lines.156,157 However, a defect in ribosome biogenesis may appear contradictory to the increased global translation reported in senescent cells,24 which could result from the activation of TOR signaling.158 Indeed, this pathway has been shown important to convert cells from a reversible quiescent state to a permanent senescent phenotype,159-162 a phenomenon called geroconversion by Blagosklonny and colleagues,163 and this could be in part due to the translational effects of TOR.164 Although further work will be required to explain how senescent cells can increase translation despite less ribosome biogenesis, it is likely that ribosome turnover decreases in these cells forcing them to use “old” ribosomes to make proteins.
Cell cycle arrest
Impaired proliferation, mainly by an arrest in the G1 phase of the cell cycle, is an established senescence feature, and the SAPD may be an important player in this process. During G1, the D-type cyclins bind the cyclin-dependent kinase (CDK) 4 and 6, and this stimulates the progression toward initiation of S phase.165 We found that the transcription coactivator Yes-associated protein YAP1 has a high turnover in Ras-expressing senescent cells (Table 1).61 Later, it was found reduced by another group during replicative senescence as well.166 YAP1 localizes to PML bodies and can regulate apoptosis via p73.167 In addition, YAP1 is the ortholog of Drosophila Yorkie that regulates organ size as part of the Hippo pathway and acts as a liver oncogene in mammals.168 Interestingly, it appears that YAP1 can circumvent senescence in some contexts by inducing the transcription of CDK6.166 Despite the fact that CDK4/6 have been shown not to be essential for proliferation, unlike CDK1,165 their downregulation in YAP1-deficient cells might interfere with cell cycle progression in a subset of specialized cells or YAP1 might play a more broad effect on CDKs. The identification of YAP1 as a potential SAPD target also suggests a role for more E3 ligases in this process. YAP1 degradation depends on a phosphodegron recognized by the E3 ligase SCF-β-TRCP169 and can also be triggered by the E3 ligase NOT4.170
The G1-to-S-phase transition is ensured by the formation of the pre-replication complexes (pre-RCs) on chromatin, which depends on the sequential recruitment of the origin recognition complexes (ORCs), CDC6, and MCM proteins.171 The DNA replication licensing factor MCM2 is an important component of the pre-RCs and was found unstable in Ras-induced senescence (Table 1). Accordingly, degradation of MCM2 could limit the initiation of DNA replication and the progression of the cell cycle.172 In addition, LRWD1/ORCA is a protein that stabilizes the origin recognition complex (ORC) on chromatin.173 LRWD1/ORCA degradation in Ras-triggered senescence (Table 1) is likely to abrogate the binding of the ORC to chromatin, consequently arresting the cells in G1. Interestingly, this protein is suspected to be polyubiquitinated by the E3 ligase complex CUL4A-DDB1,174 which has already been linked to p16INK4A upregulation in senescence.175 Hence, we can add CUL4A-DDB1 to the list of E3 ligase candidates that promote SAPD.
Since CUL4A-DDB1 has also been shown to promote proteasome-dependent degradation of MYC via the substrate receptor TRUSS (TRPC4AP),176 a role of this E3 ligase in cell cycle arrest and senescence is even more consistent considering our observation of MYC degradation in Ras-induced senescent cells (Table 1).61 MYC promotes DNA replication and is a master regulator of many cellular programs, including proliferation.177 Its downregulation is reported to contribute to senescence,178,179 and its overexpression cooperates with different oncogenes to transform cells by inhibiting cellular senescence.179-181 The downregulation of MYC levels in order to shut down the cell cycle is thus possibly at the crossroads of several senescence-promoting pathways. This not only suggests a role for the CUL4A-DDB1-TRUSS ligase in senescence, but also supports the investigation of the multiple other E3 ligases reported to target MYC to the proteasome, such as SCF-FBW7,182,183 SCF-SKP2,184,185 CHIP,186 the Mule complex (Mule/Huwe1/Arf-BP1),187 and the suggested CUL2/F–Box hybrid complex ElonginBC-CUL2-SKP2.188 Similar to MYC, JUN is another classic regulator of cell proliferation found unstable in Ras-induced senescent cells.189 Its downregulation may contribute to a block in G1-to-S-phase progression by decreasing the expression of cyclin D1190 and elevating the expression of p53 and p21.191
The degradation of the KRAB-associated protein 1 (KAP1, also known as TRIM28 or TIF1β) (Table 1), a validated SAPD target,61 may block the cell cycle by different mechanisms. First, KAP1 is known to destabilize p53, possibly explaining why high levels of KAP1 are associated with poor prognosis in gastric cancers.192 KAP1 binds and cooperates with the E3 ligase MDM2 to drive p53 degradation.193 Furthermore, the melanoma antigen (MAGE) proteins interact with KAP1 and stimulate its own E3 ligase activity to allow p53 ubiquitination and degradation in a MDM2-independent manner.194 Accordingly, downregulation of KAP1 is likely to stabilize p53, allowing the expression of key cell cycle inhibitors.195 Second, the degradation of KAP1 may relieve its transcriptional repression functions, which have been shown to directly inhibit the transcription of p53-target genes, such as the CDK inhibitor p21.196,197 Third, KAP1 depletion increases the number of PML-NBs.198 These senescence-associated nuclear structures inhibit E2F target gene expression. The latter are critical to initiate DNA synthesis, and inhibiting their transcription arrests cell proliferation.8
The T-box protein 2 (TBX2) is linked to repression of p19ARF gene expression, thereby promoting the MDM2-mediated degradation of p53 and cellular senescence suppression.199 TBX2 further antagonizes senescence by repressing the CDK inhibitors p21 and p27 (CDKN1B).200,201 Finally, TBX2 is reported to be an E2F-target gene repressed by PML, and its repression stimulates the pro-senescence functions of PML.202 Collectively, these results suggest that degradation of TBX2 (Table 1) could initiate the cell cycle arrest characterizing senescence and then reinforce the phenotype by activating a positive loop via the inhibition of its own transcription by PML.
According to a FatiGO single enrichment analysis of proteomics data with the bioinformatics platform Babelomics, the regulation of proliferation is one of the biological functions that is most enriched among unstable proteins in Ras-induced senescence (Fig. 2).61 Here, we have discussed the implication of just a few of the possible SAPD targets involved in cell proliferation. Surprisingly, after further analysis of the proteomics data, we found that the proteins corresponding to the genes identified by Chicas et al. (2010) as downregulated by RB1 in Ras-induced senescent fibroblasts are also unstable (Fig. 3).9 In this context, RB1 predominantly represses the E2F target genes implicated in DNA replication. Although this idea will need further investigation, our results suggest that bulk degradation of the same E2F-induced proteins could cooperate with transcriptional repression to safeguard cell cycle arrest. Does the SAPD cooperate with RB1 and PML-NBs to ensure a rapid and potent shutdown of E2F target genes?

Figure 3. The proteins corresponding to the genes downregulated by RB1 in Ras-induced senescent fibroblasts are also unstable. Unbiased Gene Set Enrichment Analysis (GSEA) of the proteomic data as in Figure 2. The gene set CHICAS_RB1_TARGETS_SENESCENT (Systematic name: M2125) was the second most significant result among the proteins stabilized by MG132 in Ras-induced senescent cells. The normalized enrichment score (NES), the nominal P value determined by an empirical phenotype-based permutation test procedure and the false discovery rate (FDR; Q value) are shown.
Impaired mRNA metabolism and translation
The Y-Box binding protein 1 (YB-1 or YBX1) is also unstable in Ras-induced senescent cells (Table 1), and its downregulation has been linked to the senescence phenotype,203 whereas its overexpression strongly correlates with aggressive tumors.204 However, YB-1 is a multifunctional protein, and we are still far from understanding how its functions could oppose senescence. One hypothesis is that YB-1 could stimulate the transcription of E2F-target genes by binding to multiple E2F promoters.205 Conversely, it could act as a transcriptional repressor of p53.206 These scenarios suggest that depletion of YB-1 could have a relatively direct effect on cell cycle as discussed in the previous section. Nonetheless, direct evidence also highlights critical functions in mRNA metabolism, including mRNA stability, mRNA packaging, splicing, and translational initiation.204,207 Does the regulation of p53 and E2F-target genes result from these activities? Although the answer is not clear, this could be the case at least for p53. The DNA-damaging stresses prevent YB-1-mediated splicing of MDM2, leading to an mRNA molecule lacking several exons, and resulting in a non-functional protein.208 This regulation of MDM2 may contribute to the stabilization of p53 in senescent cells experiencing DDR. Because YB-1 is a putative general regulator of mRNA maturation and translation for mRNAs with YB-1 binding sites,207 we hypothesize that suppression of these functions could promote senescence by affecting the expression of several proteins. In senescence, the YB-1 functions could be abrogated by its degradation, possibly catalyzed by the E3 ligase activity of RBBP6.209
Two other splicing regulators have an increased turnover in Ras-induced senescence (Table 1). SRm160 (SRRM1) and SRm300 (SRRM2) are splicing coactivators required for the functions of exonic splicing enhancers and for 3′-end processing of specific pre-mRNAs.210-213 These proteins are phosphorylated at multiple distinct S/T-P phosphorylation sites in senescent cells, suggesting that they may act as a sensor of ERK signaling strength.61 Perhaps an accumulation of phosphorylated sites over a given threshold controls the interaction with E3 ligases, promoting the ubiquitination and degradation of hyperphosphorylated SRm160/300 in response to oncogenic stress. Such degradation could consequently promote senescence by impeding normal mRNA maturation of a specific set of genes, including critical regulators of normal cell functions.
Key Remaining Questions
Targeting protein to SAPD: Where?
Proteasomes are widespread in cells, but can interact with some specific cellular structures. In the cytoplasm, proteasomes can bind the cytoskeleton, the outer surface of the endoplasmic reticulum, and the centrosomes.214-216 They are also found throughout the nucleoplasm, but, interestingly, they have been shown to be concentrated in PML-NBs,214,217-220 nucleoplasmic speckles,219,221 and other focal subdomains.219,222 In some particular contexts, proteasomes can also accumulate in nucleoli.222,223 Thus, the degradation of SAPD targets could use specific “proteolytic centers”. For example, Wójcik and DeMartino (2003) proposed that cytosolic proteins targeted for degradation are delivered to a master proteolytic complex located at the centrosome via microtubule-mediated transport.214 Similarly, PML-NBs and nuclear speckles could act as the proteolytic complexes for nucleoplasmic SADP targets. Speckles are enriched in splicing factors and may thus be the proteolytic center for these proteins we found unstable during Ras-induced senescence, including YBX1, SRm160, and 300 (Table 1).224,225 Also, PML-NBs might be a specialized structure for short proteins destined to be degraded vs. these that should not, thereby representing the so-called clastosome previously described as nuclear bodies enriched in proteasome-dependent degradation effectors.226 Consistent with this idea, several potential and validated SAPD targets colocalize with PML-NBs, including TERF2IP,131 YAP1,167 MYC,133,179,227 and STAT3.228,229 Furthermore, PML-NBs have been shown to be involved in the degradation of factors for which downregulation is known to mediate a senescence program, such as CREBBP (CBP)230,231 and MYC.133,178,179 Conversely, PML-NBs might also play an active role in protecting other proteins from degradation, like HIPK2,232 p73,233 TOPBP1,234 and p53.235 Thus, PML may be critical for the specificity of SAPD.
Targeting protein to SAPD: How?
The pattern of proteins degraded by the proteasome seems dramatically changed during senescence, while there is no apparent modification in total proteasome activity (Fig. 1D). Also, even if there is a large amount of unstable proteins, other key senescence mediators are stabilized (e.g., p53). These observations suggest 2 principal mechanisms explaining the proteasome-dependent degradation of a large subset of specific proteins in senescence: the upregulation of specific E3 ligases activity and the targeting of specific proteins for SAPD. Previous work and our recent observations strongly propose that post-translational modifications of proteins play a central role in SAPD. Because PML-NBs could be involved in SAPD target degradation, sumoylation is a candidate modification of particular interest. Indeed, PML-NBs are among the principal sites of sumoylation in cells, since they interact with many SUMO ligases and sumoylated proteins.220 Furthermore, sumoylation is known to lead to the subsequent ubiquitination and degradation of particular proteins.236,237 There is now accumulating evidence that sumoylation at PML-NBs is coupled with the UPP, the SUMO-dependent degradation of N4BP1135 and NRF2238 being examples. Of note, the degradation of the latter in PML-NBs could limit ROS detoxification, thereby contributing to the induction of senescence.239 Senescent cells experience oxidative stress, suggesting that protein carbonylation may serve as another modification to distinguish SAPD targets.240 This modification marks oxidized proteins for degradation, mostly via the 20S proteasome and in an ATP- and ubiquitin-independent manner.241 Despite the fact that carbonyl-mediated degradation exhibits a certain level of specificity, depending on the intrinsic susceptibility of a protein to oxidative carbonylation, this mechanism is rather unspecific and hardly explains the global proteome of senescent cells.240
Our group identified a remarkable number of phosphopeptides from proteins degraded by the proteasome in Ras-induced senescent cells, suggesting that phosphorylation is an important protein modification triggering SAPD.61 Further strengthening this hypothesis, protein phosphorylation and ubiquitination-dependent degradation are tightly linked.242 Phosphorylation can drive ubiquitination either by regulating the subcellular localization of target proteins, thereby eliminating a spatial separation between the substrate and its E3 ligase, or by creating a docking site for an E3 ligase.242 In replicative and Ras-induced senescence, hyperactivation of the ERK/MAPK pathway is essential to mediate SAPD and to maintain the senescent phenotype.61 This suggests a model where the hyperphosphorylated ERK targets are degraded, creating a negative feedback to mitogenic signaling that promotes senescence.63 In this model, the sustained phosphorylation of ERK targets is suspected to increase the chance of an interaction with an E3 ligase or to activate a phosphodegron. However, we cannot exclude that other kinases play a role in ERK-mediated SAPD. Such kinases could be either hyperactivated downstream of the ERK/MAPK pathway or contribute to the full activation of phosphodegrons. The SAPD candidates MYC and JUN are 2 examples of proteins regulated by a phosphodegron implicating multiple kinases. Both are first phosphorylated by the ERK kinases, priming them for further phosphorylation by the GSK3 kinase, which is the final act in order to recruit the E3 ligase SCF-FBW7.189,242,243 Supporting an important role for GSK3 in mediating activation of phosphodegrons during SAPD, its inhibition leads to a reduction of MYC ubiquitination.61 Furthermore, another SAPD candidate in Ras-induced senescence, namely β-catenin (CTNNB1), is a well-known protein undergoing degradation following GSK3-mediated phosphodegron activation.242 Considering that GSK3 is inactivated by the PI3K/AKT pathway,243 buffering AKT activity is maybe an important strategy employed to promote SAPD in Ras-induced senescent cells. This could explain, at less in part, why activation of AKT contributes to circumvent RAF and RAS-induced senescence.244,245 Nevertheless, we can speculate that hyperactivation of different kinases, including AKT, could also engage the degradation of their targets and thus promote a different pattern of SAPD, but with senescence as a common phenotypic output. SAPD could be a universal response to “phosphorylation stress” to avoid cellular transformation in the context of abnormal mitogenic signaling.
Kinases vs. phosphatases and E3 ligases vs. deubiquitinases: Different weapons, same fight?
When we address proteasome-dependent protein degradation, we naturally think of E3 ubiquitin ligases. However, the global picture is much more complicated and involves several players. Proteins can be dynamically ubiquitinated by E3 ligases and deubiquitinated by deubiquitinases. As we discussed in the previous section, ubiquitination can depend on phosphorylation.242 In this situation, protein degradation is also regulated by kinases and phosphatases. We can thus simplify the situation by presenting kinases and E3 ubiquitin ligases as collaborating to favor protein degradation, whereas phosphatases and deubiquitinases are their respective opponents. A similar logic can be applied for SUMO-dependent ubiquitination; while SUMO E3 ligases cooperate with E3 ubiquitin ligases, deSUMOylases antagonize the process.237 In normal conditions, a subtle equilibrium between all the players impacting on protein stability ensures determined levels for a specific protein (Fig. 4A). During SAPD, the equilibrium is displaced to favor an increased turnover leading to reduced levels of the same protein. What exactly leads to the displacement of the equilibrium? For a given protein, the process can be mediated mostly by: (1) an increased activity of its kinases/SUMO E3 ligases (Fig. 4B); (2) an increased activity of its E3 ubiquitin ligases (Fig. 4C); (3) both 1 and 2 (Fig. 4D); (4) a decreased activity of its phosphatases/deSUMOylases (Fig. 4E); (5) a decreased activity of its deubiquitinases (Fig. 4F); (6) both 4 and 5 (Fig. 4G); (7) different combinations of 2 to 6. One challenge for the coming years will be to determine how these regulators interact to affect the steady state and what the resulting dynamic is. Is the equilibrium displaced linearly or does the collaboration between different SAPD mediators rather promote switch-like mechanisms? Such switch-like responses could point to competition between regulators with opposite effects on the substrate, which has been shown in the control of the orthologous yeast ERK/MAPK pathway.246 Finally, another challenge is to evaluate whether there are master regulators of protein degradation in SAPD, allowing opportunities to target the phenotype, or whether each protein or subset of proteins is regulated via distinct machinery.

Figure 4. Modulation of protein stability for proteins regulated by phosphorylation-driven ubiquitination and proteasome-dependent degradation. (A) Under normal conditions, competition between the activity of kinases vs. phosphatases (PPases) and E3 ubiquitin ligases vs. deubiquitinases (DUBs) ensures the maintenance of appropriate levels of a specific protein. The turnover of this protein can be increased by (B) increasing the activity of its kinases; (C) increasing the activity of its E3 ubiquitin ligases; (D) both (B and C); (E) decreasing the activity of its PPases; (F) decreasing the activity of its DUBs; (G) both (E and F). Of note, different combinations of (B) to (G) can be involved. Also, a similar scenario can be applied for SUMO-dependent ubiquitination; kinases and PPases can be substituted by SUMO E3 ligases and deSUMOylase. Ub, ubiquitin; P, phosphorylation.
Is there a master senescence-associated E3 ligase?
The specificity of the UPP is conferred by E3 ubiquitin ligases, a large and complex group of proteins, with an estimated 600 to 1000 members in the human proteome.247 Based on the structure of their catalytic domain, the E3 ubiquitin ligases are generally classified into 4 main categories: the RING-finger type,248,249 the HECT type,250 the U-box type,251,252 and the less characterized PHD domain-containing type.248,253 The former is by far the most abundant and is further subdivided as single unit or multiple subunit RING-finger E3 ligases. The latter form complexes grouped into 2 principal families, the anaphase-promoting complex (APC) and the cullin-RING ligase (CRL) superfamily.248 There are 7 cullins expressed in human cells (CUL1, 2, 3, 4A, 4B, 5, and 7) and they interact with specific receptor proteins which provide target specificity, including proteins harboring F-box, SOCS-box, VHL-box, and BTB domains.254,255 These complexes are referred to by various names (reviewed in ref. 255), but the most common appellation is probably SCF for the classic complex containing CUL1 and SCF2–5 and 7 for complexes containing the corresponding cullins.
In the simplest scenario, one or few E3 ubiquitin ligases could be responsible for SAPD. Such a possibility would likely involve the regulation of the activity of specific E3 ligases. However, current evidence reviewed above points to the specificity being conferred by upstream steps targeting designated proteins for degradation. Considering these data as well as the complexity of the E3 ubiquitin ligase superfamily, we favor the view that SAPD is likely regulated by several E3 ligases, each catalyzing the ubiquitination of its specific targets. However, this more complex picture does not exclude the possibility that some E3 ligases could play a more critical role in the senescent phenotype. Indeed, as discussed previously, SCF-FBW7 is a well-known tumor suppressor and has been recently shown to contribute to senescence,243,247,256 and correspondingly many FBW7 targets are degraded in SAPD. The CUL4A–DDB1 (SCF4) complex and its interacting receptor protein DDB2 are also strong candidates, since both have been shown to drive senescence.175,257 Furthermore, the fact that phosphorylation could be a mark to distinguish SAPD targets highlights the interest in investigating the roles of the SCF complexes in senescence. Indeed, this subfamily of E3 ubiquitin ligases is primarily responsible for serine/threonine phosphorylation-dependent ubiquitination. Two classes of F-box proteins are specialized to recognize phosphodegrons, namely, the WD40 F-box proteins (e.g., FBW7 and β-TRCP1/2) and leucine-rich repeat (LRR) F-box proteins (e.g., SKP2).242 The proposed involvement of PML-NBs and SUMO-dependent degradation in SAPD also increases the interest in studying the contribution of the SUMO-targeted ubiquitin ligase (STUbL) family in senescence,258 such as RNF4, which contains a SUMO-interacting motif (SIM).259
Although many E3 ubiquitin ligases have tumor-suppressive functions, including APC, SCF-FBW7, BRCA1, VHL, and FANC, several others are clearly oncogenic and can oppose senescence.248 For example, MDM2 and MDMX are bona fide oncogenes and limit senescence by catalyzing ubiquitination and degradation of p53.152,248 Senescence is also limited by the oncogene SCF-SKP2 that targets p27 and p21 in a p53-independent manner.260 The potential role in cancer of E3 ligases that have a complex array of targets, including both tumor suppressors and oncogenes, is more difficult to ascertain. This is the case for SCF-β-TRCP, functioning primarily as an oncogene by targeting apoptotic proteins, but showing tumor-suppressive activities in some contexts.248 Since SCF-β-TRCP targets preferentially phosphorylated proteins,242 whether it acts as an oncogene or as a tumor suppressor may depend on the pool of phosphorylated substrates in a given context. In light of these dissimilar functions in tumorigenesis, it seems obvious that different members of the large family of E3 ubiquitin ligases use the UPP to compete in opposite directions. We can thus compare the effect of the E3 ligases on cell fate to a delicate balance, where the equilibrium between the activities of oncogenic vs. tumor-suppressive E3 ligases is critical to maintain cells in a normal state (Fig. 5). Under oncogenic stress, depending on whether the balance is tipped in one direction or the other, the UPP could favor transformation into cancer cells or tumor suppression, respectively.
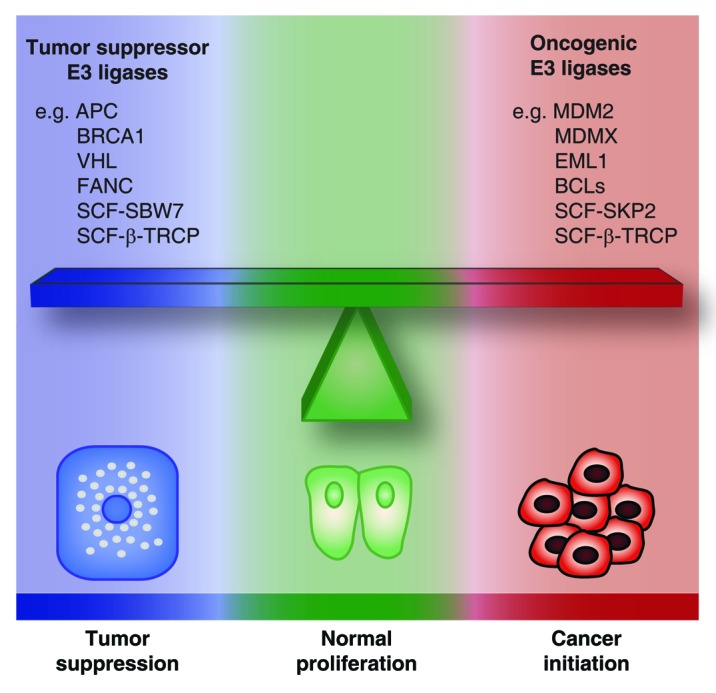
Figure 5. The balance of oncogenic vs. tumor suppressor E3 ubiquitin ligases. The activities of oncogenic vs. tumor suppressor E3 ligases are in equilibrium to maintain cells in a normal state. Tipping the balance in one direction or the other can be critical for determining whether a cell under oncogenic stress will undergo tumor suppression or neoplastic transformation.
SAPD: A coordinated proteome reprogramming?
The answer to this question first depends on how senescence should be seen. Is it a totally abnormal and non-functional cellular state initiated in response to stress, which is basically avoided in normal organisms? Is it rather one of the fundamental tools that evolution has provided as a defense against the insults inherent to organismal life? The prevailing view at present favors that latter paradigm. Indeed, not only is senescence a gatekeeper response that is acutely triggered by stress stimuli, but it now appears that the process can have important functions in non-stressed conditions, namely in embryonic patterning.261,262 Hence, considering senescence as a “normal” adaptive state, the question is now: what is the fundamental role of SAPD in senescence, and why it was selected during evolution?
Cellular senescence was first thought to underlie organismal aging, and this hypothesis steadily gained experimental support.263 The deleterious effects of senescence are caused by the accumulation of senescent cells in aging organisms. However, it is possible that this accumulation is rather the result of an abnormal senescence program, and that evolution has selected a mechanism to avoid the accumulation of senescent cells. The recent literature suggests that this mechanism may be the clearance of senescent cells by the immune system.57-60 The SASP seems critical to activate the immune response by signaling the presence of senescent cells and attracting destructive immune cells.5 If the ultimate destiny of senescent cells is their elimination, the production of signaling molecules during the SASP appears central to ensure a complete and effective senescence phenotype (Fig. 6). An abnormal SASP pattern or a defect in the capacity of the immune system to eliminate senescent cells could thus be the basis of an “abnormal” accumulation of senescent cells and age-related pathologies.263
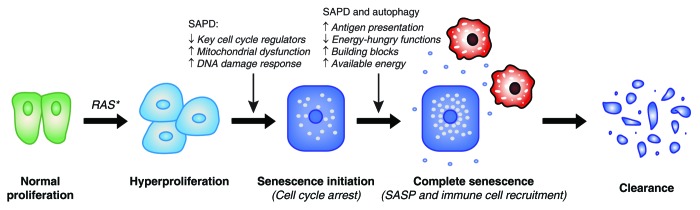
Figure 6. Theoretical purpose of oncogene-induced senescence and contribution of protein degradation. Increasing evidence suggests that the destiny of senescent cells in many organs is clearance by the immune system. This implies a central role for the cytokine production characteristic of the SASP in the recruitment of immune cells (in red). Specific protein degradation (SAPD) may contribute directly and/or indirectly to the initial cell cycle arrest, but may also cooperate with macroautophagy to produce antigenic peptides and to support the SASP. Proteolysis may redistribute cellular energy to the SASP and may supply nutrient building blocks for biosynthetic reactions.
The SASP is a costly anabolic process, and senescent cells have to deal with the limited availability of building blocks and energy to support the process. Thus, we can suppose that cells reorganize the distribution of these resources in order to favor the synthesis of cytokines. Does reorganizing the proteome mean reallocating resources? Such a link between autophagy and SASP has already been proposed.25,51,52 Is the SAPD part of this reorganization? The degradation of specific proteins by the UPP could shut down highly energy-consuming functions, such as protein repair, DNA repair, synthesis of new ribosomes, and DNA synthesis. Since senescent cells are destined for clearance, these functions are dispensable for senescent cells, and their inhibition allows more resources to support the SASP (Fig. 6). Protein degradation by UPP consumes ATP, but the resulting amino acids can be used to obtain energy or supply building blocks for anabolic reactions. Overall, the SAPD could be a better investment for senescent cells whose final destiny is to be eliminated.
Breaking down cancer?
Although SAPD could be a powerful mechanism to mediate senescence and tumor suppression, it raises many new questions for further research. The exact contribution of protein degradation to senescence, including SAPD and autophagy, is still mostly speculative. Perhaps it simply brings a balance to cells unable to divide but making more proteins. However, catabolic processes may take a central place to induce cell cycle arrest of premalignant cells and to trigger their elimination by the immune system. Not only could proteolysis redistribute the resources to support the production of cytokines by oncogene-expressing cells, but it could also generate peptides for antigen presentation to ensure their specific recognition and destruction by immune cells (Fig. 6).264,265 This may involve the production of an abnormal quantity of a self-antigen or the generation of abnormal antigens, such as pieces of activated oncogenes or damaged proteins.
A better understanding of the senescence degradome appears essential to have a more global picture of how anabolic and catabolic changes are linked together to trigger a complete senescence phenotype. This could provide insights into how cancer cells circumvent senescence and the role of metabolic changes in this process, thereby suggesting new therapeutic strategies. Targeting components of the UPP and autophagy with small-molecule inhibitors is an emerging area for the treatment of cancer.266 The clinical potential of this strategy has been highlighted by the success of the proteasome inhibitor bortezomib for the treatment of myeloma and lymphoma. Currently, most of the efforts are invested in the development of proteasome inhibitors, which have a global, and thus non-specific, effect on the UPP-mediated degradome. Such an approach can preferentially affect cancer cells where the pattern of E3 ubiquitin ligase activities and UPP-targeted substrates clearly support tumorigenesis and cancer progression (Fig. 5). However, the UPP has a fundamental role in normal cellular functions and in tumor suppression as well. This suggests caution in the clinical use of proteasome inhibitors and may explain the toxicity associated with these compounds.266 A better comprehension of SAPD and its dysfunction in cancer cells will certainly uncover new pharmacologic vulnerabilities to allow the rational development of new targeted therapies. Can we restore the advantages given by the SAPD, such as the elimination of precancerous cells by the immune system, and at the same time inhibit UPP-driven oncogenesis? In other words, can we tip the balance of protein breakdown to break down cancer?
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
X.D.-S. is a fellow of the Vanier Canada Graduate Scholarships Program and Michael Smith Foreign Study Supplements Program. F.L. is a fellow of FRQS (Fonds de recherche du Québec - Santé). N.B. is supported by grants from the National Institutes of Health (R01 CA133557-05 and P01 CA117969-07) and the Linda J. Verville Cancer Research Foundation. G.F. is a FRSQ national research fellow.
References
- 1.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–45. doi: 10.1016/S0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Pan KH, Cohen SN. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc Natl Acad Sci U S A. 2003;100:3251–6. doi: 10.1073/pnas.2627983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac-de Paillerets B, Hofman P, et al. Senescent cells develop a PARP-1 and nuclear factor-kappaB-associated secretome (PNAS) Genes Dev. 2011;25:1245–61. doi: 10.1101/gad.625811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Bégin V, Saad F, Mes-Masson AM, Ferbeyre G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011;25:41–50. doi: 10.1101/gad.1975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–87. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–4. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 11.Jackson JG, Pereira-Smith OM. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res. 2006;66:8356–60. doi: 10.1158/0008-5472.CAN-06-1752. [DOI] [PubMed] [Google Scholar]
- 12.McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–4. doi: 10.1016/S0960-9822(98)70137-X. [DOI] [PubMed] [Google Scholar]
- 13.Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–27. [PMC free article] [PubMed] [Google Scholar]
- 14.de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004;13:523–35. doi: 10.1016/S1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 15.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, Lowe SW. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev. 2012;26:1546–57. doi: 10.1101/gad.196238.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–22. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns DM, Richter JD. CPEB regulation of human cellular senescence, energy metabolism, and p53 mRNA translation. Genes Dev. 2008;22:3449–60. doi: 10.1101/gad.1697808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24:1580–9. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petroulakis E, Parsyan A, Dowling RJ, LeBacquer O, Martineau Y, Bidinosti M, Larsson O, Alain T, Rong L, Mamane Y, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–46. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 21.Scaglioni PP, Rabellino A, Yung TM, Bernardi R, Choi S, Konstantinidou G, Nardella C, Cheng K, Pandolfi PP. Translation-dependent mechanisms lead to PML upregulation and mediate oncogenic K-RAS-induced cellular senescence. EMBO Mol Med. 2012;4:594–602. doi: 10.1002/emmm.201200233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salomoni P, Dvorkina M, Michod D. Role of the promyelocytic leukaemia protein in cell death regulation. Cell Death Dis. 2012;3:e247. doi: 10.1038/cddis.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louria-Hayon I, Alsheich-Bartok O, Levav-Cohen Y, Silberman I, Berger M, Grossman T, Matentzoglu K, Jiang YH, Muller S, Scheffner M, et al. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 2009;16:1156–66. doi: 10.1038/cdd.2009.31. [DOI] [PubMed] [Google Scholar]
- 24.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–27. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 32.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–83. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 37.Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059–63. doi: 10.1016/j.humpath.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 38.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–13. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He YW, Ratnayaka I, Zhong S, Goldin RD, Goemans CG, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A. 2012;109:13325–30. doi: 10.1073/pnas.1120193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerland LM, Peyrol S, Lallemand C, Branche R, Magaud JP, Ffrench M. Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp Gerontol. 2003;38:887–95. doi: 10.1016/S0531-5565(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 42.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28:889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh K, Matsuyama S, Drazba JA, Almasan A. Autophagy-dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy. 2012;8:236–51. doi: 10.4161/auto.8.2.18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90:835–43. doi: 10.1038/labinvest.2010.56. [DOI] [PubMed] [Google Scholar]
- 46.Mosieniak G, Adamowicz M, Alster O, Jaskowiak H, Szczepankiewicz AA, Wilczynski GM, Ciechomska IA, Sikora E. Curcumin induces permanent growth arrest of human colon cancer cells: link between senescence and autophagy. Mech Ageing Dev. 2012;133:444–55. doi: 10.1016/j.mad.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, Goligorsky MS. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol. 2008;294:H1119–29. doi: 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- 48.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe. 2012;11:167–80. doi: 10.1016/j.chom.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol. 2013;202:129–43. doi: 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–5. doi: 10.1038/nature12437. [DOI] [PubMed] [Google Scholar]
- 52.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 53.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 55.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 56.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–60. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 60.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deschênes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette FA, Saba-El-Leil MK, Meloche S, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27:900–15. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Jong A, Schuurman KG, Rodenko B, Ovaa H, Berkers CR. Fluorescence-based proteasome activity profiling. Methods Mol Biol. 2012;803:183–204. doi: 10.1007/978-1-61779-364-6_13. [DOI] [PubMed] [Google Scholar]
- 63.Deschênes-Simard X, Kottakis F, Meloche S, Ferbeyre G. ERKs in cancer: friends or foes? Cancer Res. 2014;74:412–9. doi: 10.1158/0008-5472.CAN-13-2381. [DOI] [PubMed] [Google Scholar]
- 64.Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Ballmoos C, Wiedenmann A, Dimroth P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu Rev Biochem. 2009;78:649–72. doi: 10.1146/annurev.biochem.78.081307.104803. [DOI] [PubMed] [Google Scholar]
- 67.Zwerschke W, Mazurek S, Stöckl P, Hütter E, Eigenbrodt E, Jansen-Dürr P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J. 2003;376:403–11. doi: 10.1042/BJ20030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hutter E, Renner K, Pfister G, Stöckl P, Jansen-Dürr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004;380:919–28. doi: 10.1042/BJ20040095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem. 2013;288:4723–32. doi: 10.1074/jbc.M112.378984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elschami M, Scherr M, Philippens B, Gerardy-Schahn R. Reduction of STAT3 expression induces mitochondrial dysfunction and autophagy in cardiac HL-1 cells. Eur J Cell Biol. 2013;92:21–9. doi: 10.1016/j.ejcb.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, Derecka M, Chen Q, Hu Y, Sisler J, et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem. 2013;288:31280–8. doi: 10.1074/jbc.M113.505057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faou P, Hoogenraad NJ. Tom34: a cytosolic cochaperone of the Hsp90/Hsp70 protein complex involved in mitochondrial protein import. Biochim Biophys Acta. 2012;1823:348–57. doi: 10.1016/j.bbamcr.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 75.Rao S, Gerbeth C, Harbauer A, Mikropoulou D, Meisinger C, Schmidt O. Signaling at the gate: phosphorylation of the mitochondrial protein import machinery. Cell Cycle. 2011;10:2083–90. doi: 10.4161/cc.10.13.16054. [DOI] [PubMed] [Google Scholar]
- 76.Gabai VL, Yaglom JA, Waldman T, Sherman MY. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol. 2009;29:559–69. doi: 10.1128/MCB.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–38. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–13. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–12. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 80.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–6. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 81.Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta. 2005;1703:93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 82.Dai C, Dai S, Cao J. Proteotoxic stress of cancer: implication of the heat-shock response in oncogenesis. J Cell Physiol. 2012;227:2982–7. doi: 10.1002/jcp.24017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kalmar B, Greensmith L. Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev. 2009;61:310–8. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 84.Sherman M. Major heat shock protein Hsp72 controls oncogene-induced senescence. Ann N Y Acad Sci. 2010;1197:152–7. doi: 10.1111/j.1749-6632.2010.05196.x. [DOI] [PubMed] [Google Scholar]
- 85.Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jäättelä M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005;19:570–82. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yaglom JA, Gabai VL, Sherman MY. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007;67:2373–81. doi: 10.1158/0008-5472.CAN-06-3796. [DOI] [PubMed] [Google Scholar]
- 87.Kim G, Meriin AB, Gabai VL, Christians E, Benjamin I, Wilson A, Wolozin B, Sherman MY. The heat shock transcription factor Hsf1 is downregulated in DNA damage-associated senescence, contributing to the maintenance of senescence phenotype. Aging Cell. 2012;11:617–27. doi: 10.1111/j.1474-9726.2012.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonelli MA, Alfieri RR, Petronini PG, Brigotti M, Campanini C, Borghetti AF. Attenuated expression of 70-kDa heat shock protein in WI-38 human fibroblasts during aging in vitro. Exp Cell Res. 1999;252:20–32. doi: 10.1006/excr.1999.4614. [DOI] [PubMed] [Google Scholar]
- 89.O’Callaghan-Sunol C, Gabai VL, Sherman MY. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res. 2007;67:11779–88. doi: 10.1158/0008-5472.CAN-07-2441. [DOI] [PubMed] [Google Scholar]
- 90.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, Maertens O, Jeong SM, Bronson RT, Lebleu V, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–13. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34:1181–8. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 94.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 96.Marques C, Guo W, Pereira P, Taylor A, Patterson C, Evans PC, Shang F. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006;20:741–3. doi: 10.1096/fj.05-5080fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sisoula C, Gonos ES. CHIP E3 ligase regulates mammalian senescence by modulating the levels of oxidized proteins. Mech Ageing Dev. 2011;132:269–72. doi: 10.1016/j.mad.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 98.Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. 2011;10:006924. doi: 10.1074/mcp.M110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/S0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 100.Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol. 2012;14:355–65. doi: 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodier F, Muñoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppé JP, Campeau E, Beauséjour CM, Kim SH, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 103.Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007;6:1831–6. doi: 10.4161/cc.6.15.4516. [DOI] [PubMed] [Google Scholar]
- 104.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 105.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–13. doi: 10.1016/S1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 106.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 107.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 108.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–8. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Merolla F, Pentimalli F, Pacelli R, Vecchio G, Fusco A, Grieco M, Celetti A. Involvement of H4(D10S170) protein in ATM-dependent response to DNA damage. Oncogene. 2007;26:6167–75. doi: 10.1038/sj.onc.1210446. [DOI] [PubMed] [Google Scholar]
- 110.Zhao J, Tang J, Men W, Ren K. FBXW7-mediated degradation of CCDC6 is impaired by ATM during DNA damage response in lung cancer cells. FEBS Lett. 2012;586:4257–63. doi: 10.1016/j.febslet.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 111.Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu ZX, Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–40. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 113.Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–22. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–80. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 116.Blander G, de Oliveira RM, Conboy CM, Haigis M, Guarente L. Superoxide dismutase 1 knock-down induces senescence in human fibroblasts. J Biol Chem. 2003;278:38966–9. doi: 10.1074/jbc.M307146200. [DOI] [PubMed] [Google Scholar]
- 117.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 118.Bermejo R, Doksani Y, Capra T, Katou YM, Tanaka H, Shirahige K, Foiani M. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007;21:1921–36. doi: 10.1101/gad.432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cuvier O, Stanojcic S, Lemaitre JM, Mechali M. A topoisomerase II-dependent mechanism for resetting replicons at the S-M-phase transition. Genes Dev. 2008;22:860–5. doi: 10.1101/gad.445108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, Kanoh Y, Shirahige K, Azvolinsky A, Zakian VA, Foiani M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell. 2010;39:595–605. doi: 10.1016/j.molcel.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baxter J, Diffley JF. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell. 2008;30:790–802. doi: 10.1016/j.molcel.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 122.Holm C, Stearns T, Botstein D. DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol Cell Biol. 1989;9:159–68. doi: 10.1128/mcb.9.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bermejo R, Capra T, Gonzalez-Huici V, Fachinetti D, Cocito A, Natoli G, Katou Y, Mori H, Kurokawa K, Shirahige K, et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell. 2009;138:870–84. doi: 10.1016/j.cell.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 124.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 125.Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–83. doi: 10.1016/S0092-8674(00)80858-2. [DOI] [PubMed] [Google Scholar]
- 126.Crabbe L, Cesare AJ, Kasuboski JM, Fitzpatrick JA, Karlseder J. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2012;2:1521–9. doi: 10.1016/j.celrep.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bae NS, Baumann P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol Cell. 2007;26:323–34. doi: 10.1016/j.molcel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 128.Sarthy J, Bae NS, Scrafford J, Baumann P. Human RAP1 inhibits non-homologous end joining at telomeres. EMBO J. 2009;28:3390–9. doi: 10.1038/emboj.2009.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, Boccardi V, Fumagalli M, Di Micco R, Mirani N, Gurung RL, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012;31:2839–51. doi: 10.1038/emboj.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee OH, Kim H, He Q, Baek HJ, Yang D, Chen LY, Liang J, Chae HK, Safari A, Liu D, et al. Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol Cell Proteomics. 2011;10:001628. doi: 10.1074/mcp.M110.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honoré N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P, et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med. 2001;193:1361–71. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buschbeck M, Uribesalgo I, Ledl A, Gutierrez A, Minucci S, Muller S, Di Croce L. PML4 induces differentiation by Myc destabilization. Oncogene. 2007;26:3415–22. doi: 10.1038/sj.onc.1210128. [DOI] [PubMed] [Google Scholar]
- 134.Vennemann A, Hofmann TG. SUMO regulates proteasome-dependent degradation of FLASH/Casp8AP2. Cell Cycle. 2013;12:1914–21. doi: 10.4161/cc.24943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sharma P, Murillas R, Zhang H, Kuehn MR. N4BP1 is a newly identified nucleolar protein that undergoes SUMO-regulated polyubiquitylation and proteasomal turnover at promyelocytic leukemia nuclear bodies. J Cell Sci. 2010;123:1227–34. doi: 10.1242/jcs.060160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moss T. At the crossroads of growth control; making ribosomal RNA. Curr Opin Genet Dev. 2004;14:210–7. doi: 10.1016/j.gde.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 137.Yang Y, Isaac C, Wang C, Dragon F, Pogacic V, Meier UT. Conserved composition of mammalian box H/ACA and box C/D small nucleolar ribonucleoprotein particles and their interaction with the common factor Nopp140. Mol Biol Cell. 2000;11:567–77. doi: 10.1091/mbc.11.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tsai YT, Lin CI, Chen HK, Lee KM, Hsu CY, Yang SJ, Yeh NH. Chromatin tethering effects of hNopp140 are involved in the spatial organization of nucleolus and the rRNA gene transcription. J Biomed Sci. 2008;15:471–86. doi: 10.1007/s11373-007-9226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lechertier T, Grob A, Hernandez-Verdun D, Roussel P. Fibrillarin and Nop56 interact before being co-assembled in box C/D snoRNPs. Exp Cell Res. 2009;315:928–42. doi: 10.1016/j.yexcr.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 140.Srivastava L, Lapik YR, Wang M, Pestov DG. Mammalian DEAD box protein Ddx51 acts in 3′ end maturation of 28S rRNA by promoting the release of U8 snoRNA. Mol Cell Biol. 2010;30:2947–56. doi: 10.1128/MCB.00226-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Utama B, Kennedy D, Ru K, Mattick JS. Isolation and characterization of a new nucleolar protein, Nrap, that is conserved from yeast to humans. Genes Cells. 2002;7:115–32. doi: 10.1046/j.1356-9597.2001.00507.x. [DOI] [PubMed] [Google Scholar]
- 142.Wu J, Zhang Y, Wang Y, Kong R, Hu L, Schuele R, Du X, Ke Y. Transcriptional repressor NIR functions in the ribosome RNA processing of both 40S and 60S subunits. PLoS One. 2012;7:e31692. doi: 10.1371/journal.pone.0031692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abdelmohsen K, Gorospe M. RNA-binding protein nucleolin in disease. RNA Biol. 2012;9:799–808. doi: 10.4161/rna.19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Artero-Castro A, Kondoh H, Fernández-Marcos PJ, Serrano M, Ramón y Cajal S, Lleonart ME. Rplp1 bypasses replicative senescence and contributes to transformation. Exp Cell Res. 2009;315:1372–83. doi: 10.1016/j.yexcr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 145.Ma L, Chang N, Guo S, Li Q, Zhang Z, Wang W, Tong T. CSIG inhibits PTEN translation in replicative senescence. Mol Cell Biol. 2008;28:6290–301. doi: 10.1128/MCB.00142-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Meng L, Yasumoto H, Tsai RY. Multiple controls regulate nucleostemin partitioning between nucleolus and nucleoplasm. J Cell Sci. 2006;119:5124–36. doi: 10.1242/jcs.03292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Romanova L, Grand A, Zhang L, Rayner S, Katoku-Kikyo N, Kellner S, Kikyo N. Critical role of nucleostemin in pre-rRNA processing. J Biol Chem. 2009;284:4968–77. doi: 10.1074/jbc.M804594200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mialon A, Thastrup J, Kallunki T, Mannermaa L, Westermarck J, Holmström TH. Identification of nucleolar effects in JNK-deficient cells. FEBS Lett. 2008;582:3145–51. doi: 10.1016/j.febslet.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 149.Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DL. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol Cell. 2013;51:539–51. doi: 10.1016/j.molcel.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 150.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–7. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 151.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–64. doi: 10.1016/S1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 154.Tago K, Chiocca S, Sherr CJ. Sumoylation induced by the Arf tumor suppressor: a p53-independent function. Proc Natl Acad Sci U S A. 2005;102:7689–94. doi: 10.1073/pnas.0502978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lessard F, Morin F, Ivanchuk S, Langlois F, Stefanovsky V, Rutka J, Moss T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol Cell. 2010;38:539–50. doi: 10.1016/j.molcel.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 156.Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–30. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 157.Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD. Targeting the nucleolus for cancer intervention. Biochim Biophys Acta. 2014;1842:802–16. doi: 10.1016/j.bbadis.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 158.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–95. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 160.Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012;109:13314–8. doi: 10.1073/pnas.1205690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20:1241–9. doi: 10.1038/cdd.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013;9:260–2. doi: 10.4161/auto.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012;4:159–65. doi: 10.18632/aging.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 166.Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y, Huang L, Li J, Huang J, Meng S, et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013;73:3615–24. doi: 10.1158/0008-5472.CAN-12-3793. [DOI] [PubMed] [Google Scholar]
- 167.Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G, Pandolfi PP, Givol D, Strano S, Lu X, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell. 2008;32:803–14. doi: 10.1016/j.molcel.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 168.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–38. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gulshan K, Thommandru B, Moye-Rowley WS. Proteolytic degradation of the Yap1 transcription factor is regulated by subcellular localization and the E3 ubiquitin ligase Not4. J Biol Chem. 2012;287:26796–805. doi: 10.1074/jbc.M112.384719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fernández-Cid A, Riera A, Tognetti S, Herrera MC, Samel S, Evrin C, Winkler C, Gardenal E, Uhle S, Speck C. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol Cell. 2013;50:577–88. doi: 10.1016/j.molcel.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 172.Labib K, Tercero JA, Diffley JF. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science. 2000;288:1643–7. doi: 10.1126/science.288.5471.1643. [DOI] [PubMed] [Google Scholar]
- 173.Shen Z, Sathyan KM, Geng Y, Zheng R, Chakraborty A, Freeman B, Wang F, Prasanth KV, Prasanth SG. A WD-repeat protein stabilizes ORC binding to chromatin. Mol Cell. 2010;40:99–111. doi: 10.1016/j.molcel.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shen Z, Prasanth SG. Orc2 protects ORCA from ubiquitin-mediated degradation. Cell Cycle. 2012;11:3578–89. doi: 10.4161/cc.21870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kotake Y, Zeng Y, Xiong Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009;69:1809–14. doi: 10.1158/0008-5472.CAN-08-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev. 2010;24:1236–41. doi: 10.1101/gad.1920310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028–33. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mallette FA, Gaumont-Leclerc MF, Huot G, Ferbeyre G. Myc down-regulation as a mechanism to activate the Rb pathway in STAT5A-induced senescence. J Biol Chem. 2007;282:34938–44. doi: 10.1074/jbc.M707074200. [DOI] [PubMed] [Google Scholar]
- 180.Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A. 2010;107:58–63. doi: 10.1073/pnas.0900121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, Berman AE, Giordano TJ, Prochownik EV, Soengas MS, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008;27:6623–34. doi: 10.1038/onc.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–200. doi: 10.1016/S1097-2765(03)00193-X. [DOI] [PubMed] [Google Scholar]
- 185.Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–88. doi: 10.1016/S1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- 186.Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene. 2013;32:1284–95. doi: 10.1038/onc.2012.144. [DOI] [PubMed] [Google Scholar]
- 187.Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L, Silvester J, Snow B, Harris IS, Sasaki M, Li WY, et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013;27:1101–14. doi: 10.1101/gad.214577.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kimura Y, Nagao A, Fujioka Y, Satou A, Taira T, Iguchi-Ariga SM, Ariga H. MM-1 facilitates degradation of c-Myc by recruiting proteasome and a novel ubiquitin E3 ligase. Int J Oncol. 2007;31:829–36. [PubMed] [Google Scholar]
- 189.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 190.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–97. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schreiber M, Kolbus A, Piu F, Szabowski A, Möhle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–19. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 193.Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher FJ, 3rd, Chen J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279–90. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 2010;39:963–74. doi: 10.1016/j.molcel.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 196.Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007;282:36177–89. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
- 197.Lee YK, Thomas SN, Yang AJ, Ann DK. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J Biol Chem. 2007;282:1595–606. doi: 10.1074/jbc.M606306200. [DOI] [PubMed] [Google Scholar]
- 198.Kepkay R, Attwood KM, Ziv Y, Shiloh Y, Dellaire G. KAP1 depletion increases PML nuclear body number in concert with ultrastructural changes in chromatin. Cell Cycle. 2011;10:308–22. doi: 10.4161/cc.10.2.14551. [DOI] [PubMed] [Google Scholar]
- 199.Jacobs JJ, Keblusek P, Robanus-Maandag E, Kristel P, Lingbeek M, Nederlof PM, van Welsem T, van de Vijver MJ, Koh EY, Daley GQ, et al. Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet. 2000;26:291–9. doi: 10.1038/81583. [DOI] [PubMed] [Google Scholar]
- 200.Vance KW, Carreira S, Brosch G, Goding CR. Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res. 2005;65:2260–8. doi: 10.1158/0008-5472.CAN-04-3045. [DOI] [PubMed] [Google Scholar]
- 201.Lüdtke TH, Farin HF, Rudat C, Schuster-Gossler K, Petry M, Barnett P, Christoffels VM, Kispert A. Tbx2 controls lung growth by direct repression of the cell cycle inhibitor genes Cdkn1a and Cdkn1b. PLoS Genet. 2013;9:e1003189. doi: 10.1371/journal.pgen.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Martin N, Benhamed M, Nacerddine K, Demarque MD, van Lohuizen M, Dejean A, Bischof O. Physical and functional interaction between PML and TBX2 in the establishment of cellular senescence. EMBO J. 2012;31:95–109. doi: 10.1038/emboj.2011.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lu ZH, Books JT, Ley TJ. YB-1 is important for late-stage embryonic development, optimal cellular stress responses, and the prevention of premature senescence. Mol Cell Biol. 2005;25:4625–37. doi: 10.1128/MCB.25.11.4625-4637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dolfini D, Mantovani R. Targeting the Y/CCAAT box in cancer: YB-1 (YBX1) or NF-Y? Cell Death Differ. 2013;20:676–85. doi: 10.1038/cdd.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lasham A, Samuel W, Cao H, Patel R, Mehta R, Stern JL, Reid G, Woolley AG, Miller LD, Black MA, et al. YB-1, the E2F pathway, and regulation of tumor cell growth. J Natl Cancer Inst. 2012;104:133–46. doi: 10.1093/jnci/djr512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lasham A, Moloney S, Hale T, Homer C, Zhang YF, Murison JG, Braithwaite AW, Watson J. The Y-box-binding protein, YB1, is a potential negative regulator of the p53 tumor suppressor. J Biol Chem. 2003;278:35516–23. doi: 10.1074/jbc.M303920200. [DOI] [PubMed] [Google Scholar]
- 207.Lyabin DN, Eliseeva IA, Ovchinnikov LP. YB-1 protein: functions and regulation. Wiley Interdiscip Rev RNA. 2014;5:95–110. doi: 10.1002/wrna.1200. [DOI] [PubMed] [Google Scholar]
- 208.Dutertre M, Sanchez G, De Cian MC, Barbier J, Dardenne E, Gratadou L, Dujardin G, Le Jossic-Corcos C, Corcos L, Auboeuf D. Cotranscriptional exon skipping in the genotoxic stress response. Nat Struct Mol Biol. 2010;17:1358–66. doi: 10.1038/nsmb.1912. [DOI] [PubMed] [Google Scholar]
- 209.Chibi M, Meyer M, Skepu A, G Rees DJ, Moolman-Smook JC, Pugh DJ. RBBP6 interacts with multifunctional protein YB-1 through its RING finger domain, leading to ubiquitination and proteosomal degradation of YB-1. J Mol Biol. 2008;384:908–16. doi: 10.1016/j.jmb.2008.09.060. [DOI] [PubMed] [Google Scholar]
- 210.Blencowe BJ, Issner R, Nickerson JA, Sharp PA. A coactivator of pre-mRNA splicing. Genes Dev. 1998;12:996–1009. doi: 10.1101/gad.12.7.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Eldridge AG, Li Y, Sharp PA, Blencowe BJ. The SRm160/300 splicing coactivator is required for exon-enhancer function. Proc Natl Acad Sci U S A. 1999;96:6125–30. doi: 10.1073/pnas.96.11.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Szymczyna BR, Bowman J, McCracken S, Pineda-Lucena A, Lu Y, Cox B, Lambermon M, Graveley BR, Arrowsmith CH, Blencowe BJ. Structure and function of the PWI motif: a novel nucleic acid-binding domain that facilitates pre-mRNA processing. Genes Dev. 2003;17:461–75. doi: 10.1101/gad.1060403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Blencowe BJ, Baurén G, Eldridge AG, Issner R, Nickerson JA, Rosonina E, Sharp PA. The SRm160/300 splicing coactivator subunits. RNA. 2000;6:111–20. doi: 10.1017/S1355838200991982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wójcik C, DeMartino GN. Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003;35:579–89. doi: 10.1016/S1357-2725(02)00380-1. [DOI] [PubMed] [Google Scholar]
- 215.Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–90. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Activity and regulation of the centrosome-associated proteasome. J Biol Chem. 2000;275:409–13. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- 217.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Interferon gamma regulates accumulation of the proteasome activator PA28 and immunoproteasomes at nuclear PML bodies. J Cell Sci. 2001;114:29–36. doi: 10.1242/jcs.114.1.29. [DOI] [PubMed] [Google Scholar]
- 218.Mattsson K, Pokrovskaja K, Kiss C, Klein G, Szekely L. Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc Natl Acad Sci U S A. 2001;98:1012–7. doi: 10.1073/pnas.98.3.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.von Mikecz A. The nuclear ubiquitin-proteasome system. J Cell Sci. 2006;119:1977–84. doi: 10.1242/jcs.03008. [DOI] [PubMed] [Google Scholar]
- 220.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 221.Chen M, Rockel T, Steinweger G, Hemmerich P, Risch J, von Mikecz A. Subcellular recruitment of fibrillarin to nucleoplasmic proteasomes: implications for processing of a nucleolar autoantigen. Mol Biol Cell. 2002;13:3576–87. doi: 10.1091/mbc.02-05-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rockel TD, Stuhlmann D, von Mikecz A. Proteasomes degrade proteins in focal subdomains of the human cell nucleus. J Cell Sci. 2005;118:5231–42. doi: 10.1242/jcs.02642. [DOI] [PubMed] [Google Scholar]
- 223.Arabi A, Rustum C, Hallberg E, Wright AP. Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J Cell Sci. 2003;116:1707–17. doi: 10.1242/jcs.00370. [DOI] [PubMed] [Google Scholar]
- 224.Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–12. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 225.Baldin V, Militello M, Thomas Y, Doucet C, Fic W, Boireau S, Jariel-Encontre I, Piechaczyk M, Bertrand E, Tazi J, et al. A novel role for PA28gamma-proteasome in nuclear speckle organization and SR protein trafficking. Mol Biol Cell. 2008;19:1706–16. doi: 10.1091/mbc.E07-07-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lafarga M, Berciano MT, Pena E, Mayo I, Castaño JG, Bohmann D, Rodrigues JP, Tavanez JP, Carmo-Fonseca M. Clastosome: a subtype of nuclear body enriched in 19S and 20S proteasomes, ubiquitin, and protein substrates of proteasome. Mol Biol Cell. 2002;13:2771–82. doi: 10.1091/mbc.E02-03-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Smith KP, Byron M, O’Connell BC, Tam R, Schorl C, Guney I, Hall LL, Agrawal P, Sedivy JM, Lawrence JB. c-Myc localization within the nucleus: evidence for association with the PML nuclear body. J Cell Biochem. 2004;93:1282–96. doi: 10.1002/jcb.20273. [DOI] [PubMed] [Google Scholar]
- 228.Herrmann A, Sommer U, Pranada AL, Giese B, Küster A, Haan S, Becker W, Heinrich PC, Müller-Newen G. STAT3 is enriched in nuclear bodies. J Cell Sci. 2004;117:339–49. doi: 10.1242/jcs.00833. [DOI] [PubMed] [Google Scholar]
- 229.Kawasaki A, Matsumura I, Kataoka Y, Takigawa E, Nakajima K, Kanakura Y. Opposing effects of PML and PML/RAR alpha on STAT3 activity. Blood. 2003;101:3668–73. doi: 10.1182/blood-2002-08-2474. [DOI] [PubMed] [Google Scholar]
- 230.St-Germain JR, Chen J, Li Q. Involvement of PML nuclear bodies in CBP degradation through the ubiquitin-proteasome pathway. Epigenetics. 2008;3:342–9. doi: 10.4161/epi.3.6.7203. [DOI] [PubMed] [Google Scholar]
- 231.Bandyopadhyay D, Okan NA, Bales E, Nascimento L, Cole PA, Medrano EE. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002;62:6231–9. [PubMed] [Google Scholar]
- 232.Shima Y, Shima T, Chiba T, Irimura T, Pandolfi PP, Kitabayashi I. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol Cell Biol. 2008;28:7126–38. doi: 10.1128/MCB.00897-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bernassola F, Salomoni P, Oberst A, Di Como CJ, Pagano M, Melino G, Pandolfi PP. Ubiquitin-dependent degradation of p73 is inhibited by PML. J Exp Med. 2004;199:1545–57. doi: 10.1084/jem.20031943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Xu ZX, Timanova-Atanasova A, Zhao RX, Chang KS. PML colocalizes with and stabilizes the DNA damage response protein TopBP1. Mol Cell Biol. 2003;23:4247–56. doi: 10.1128/MCB.23.12.4247-4256.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–72. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- 236.Miteva M, Keusekotten K, Hofmann K, Praefcke GJ, Dohmen RJ. Sumoylation as a signal for polyubiquitylation and proteasomal degradation. Subcell Biochem. 2010;54:195–214. doi: 10.1007/978-1-4419-6676-6_16. [DOI] [PubMed] [Google Scholar]
- 237.Praefcke GJ, Hofmann K, Dohmen RJ. SUMO playing tag with ubiquitin. Trends Biochem Sci. 2012;37:23–31. doi: 10.1016/j.tibs.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 238.Malloy MT, McIntosh DJ, Walters TS, Flores A, Goodwin JS, Arinze IJ. Trafficking of the transcription factor Nrf2 to promyelocytic leukemia-nuclear bodies: implications for degradation of NRF2 in the nucleus. J Biol Chem. 2013;288:14569–83. doi: 10.1074/jbc.M112.437392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Nyström T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–7. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–10. doi: 10.1016/S0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 242.Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–8. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 243.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 244.Vredeveld LC, Possik PA, Smit MA, Meissl K, Michaloglou C, Horlings HM, Ajouaou A, Kortman PC, Dankort D, McMahon M, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012;26:1055–69. doi: 10.1101/gad.187252.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42:36–49. doi: 10.1016/j.molcel.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Malleshaiah MK, Shahrezaei V, Swain PS, Michnick SW. The scaffold protein Ste5 directly controls a switch-like mating decision in yeast. Nature. 2010;465:101–5. doi: 10.1038/nature08946. [DOI] [PubMed] [Google Scholar]
- 247.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 248.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11:629–43. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 250.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 251.Hatakeyama S, Nakayama KI. U-box proteins as a new family of ubiquitin ligases. Biochem Biophys Res Commun. 2003;302:635–45. doi: 10.1016/S0006-291X(03)00245-6. [DOI] [PubMed] [Google Scholar]
- 252.Aravind L, Koonin EV. The U box is a modified RING finger - a common domain in ubiquitination. Curr Biol. 2000;10:R132–4. doi: 10.1016/S0960-9822(00)00398-5. [DOI] [PubMed] [Google Scholar]
- 253.Coscoy L, Ganem D. PHD domains and E3 ubiquitin ligases: viruses make the connection. Trends Cell Biol. 2003;13:7–12. doi: 10.1016/S0962-8924(02)00005-3. [DOI] [PubMed] [Google Scholar]
- 254.Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, Conaway JW, Nakayama KI. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18:3055–65. doi: 10.1101/gad.1252404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 256.Ishikawa Y, Hosogane M, Okuyama R, Aoyama S, Onoyama I, Nakayama KI, Nakayama K. Opposing functions of Fbxw7 in keratinocyte growth, differentiation and skin tumorigenesis mediated through negative regulation of c-Myc and Notch. Oncogene. 2013;32:1921–32. doi: 10.1038/onc.2012.213. [DOI] [PubMed] [Google Scholar]
- 257.Roy N, Stoyanova T, Dominguez-Brauer C, Park HJ, Bagchi S, Raychaudhuri P. DDB2, an essential mediator of premature senescence. Mol Cell Biol. 2010;30:2681–92. doi: 10.1128/MCB.01480-09. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 258.Prudden J, Pebernard S, Raffa G, Slavin DA, Perry JJ, Tainer JA, McGowan CH, Boddy MN. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 2007;26:4089–101. doi: 10.1038/sj.emboj.7601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sun H, Leverson JD, Hunter T. Conserved function of RNF4 family proteins in eukaryotes: targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007;26:4102–12. doi: 10.1038/sj.emboj.7601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–9. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104–18. doi: 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 262.Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–30. doi: 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 263.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999;17:739–79. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 265.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–18. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science. 2010;327:1657–61. doi: 10.1126/science.1185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Martinez P, Thanasoula M, Carlos AR, Gómez-López G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12:768–80. doi: 10.1038/ncb2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hu C, Li H, Li J, Zhu Z, Yin S, Hao X, Yao M, Zheng S, Gu J. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis. 2008;29:2289–97. doi: 10.1093/carcin/bgn223. [DOI] [PubMed] [Google Scholar]