

Abstract
HAT HBO1 interacts with 2 isoforms of JADE1: JADE1S and JADE1L. JADE1 promotes acetylation of nucleosomal histones by HBO1. HBO1–JADE1 complex facilitates cell proliferation by unclear mechanisms. Here we report intracellular chromatin shuttling of HBO1–JADE1 complex during mitosis coupled to phosphorylation of JADE1. In interphase of dividing cells JADE1S was localized to the nucleus and associated with chromatin. As cells approached mitosis, specifically prophase, JADE1S dissociated from chromatin and associated with cytoplasm. JADE1S chromatin re-association began in telophase and paralleled nuclear envelope membrane reassembly. By early G1, JADE1S was re-associated with chromatin and localized to the nucleus. Importantly, cytoplasmic but not chromatin-associated JADE1 protein was phosphorylated. Mass-Spectrometric analysis of JADE1S protein isolated from G2/M-arrested cells identified 6 phosphorylated amino acid residues: S89, T92, S102, S121, S392, and T468, including 3 novel sites. Temporally, JADE1S phosphorylation and dephosphorylation during mitosis correlated with JADE1S chromatin dissociation and recruitment. JADE1S chromatin recruitment was accompanied by the global histone H4 acetylation. Pharmacological inhibitor of Aurora A kinase prevented JADE1S protein band shift and chromatin dissociation, suggesting regulatory function for phosphorylation. In vivo experiments supported our in vitro results. In mouse kidneys, JADE1S transiently accumulated in the cytoplasm of tubular epithelial cells during kidney regeneration. The transient increase in the number of cells with cytoplasmic JADE1S directly correlated with activation of tubular cell proliferation and inversely correlated with the number of cells with nuclear JADE1S staining, supporting biological role of HBO1–JADE1 shuttling during organ regeneration.
Keywords: PHF17, MYST2, KAT7, regeneration, epithelial cell proliferation, chromatin shuttling, phosphorylation
Introduction
Acetylation of histones within the chromatin context is a post-translational modification that can regulate DNA replication, repair, and gene transcription.1-10 Currently it is accepted that during DNA replication and transcription, coordinated bulk histone acetylation and deacetylation are required for proper remodeling of chromatin. In addition, targeted spatial and temporal acetylation of histones within the chromosomes can provide epigenetic mechanisms to fine-tune the regulation of DNA transcription and replication.2,11-13 Acetylation of specific histone lysine residues is executed by several families of histone acetyl transferase complexes.14-16 Most histone acetyl transferases (HATs) require protein partners and function within protein complexes to perform their specific actions. The cooperative interactions between proteins in HAT complexes and other nuclear proteins regulate enzymes’ specificities and activities.17-21
JADE1 (gene for apoptosis and differentiation-1) protein, also known as PHF17 (PHD zinc finger factor 17) has been originally identified in a yeast 2-hybrid screen as a protein partner of pVHL,22 the key regulator of the cellular oxygen-sensing pathway. JADE1 contains one canonical Cys4HisCys3 plant homeo domain (PHD) followed by a non-canonical extended PHD domain, which are zinc-binding motifs. JADE1 mRNA gives rise to 2 protein products: a full-length JADE1L consisting of 842 amino acids, and a truncated splice variant, JADE1S, that lacks a large C-terminal fragment of 333 amino acids. The short isoform of JADE1 is the most molecularly described JADE1 form so far. JADE1S is a nuclear protein with intrinsic transcriptional activity.18 JADE1 promotes histone H4 acetylation in chromatin context by associating with a histone H4-specific endogenous HAT in cultured cells and cell-free in vitro assays.18 This activity required intact PHD zinc fingers, suggesting a chromatin-targeting role for PHD zinc fingers of JADE1.
A novel long non-coding RNA, lncRNA-JADE1, that is induced after DNA damage, has been identified very recently.23 lncRNA-JADE1 transcriptionally activated JADE1 and induced histone H4 acetylation in the double-strand DNA repair model. The study suggested link between lncRNA-JADE1, DDR, and histone H4 acetylation, as well as potential contribution of JADE1 dis-regulation to breast tumorigenesis.23
HBO1 (MYST2, KAT714) was originally identified using a yeast two-hybrid screen as a HAT binding origin recognition complex-1 (Orc1).2,3,24 Histone H4-specific HAT HBO1 has been implicated in the pre-replication complex assembly and transcriptional regulation, as well as linked to the cellular stress response and carcinogenesis.2,4,19,24-30
We previously reported cooperative interactions between HBO1, JADE1, and ING4/5. JADE1 is crucial for HBO1 to acetylate histone H4 in chromatin context, and it requires intact PHD zinc fingers.17,18 HBO1 interacts in vivo with another PHD zinc finger protein BRD1/BRPF2.20 The cellular activities of the HBO1 complex might be controlled by the presence of PHD zinc finger-targeting proteins.17,18,20,31 JADE1 deficiency led to the downregulation of HBO1 protein and diminished rates of DNA synthesis in cultured epithelial cells.32 Our recent study showed that JADE1 is required for expression and chromatin recruitment of replication factors during the cell cycle.32 Spatial and temporal distribution of JADE1 and its association with chromatin during the cell cycle have not been studied.
Very few studies have investigated the biological role of HBO1 and JADE1 in animal models or human subjects. Non-conditional total body HBO1 knockout mice are not viable, presumably due to the requirement of HBO1 for the transcriptional regulation of key developmental genes responsible for embryonic patterning.33 Interestingly, while HBO1 downregulation inhibited DNA synthesis in cultured cell lines,49 the primary embryonic fibroblasts isolated from HBO1 KO mice embryo had no defects in DNA replication or cell proliferation.33 Moreover, HBO1 gene deletion did not affect the levels of bulk histone H4 acetylation in cultured embryonic cells, while it resulted in decreased histone H3K14 acetylation levels. These data argued against the role of HBO1 in DNA replication and suggested histone H3 specificity. In a study done with JADE1 hypomorphic mice generated by gene trap,34 the resultant JADE1 promoter-driven fusion protein was highly expressed in a regulated manner during embryogenesis, suggesting a developmental role for JADE1. We recently reported that JADE1 and HBO1 may play a role in organ regeneration. Thus, in vivo, the fraction of kidney tubular epithelial cells expressing JADE1 and HBO1 proteins in the cell nuclei decreased during kidney ischemia/reperfusion injury and accumulated during kidney recovery. Temporal and spatial expression of nuclear JADE1S and HBO1 correlated with that of cell proliferation marker Ki67 and bulk histone H4 acetylation, suggesting a role for JADE1S and HBO1 during organ regeneration.32
Interactions between protein partners of HBO1 complex and a link to DNA replication have been reported.2,3,17,19,30,32 However, little is known about the mechanism by which JADE1–HBO1 complex controls the cell cycle. In this study, we investigated the properties of HBO1-JADE1 in dividing cultured cells and in regenerating tubular epithelial cells of mouse kidneys. Here we report cell cycle-dependent chromatin shuttling of the HBO1 complex during mitosis and cell cycle-dependent phosphorylation of JADE1. Our data suggest a functional link between JADE1 phosphorylation status and chromatin recruitment. We identified 6 amino acid residues phosphorylated in response to drug-mediated mitotic cell cycle arrest including 3 novel phosphorylation sites. The biological relevance of this phenomenon is supported by our in vivo studies.
Results
Cellular compartmentalization of JADE1 and HBO1 during the cell cycle
Previously we reported that JADE1S and HBO1 proteins are localized to the nucleus of cultured cells in vitro and renal tubular epithelial cells in vivo.18,32 Here we analyzed cellular compartmentalization of the endogenous HBO1–JADE1 complex in interphase vs. mitotic cells and noticed marked cell cycle stage-dependent differences. In interphase cells, JADE1S, JADE1L, and HBO1 were localized to the nucleus (Fig. 1A). In contrast, all 3 endogenous proteins of the HBO1 complex were excluded from chromatin and associated with the cytoplasm in mitotic cells (Fig. 1B; representative images).

Figure 1. Endogenous HBO1 complex is excluded from chromatin in mitosis. HeLa cells were processed for IF; proteins were visualized with indicated antibodies. (A) The endogenous HBO1 and JADE1 localize to the nucleus in interphase cells. (B) In mitotic cells, endogenous HBO1 and JADE1 are excluded from condensed chromatin (arrow). Identical results were obtained in 293T/17 and H1299 cells. Scale Bars: 10 µm
The nuclear membrane disintegrates around late prophase and re-assembles around telophase. Using indirect immunofluorescence and confocal microscopy we analyzed asynchronously dividing cells captured in various stages of mitosis. This analysis confirmed that in interphase cells JADE1S was localized to the nucleus (Fig. 2A and K). JADE1S cytoplasmic localization correlated with mitosis and was first noticeable in prophase cells (Fig. 2B–D and L–O). In metaphase and anaphase cells JADE1S was entirely cytoplasmic and fully excluded from condensed chromatin (Fig. 2E–H and P–R). Conversely, JADE1S disappearance from the cytoplasm and concentration around chromatin was noticeable in telophase cells (Fig. 2J, S, and T). JADE1S was fully excluded from the cytoplasm and localized to the nucleus in cells approaching the end of mitosis (Fig. 2J, S, and T), which is the stage of nuclear envelope re-assembly. Morphological analysis of cells co-stained with nuclear envelope marker lamin B2 and JADE1S showed similarity between dynamics of nuclear envelope remodeling and JADE1S shuttling (Fig. 2K–T). These data confirm that JADE1S is a dynamic protein and suggest JADE1S chromatin shuttling during mitosis.

Figure 2. JADE1S subcellular localization in various stages of mitosis. Asynchronously dividing HeLa cells were processed for IF with indicated antibody and mitotic stages assigned according to cell morphology. Cells co-stained with JADE1S and α-Tubulin (A–J) or JADE1S and Lamin B2 (K–T). JADE1S shuttling mostly correlates with nuclear envelope remodeling. Scale Bars: 10 µm
We aimed to examine whether co-transfected proteins recapitulate their endogenous counterparts in dividing cells. While determining experimental conditions we found that, unlike endogenous proteins, overexpressed JADE1S and JADE1L are not localized to the nucleus but are localized to the cytoplasm even in interphase cells (Fig. 3A). Note that overexpressed HBO1 resembled endogenous protein and localized to the nucleus of interphase cells (Fig. 3A). Interestingly, co-expression with HBO1 resulted in JADE1S and JADE1L nuclear localization and resembled endogenous proteins (Figs. 3B and 1A). Thus, nuclear localization of overexpressed JADE1S and JADE1L depends on HBO1. Fully recapitulating their endogenous counterparts JADE1S and JADE1L proteins were transiently excluded from chromatin and localized to the cytoplasm in mitotic cells when co-expressed with HBO1 (Figs. 3C and 1B). Taken together, these data demonstrate that cellular compartmentalization of the HBO1 complex is dynamic, and that all 3 proteins are transiently localized to the cytoplasm during mitosis. Therefore, to study overexpressed JADE1 and mimic the endogenous counterpart, JADE1 was co-transfected with cDNA of HBO1.
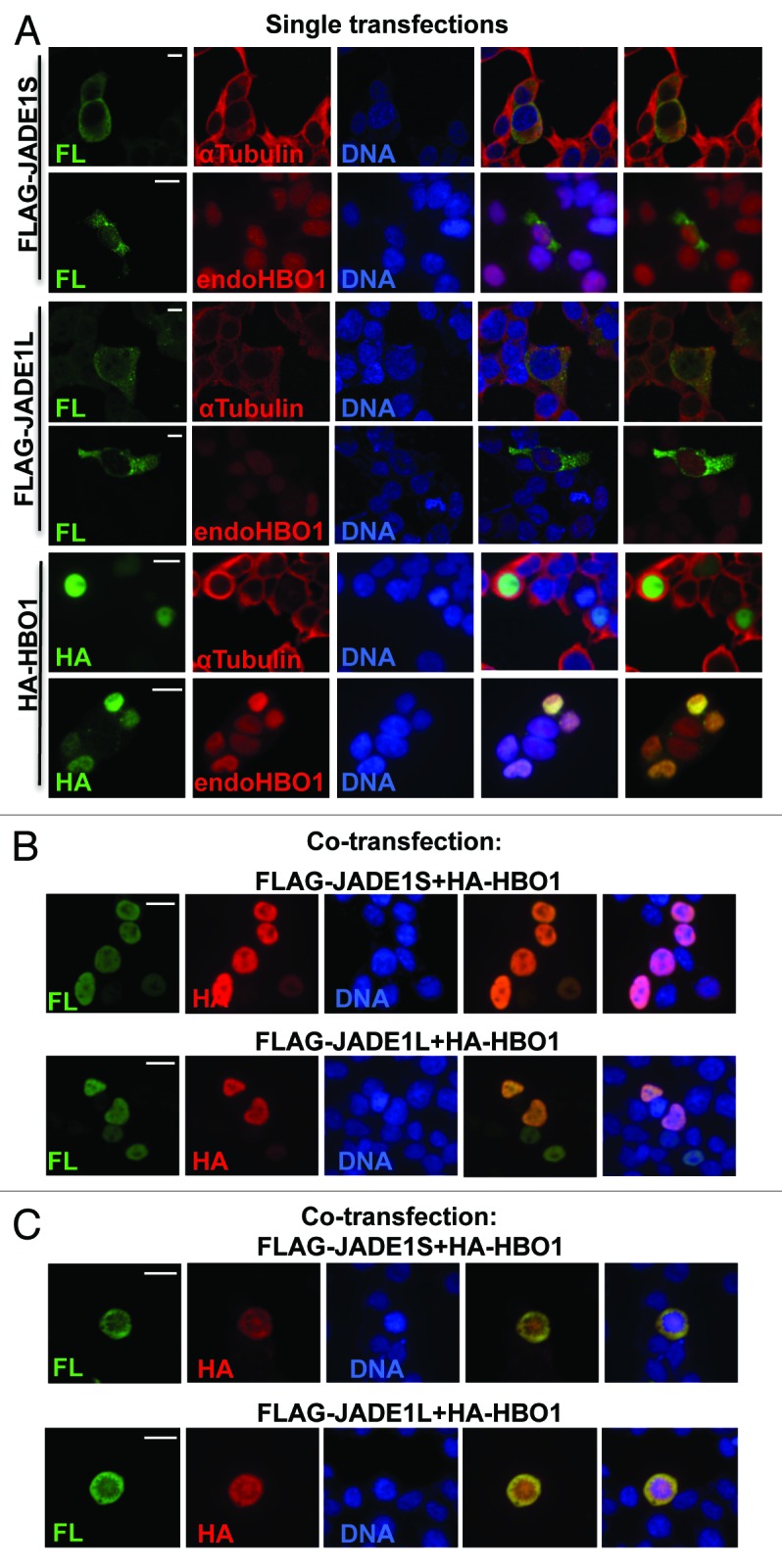
Figure 3. Nuclear localization of ectopically expressed JADE1 proteins depends on HBO1. (A) In interphase cells, singly transfected JADE1 and HBO1 localize to the cytoplasm and the nucleus, respectively. 293T/17 cells were transfected with FLAG-JADE1S, FLAG-JADE1L, or HA-HBO1 cDNA plasmids, 48 h post-transfection cells were processed for IF with indicated antibodies. (B) Co-expression with HBO1 results in nuclear localization of JADE1 proteins in interphase cells. Cells (293T/17) were co-transfected with FLAG-JADE1 and HA-HBO1 cDNA plasmids. (C) Co-expressed HBO1 and JADE1 proteins are excluded from chromatin and localized to the cytoplasm in mitotic cells. Scale Bars: 10 µm
To examine the temporal and spatial expression of HBO1 and JADE1 we used dividing HeLa cells synchronized by nocodazole. Cell cycle arrest and release after the drug removal was confirmed and monitored by FACS analysis (Fig. 4A). Cell cycle arrest in the G2/M phase by nocodazole resulted in complete exclusion of overexpressed proteins from the condensed DNA and in their association with cytoplasm (Fig. 4B). Nocodazole removal and cell cycle release resulted in a time-dependent re-association of JADE1S and HBO1 with the nucleus. Interestingly, even after G2/M arrest, cytoplasmic JADE1S and HBO1 were co-localized, and proteins co-immunoprecipitated each other (Fig. 4B and C), demonstrating that G2/M arrest and change of localization did not affect the interaction between JADE1S and HBO1. Similarly to overexpressed proteins, at mitotic arrest, endogenous JADE1S and HBO1 proteins were excluded from condensed DNA and localized to the cytoplasm, while cell cycle recovery resulted in re-association of both proteins with the nucleus (Fig. 4D). These data suggest that JADE1S and HBO1 undergo reversible cell cycle-dependent chromatin shuttling.

Figure 4. JADE1S–HBO1 complex chromatin shuttling. (A) FACS analysis of HeLa cells synchronized by nocodazole. Hours indicate time after nocodazole removal. (B) Cells (293T/17) were co-transfected with FLAG-JADE1S and HA-HBO1 cDNA, synchronized with nocodazole and processed for IF. (C) H1299 cells were transfected with empty vectors (1) or FLAG-JADE1S and HA-HBO1 (2), and cells synchronized as in (A). Proteins were immunoprecipitated as shown and analyzed by WB. Note that JADE1S co-precipitates HBO1 at both times; 0 h (G2/M) and 4 h (G1) after cell cycle recovery. (D) HeLa cells were treated as in (A). Endogenous JADE1S and HBO1 visualized by IF. (E) HeLa cells were treated as in (A). Soluble (S) and chromatin-enriched fractions (Ch) were analyzed for endogenous JADE1S protein. Soluble and chromatin fractions were extracted with equal volumes of appropriate buffers (see “Materials and Methods”) to enable comparison of protein abundance between the 2 different fractions. Note that JADE1S levels decrease in the soluble fraction and increase in the chromatin as cells recover from G2/M growth arrest. Note the band shift corresponding to JADE1S in the total extract. Cyclin D1 and E1 represent cell cycle markers. The global acetylation levels of histone H4 correlates with JADE1S chromatin recruitment. Scale Bars: 10 µm (F) JADE1L is excluded from chromatin and is posttranslationally modified in G2/M-arrested cells. HeLa cells were treated with nocodazole or vehicle control for 16 h. Soluble and chromatin fractions were generated by using equal volumes of appropriate extraction buffers (see “Materials and Methods”) to enable comparison of protein abundance between the 2 different fractions. JADE1L was visualized with specific antibody. (G) Verification of JADE1L antibody specificity. HeLa cells were transfected with siRNA for JADE1 or siControl, cellular fractions were prepared and JADE1L and JADE1S were visualized with JADE1 antibody. α-Tubulin and nucleolin visualization detected no cross-contamination between cytoplasm and chromatin fractions. *Nucleolin appears in the second lane of the soluble fraction due to the effect of nocodazole.
We assessed biochemically the dynamics of JADE1S chromatin association during mitosis, using a cell fractionation and chromatin recruitment assay. Synchronized HeLa cells were harvested at different time points after cell cycle arrest and release, and cellular proteins were separated into chromatin-enriched and soluble fractions. JADE1S protein levels were assessed by western blot using a specific antibody. In agreement with the IF data, cell growth arrest and recovery resulted in dramatic alteration of JADE1S distribution between cell fractions (Fig. 4E). After 16 h treatment of cells with nocodazole, endogenous JADE1S protein was dissociated from the chromatin-enriched fraction into the soluble fraction of cells. Drug removal and the following cell cycle recovery resulted in a time-dependent reversal of this process (Fig. 4E). By early G1, JADE1 mostly re-associated with the chromatin fraction (Fig. 4E). Interestingly, levels of global histone H4 acetylation correlated with chromatin recruitment of JADE1S (Fig. 4E).
Cell cycle-dependent phosphorylation of JADE1S
Analysis of the JADE1S protein band in chromatin-enriched, soluble, and total protein fractions of synchronized cycling cells revealed a reversible JADE1S protein band shift, suggesting posttranslational protein modification via phosphorylation (Fig. 4E). Reversal of the band shift occurred between 1.5 and 4 h after cell cycle release and correlated with recruitment of JADE1S to chromatin. Similarly, nocodazole caused dramatic re-distribution of JADE1L isoform between the cytoplasmic and chromatin fractions (Fig. 4F, compare lanes 1 and 3 vs. 2 and 4). In addition, like JADE1S, cytoplasmic JADE1L appeared to be posttranslationally modified, as evidenced by the JADE1L protein band shift (Fig. 4F, compare lanes 2 and 3). JADE1L antibody specificity was verified by using positive and negative (siJADE1) control samples (Fig. 4G). To our knowledge, this is the first experimental visualization of endogenous JADE1L protein by the western blot technique performed in a controlled functional assay.
To this end we show that JADE1S is localized to the cytoplasmic fraction and appears to be post-transnationally modified in cells arrested in mitosis (Fig. 4). We determined kinetics of JADE1 chromatin dissociation and protein band shift during the cell cycle. Dividing HeLa cells were synchronized by arresting cell cycle in late G1. At time 0 h the cell cycle was released by removing the drug and replacing the media with fresh media supplemented with nocodazole (Fig. 5A). Chromatin-enriched and soluble cellular fractions were harvested at indicated time points and analyzed for expression of JADE1S protein, appropriate cell cycle markers, as well as loading controls (Fig. 5B). FACS analysis confirmed cell cycle synchronization and progression (Fig. 5C). At 10 h after mimosine removal and cell cycle recovery (S-phase) JADE1S was still prevalent in chromatin-enriched fraction (Fig. 5B and C). At time point 16 h after cell cycle recovery, when cells are approaching G2/M transition, JADE1S started to accumulate in soluble fraction, and this correlated with discrete protein band shift, suggesting posttranslational modification (Fig. 5B). JADE1S protein dissociation from chromatin to cytoplasm continued as more cells reached G2/M phase (Fig. 5B). Combined, our data show that JADE1S undergoes chromatin shuttling during mitosis, and that it correlates with JADE1S posttranslational modification.

Figure 5. JADE1S dissociates from chromatin and is posttranslationally modified in late G2/ early mitosis. (A) Experimental design: HeLa cells were synchronized by arresting cell cycle at late G1 with mimosine. At 0 h cell cycle was released by refreshing media supplemented with nocodazole. At indicated time points (10, 13, 16, 18, 20, and 22 h) samples were collected for analysis as described in Figure 4, except at all time points the total cell population was collected (no mitotic shake off). (B) Soluble (S) and chromatin-enriched (Ch) cellular fractions were analyzed for endogenous JADE1S by western blots. Soluble fraction after nocodazole (S) and chromatin fraction of asynchronous cells (Ch) was used as a reference for high and low molecular mass specie of JADE1S, respectively. Note the band shift corresponding to JADE1S at the 16 h time point. JADE1S levels increase in the soluble fraction and decrease in the chromatin fraction as the cell cycle enters late G2/early M. Blots were probed with β-Actin (loading control), nucleolin (nuclear marker), and cell cycle markers cyclin B1 (G2/M) and cyclin E (G1). At 0 h of cell cycle release, when cells are in the G1/S border, JADE1S band intensity appears slightly lower in chromatin-enriched fraction due to the loading as reflected by nucleolin. (C) Cell cycle profiles of the samples by FACS analysis. Note the prominent G2/M cell cycle profile of the 16 h and later time points. *Cells were treated for 16 h with nocodazole only.
We employed several experimental approaches to examine whether the JADE1S band shift was due to phosphorylation. If JADE1S is phosphorylated, treatment with a phosphatase would reverse protein band shift. Cell extracts, enriched with a high molecular weight specie of endogenous JADE1S were incubated in the presence of λ-phosphatase, phosphatase inhibitors, or a combination of both. Treatment with λ-phosphatase resulted in increased apparent mobility of the endogenous JADE1S protein band, suggesting de-phosphorylation (Fig. 6A, upper panel). Confirming the specificity of this effect, JADE1S protein band shift was prevented by the addition of phosphatase inhibitors. The effects of λ-phosphatase on endogenous JADE1S band shift was totally recapitulated when overexpressed JADE1S was used (Fig. 6A, lower panel). Moreover, as expected, only JADE1S protein from nocodazole-treated but not vehicle-treated cells was sensitive to the treatment with phosphatase (Fig. 6B, compare samples 1 and 3 vs. 2 and 4). These data strongly suggest that the JADE1S band shift in G2/M-arrested cells is due to phosphorylation.
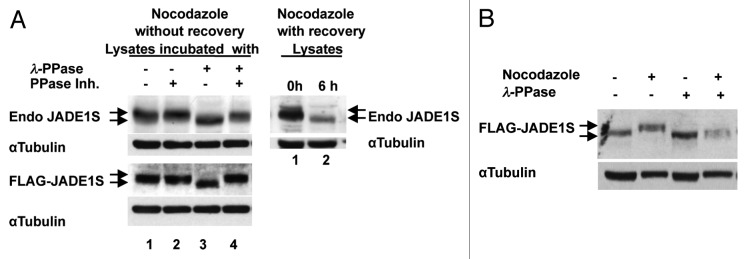
Figure 6. Nocodazole-dependent JADE1S gel band shift collapses after treatment with λ-phosphatase. (A) Left panel: lysates from synchronized HeLa cells enriched with high molecular weight specie of endogenous (upper panel) or overexpressed (lower panel) JADE1S (lane 1) were incubated with phosphatase (lane 3), phosphatase inhibitors (lane 2) or both (lane 4). Right panel: lysates from cells synchronized with nocodazole (lane 1) and after cell cycle release (lane 2) as in Figure 4. Endogenous and overexpressed JADE1S proteins were analyzed by western blot with JADE1S or FLAG antibodies. (B) JADE1S protein from cells treated with nocodazole but not vehicle collapses after treatment with phosphatase. JADE1S and HBO1 cDNA were co-transfected into H1299 cells, lysates incubated with phosphatase as in (A). Note that, protein amounts analyzed were adjusted for better bands resolution and do not represent the effect of nocodazole on JADE1S protein enrichment in soluble fraction.
JADE1 apparent phosphorylation and chromatin dissociation could be due to other effects of nocodazole (for example, microtubule de-polymerization),35 not G2/M arrest. To examine this possibility we used the cdk2 inhibitor roscovitine, which is known to interfere with G1/S transition of the cell cycle.35-37 Cells were conditioned for 2 h with roscovitine or vehicle, followed by addition of nocodazole, and effects of roscovitine on nocodazole-mediated JADE1S phosphorylation and chromatin dissociation were examined (Fig. 7). Indeed, in the presence of roscovitine, nocodazole failed to cause accumulation of cells in G2/M, presumably due to the inhibition of G1/S transition by roscovitine (Fig. 7A). Correlating with this, roscovitine prevented nocodazole-mediated JADE1S protein gel shift (Fig. 7B, compare lanes 2 and 3). Further confirming these results, the IF experiments show that roscovitine prevented nocodazole from inducing characteristic round up mitotic cells, as well as prevented nocodazole from inducing JADE1S chromatin dissociation (Fig. 7C, compare second and third columns). Note that, even in double drug-treated cells with depolymerized microtubules, JADE1S is localized to the nuclei (Fig. 7C, third column). Thus, G2/M arrest by nocodazole is required for JADE1S band shift and chromatin dissociation, suggesting a link between mitotic arrest, JADE1S phosphorylation, and chromatin dissociation.
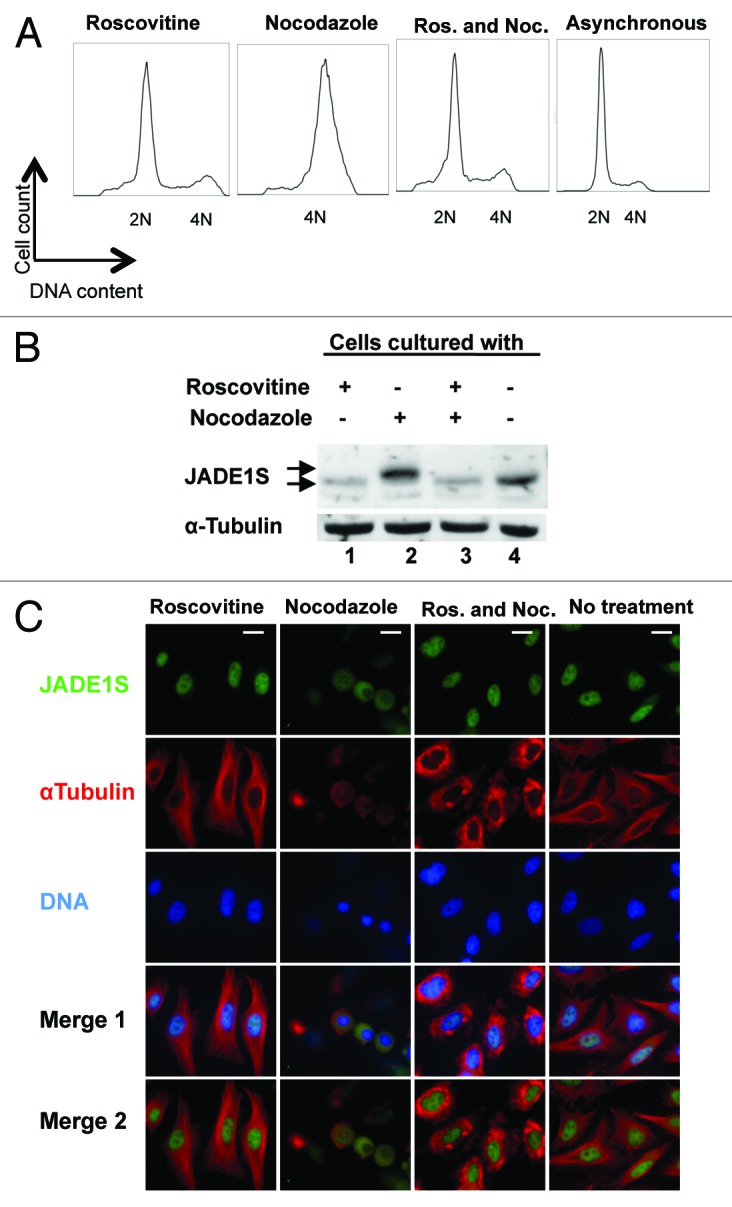
Figure 7. Roscovitine prevents nocodazole-induced accumulation of cells in G2/M, JADE1S protein phosphorylation and chromatin dissociation. Cells were conditioned for 2 h with cdk2 inhibitor roscovitine. Nocodazole was added and cells were conditioned for additional 16 h with both reagents. (A) Cell cycle profiles by FACS analysis. (B) Soluble fractions of proteins were extracted and endogenous JADE1S was analyzed. (C) HeLa cells were treated with drugs as in (A), and IF analysis performed with indicated antibodies. Scale Bars: 10 µm
Activities of Aurora A/B kinase family are elevated in mitosis and are crucial for proper mitotic progression.38-40 Phosphorylation of JADE1S occurs between 13th and 16th hours of cell cycle release, which falls to late G2/early mitosis (Fig. 5). We examined effects of pharmacological inhibitor of Aurora A kinase on JADE1S protein band shift and chromatin dissociation in cultured cells undergoing mitosis using cell cycle synchronization approach (see experimental design in Fig. 8A). We reasoned that if JADE1S phosphorylation and chromatin dissociation depend on Aurora A kinase activity, then addition of VX-680 to the growth media while cells are in G2 phase will inhibit kinase activity and subsequently JADE1S phosphorylation and chromatin dissociation. Indeed, addition of pharmacological inhibitor of Aurora A kinase between the 14th and 15.5th hours of cell cycle release prevented JADE1S band shift and chromatin dissociation (Fig. 8B; Fig. S1). Judging by cell phenotypes, the VX-680 treatment interfered with spindle apparatus assembly (not shown) and led to the accumulation of 4N cells, suggesting G2/M arrest (Fig. 8C). These data strongly suggest that JADE1S phosphorylation in mitosis is required for its chromatin dissociation. It also raises a possibility that JADE1S might be a novel direct or indirect target of Aurora A kinase.

Figure 8. JADE1S phosphorylation and chromatin dissociation is hindered by pharmacological inhibitor of Aurora A kinase. (A) Experimental design: HeLa cells were synchronized by arresting cell cycle at late G1 with mimosine. At 0 h cell cycle was released by refreshing media supplemented with nocodazole. At 15 h, inhibitor of Aurora A, VX-680 was added into the media, and at 18 h samples were collected for analysis as described in Figure 4, except here the total cell population was collected (no mitotic shake off). (B) Soluble (S) and chromatin-enriched (Ch) fractions were analyzed for endogenous JADE1S, β-Actin (loading control) and nucleolin (nuclear marker) by western blots. Samples 1–4: (1) cells treated with mimosine only, (2) cells treated with mimosine, nocodazole, and VX-680 added at 15 h, (3) cells treated with nocodazole only, (4) asynchronous cells. Soluble fraction after nocodazole (3) and chromatin fraction of asynchronous cells (4) was used as a reference for high and low molecular mass specie of JADE1S, respectively. Note that the band shift and the chromatin dissociation corresponding to JADE1S were inhibited by the addition of VX-680. (C) Cell cycle profiles of the samples 1–4 by FACS analysis.
We used mass spectrometry to directly assess JADE1S protein phosphorylation and to identify amino acid residues that are phosphorylated in JADE1S polypeptide during the nocodazole-induced G2/M cell growth arrest. JADE1S and HBO1 cDNAs were co-transfected into H1299 cells. Cells were treated with vehicle or nocodazole to induce JADE1S phosphorylation, and soluble fractions of proteins were prepared. Next, protein complexes were isolated from these lysates by affinity purification, and small aliquots of samples were separated on 7.5% SDS-PAGE gel, transferred to nitrocellulose membrane, and analyzed by western blot to assess purification and verify protein band identity (Fig. 9A). The rest of these samples were subjected to nano-LC/MS/MS analysis. Given the stoichiometry of the phosphorylation events between the treated and the control samples, 6 phosphorylated amino acid residues were identified and manually validated from this analysis (see Methods): S89, T92, S102, S121, S392, and T468. Of these sites, S121 and S392 were found only in the nocodazole-treated sample, and S89, T92, S102, and T468 were highly upregulated in the treated sample (Fig. 9B). These sites are located in either the N- or C-terminal regions of the protein and not in the PHD domains (Fig. 9C). A few sites identified are putative sites for cyclin-dependent kinases (Fig. 9B and D). To our knowledge, this is the first report of JADE1S protein phosphorylation identified and confirmed in a functional cellular experimental model. Moreover, phosphosite database and literature analysis of PHF17 protein (http://www.phosphosite.org) revealed that out of the 6 JADE1S amino acid residues phosphorylated in nocodazole-induced G2/M-arrested cells, S102, S121, and T468 phosphorylation sites have never been reported elsewhere (Fig. 9C, red font).41-47

Figure 9. Mass spectrometry analysis of JADE1S during nocodazole-induced G2/M cell growth arrest identified 6 amino acid residues that are phosphorylated. H1299 cells were co-transfected with FLAG-JADE1S and HA-HBO1, and subsequently treated with nocodazole or vehicle. (A) JADE1S-HBO1 complex affinity purification. Sample IP, 7.5% SDS-PAGE. Lane 1; JADE1S protein bound to A/G agarose beads, lane 2; JADE1S protein eluted from beads. (B) Phosphorylation sites were identified in JADE1S by nano-LC/MS/MS. Using peak areas of the unphosphorylated peptide and the phosphorylated peptide the percentage of phosphorylation was calculated, and the fold change ratio was determined. Note that phosphorylation of S121 and S392 was not detected in the control sample (red asterisk). (C) In silico analysis of JADE1 phosphorylation sites (black and green; http://www.phosphosite.org) and sites identified through MS analysis (red and green). (D) Sequence alignment of JADE1 protein in various species showing the conservation of the identified phosphorylation sites (ClustalW2). Hs, Homo sapiens; Mm, Mus musculus; Pt, Pan troglodytes; Dr, Danio rerio; Xl, Xenopus laevis. Yellow highlights indicate the identified amino acid residues. Note that lined region contains CDK binding site sequence (S/T-P-X-R/K) and consensus cyclin binding motif RRL (red box).
JADE1S is transiently excluded from tubular epithelial cell nuclei during acute kidney injury (AKI) caused by ischemia and reperfusion
We questioned whether, similarly to cells in cultures, JADE1S undergoes cell cycle-dependent chromatin shuttling in vivo. JADE1S subcellular localization was examined in a mouse model of renal ischemia reperfusion where the cell cycle is reactivated after kidney epithelial cell injury. The pathogenesis of murine ischemic AKI has been well characterized and is similar to human ischemic kidney injury.48 It has been established that the initial injury is followed by regeneration of kidney tubules that is required for recovery of kidney function. On the cellular level, the initial injury leads to loss of epithelial cells and cell growth arrest. Thereafter, surviving tubular epithelial cells reconstruct the tubule in a process requiring activation, dedifferentiation, proliferation, and re-differentiation. JADE1S cellular compartmentalization was assessed in quiescent tubular epithelial cells of normal uninjured kidneys and activated tubular cells of kidneys undergoing repair after injury.
Kidneys were subjected to sham surgery or 28 min of ischemia by bilateral renal artery clamping and were followed up to 7 d post-injury. Kidneys were removed, and the percentage of positive cells was determined by immunohistochemistry (IHC) with antibodies specific for JADE1S and Ki67. The percentage of cells expressing nuclear or cytoplasmic JADE1S protein was compared with that of the cell proliferation marker Ki67 (Fig. 10B). Agreeing with our previous results,32 in sham-operated animals, JADE1S protein was localized predominantly to the nuclei of proximal and distal epithelial tubular cells (Fig. 10A and B, sham). Ischemia reperfusion resulted in significant downregulation of the percentage of cells with nuclear JADE1S (Fig. 10A and B, 1D–3D of IR; ref. 32). Strikingly, at day 1 after IR, the percentage of cells with cytoplasmic JADE1S began to increase, reaching maximum at day 3 and decreasing to the baseline by day 7 of recovery (Fig. 10A and B).

Figure 10. JADE1S transiently relocates from the nuclei to the cytoplasm of proliferating kidney tubular epithelial cells in vivo. Ischemia reperfusion (IR) was induced by transient bilateral renal artery clamping followed by reperfusion. Kidney tissue sections from sham, 1 d (1D), 2 d (2D), 3 d (3D), 5 d (5D), and 7 d (7D) post-operative. Sections were processed for immunohistochemistry with JADE1S and Ki67 antibodies as described.32 Nuclei were visualized with hematoxylin counterstain. (A) Representative fields are shown. (B) Temporal expression profiles of JADE1S and Ki67 proteins in tubular epithelium during kidney injury and recovery time course. Upper panel, percent of cells positive only for cytoplasmic JADE1S; middle panel, cells positive for JADE1S in the nuclei; lower panel, cells positive for Ki67.
Thus, our analyses show that during kidney regeneration the transient increase in the proportion of cells with cytoplasmic JADE1S inversely correlated with the proportion of cells with nuclear JADE1S. Importantly, the transient increase in proportion of cells with cytoplasmic JADE1S correlated with that of Ki67 protein (Fig. 10B). Thus, the dynamic changes in JADE1S subcellular localization correlated with proliferative status of regenerating kidneys. Our in vivo data signify the results from cultured cell models and confirm a potential role of JADE1S chromatin shuttling during the cell cycle in a regenerating organ.
Discussion
JADE1S protein is a candidate transcription factor and a member of HAT HBO1 complex.17-19,32,49 We recently reported that JADE1 and HBO1 interact in a cooperative manner and suggested that JADE1 might regulate chromatin affinity and activities of HBO1 during DNA synthesis and transcription.17,32
In this study we investigated properties of HBO1-JADE1 during the cell cycle in cell cultures and in regenerating kidney tubular epithelial cells. Our results demonstrate that the HBO1 complex is dynamic and undergoes chromatin shuttling during the cell cycle. We demonstrated for the first time that JADE1S, JADE1L, and HBO1 are excluded from chromatin and localized to the cytoplasm in late G2/ early mitosis, while they re-associate with chromatin in the end of mitosis/early G1 (Figs. 1, 2, and 4). Both HBO1 and JADE1 are required for bulk histone H4 acetylation and cell proliferation in cultured cells.17,49 Interestingly, the global acetylation levels of histone H4 correlated with JADE1–HBO1 chromatin recruitment during the cell cycle arrest and following cell cycle release (Fig. 4), suggesting function.
While chromatin shuttling of HBO1 complex during the cell cycle is evident, at this point it is unclear whether HBO1 complex undergoes nuclear transport. In asynchronous cultured cells the dynamics of JADE1S chromatin shuttling parallels the dynamics of the nuclear envelope integrity (as assessed by lamin B2 staining, Fig. 2). However, we do not rule out the possibility of a nucleocytoplasmic transport of the HBO1 complex due to the following reasons: (1) in interphase cells, co-expression of HBO1 is required for overexpressed JADE1 to localize to the nucleus and not to the cytoplasm (Fig. 3); (2) in asynchronous mitotic cells JADE1S was already partially cytoplasmic before nuclear envelope disassembly (Fig. 2M) and was still partially cytoplasmic after nuclear envelope re-assembly (Fig. 2S, the lower couple of cells); (3) singly overexpressed JADE1S accumulates in the nuclei of cells treated with leptomycin B, an inhibitor of nuclear export (Fig. S2).
Our results demonstrate that JADE1 chromatin dissociation is accompanied by JADE1 phosphorylation. Based on these data, phosphorylation and chromatin dissociation of JADE1 occurs at G2/M boundary. On the other hand, the de-phosphorylation of endogenous JADE1S protein correlated with the chromatin recruitment and occurred around late mitosis to early G1 (Fig. 4). The temporal and spatial correlation between JADE1S phosphorylation status and chromatin association status suggests a functional link. This link is also supported by the fact that both JADE1S phosphorylation and chromatin dissociation require G2/M arrest by nocodazole (Fig. 7). Most importantly, effects of pharmacological inhibition of JADE1S phosphorylation further confirmed the requirement of phosphorylation for JADE1S dissociation from chromatin (Fig. 8).
Although presumably VX-680 is more specific for Aurora kinase A, this agent might inhibit other kinases. It is tempting to speculate that Aurora A is a candidate kinase that could be directly or indirectly responsible for JADE1 phosphorylation. Aurora A kinase is required for G2/M transition and is highly activated in late G2, which is about the time of JADE1S phosphorylation. Determining JADE1 kinase awaits further investigations involving in vitro kinase assays and more extensive mass spectrometry analysis. Other mitosis-specific kinases, such as, for example, cdk1, should be considered. In addition interactions with other factors might be involved in JADE1 phosphorylation and chromatin dissociation. Clearly results of experiments employing VX-680 established a link between JADE1S phosphorylation and chromatin association status.
Large-scale phosphoproteome screening studies have identified a number of phosphorylated amino acid residues in JADE1 (Fig. 9B and C). Here, we reported for the first time that cell cycle arrest induced phosphorylation of 6 individual amino acid residues within JADE1S polypeptide. All of the residues modified by phosphorylation are highly conserved in other JADE1 mammalian orthologs (Fig. 9D), suggesting functional importance. The 3 residues identified by mass spectral analysis are putative substrates for cdks (Fig. 9B and D). The substrate specificity of Aurora A kinase has not been fully defined, but based on data available, none of the sites phosphorylated in JADE1S in response to mitotic arrest match Aurora A consensus motif, arguing against JADE1 being a direct target of this kinase.39,50 To our knowledge, this study is the first functional study identifying phosphorylation of JADE1S in relationship to the cell cycle.
The role of JADE1–HBO1 chromatin shuttling and phosphorylation during mitosis is intriguing. In general, the global deacetylation of histones H4 and H3 facilitates chromatin condensation during mitosis, which help to prevent erroneous chromosome segregation. Based on our data, the removal of HAT HBO1 complex from chromatin might be driven by JADE1 phosphorylation and is likely to aid histone deacetylation in early mitosis. Chromatin re-association of JADE1 was timed to late mitosis and might serve several possible functions, including re-establishing histone acetylation marks on chromatin of the newly divided cells, pre-replication complex assembly, or, at later stages, histone marking during chromatin replication.10,51 In either case, the specific kinetics of JADE1-HBO1 re-association with chromatin would have to coordinate with cell cycle progression to serve specific function.52-55 More studies would have to address these possibilities.
It has been reported that HBO1 is associated with the origins of replication and is required for the recruitment of MCM potentially promoting the licensing step before initiation of DNA synthesis.2,13,28,56,57 PHD zinc finger protein JADE1 is required for histone H4 acetylation function, DNA synthesis, and cell proliferation.17,23,32 PHD zinc fingers of other chromatin-binding proteins may recognize and bind several specific methylated lysine residues of histone H3.19,58 Affinities of JADE1 PHDs are less defined19 but could potentially recognize methylated histone H3 methyl marks at origins of replication. Supporting this, JADE1L is associated with genomic methylated H3K4me2/3 and this association depends on intact PHD zinc fingers, while JADE1S is found associated with histone H3K36me2/3.19 Genomic study of replication initiation in human chromosome revealed that the chromatin signatures around the origins were enriched in H3K4me2/3 and histone H3 acetylation modifications.59 Interestingly, according to our study, endogenous JADE1S re-association with chromatin begins around the M/G1 border (Figs. 2, 4D, and E), the time when the preRC complex begins to assemble. It is possible that JADE1S de-phosphorylation in late mitosis might promote timely and precise targeting of HBO1 complex to those origins of replication, enabling HBO1 to acetylate histones or other targets important for DNA replication.
A few known factors, including some histone-modifying proteins or their adaptors remain associated with condensed chromatin throughout mitosis. Chromatin association of these factors during mitosis may play a role in holding epigenetic mitotic memory and consequently cell identity.54,55,60-62 According to our results, condensed chromatin in mitosis did not retain significant quantities of JADE1 protein. Small quantities of chromatin-associated JADE1 were detected in early mitosis only by biochemical means (Fig. 4) are most likely due to some inevitable heterogeneity of cell population and incomplete G2/M synchronization by nocodazole. In addition, the results of IF experiments clearly show full chromatin exclusion of JADE1 from late prophase to early telophase (Fig. 2). However we do not exclude a possibility that small quantities of mitotic JADE1 pool that appear to be associated with chromatin are qualitatively different and serve a function.
We detected JADE1 chromatin shuttling in vitro in asynchronously dividing as well as synchronized cultured cells. Importantly, the cell cycle-dependent dynamic properties of JADE1S were recapitulated in our in vivo studies. We demonstrate the transient translocation of JADE1S from nuclei to the cytoplasm of renal tubular cells undergoing repair after injury (Fig. 10). According to the literature, the effect of IR on the progression of kidney tubular cell cycle depends on the specific injury model.63 In our study the transient increase in the proportion of cells with cytoplasmic JADE1S preceded activation of cell proliferation (Ki67 staining). We predict that the cells with cytoplasmic JADE1 are actively proliferating tubular epithelial cells and are in the mitotic phase of the cell cycle. These in vivo observations combined with our recent report32 support the biological role of JADE1 in kidney tubular cell proliferation during tissue regeneration.
Until recently the HBO1 complex was the only HAT complex known to regulate DNA replication. This study demonstrates a novel regulatory pathway of HBO1 complex in relation to the cell cycle. Chromatin shuttling of the HBO1 complex coupled to JADE1S phosphorylation during the cell cycle uncover part of the mechanism by which the HBO1 complex participates in epithelial cell proliferation. Results of our in vitro and in vivo studies support the role of HBO1–JADE1 complex in organ regeneration.
Materials and Methods
Cell lines
H1299, HeLa, and 293T/17 were obtained from ATCC. All cells were grown in Dulbecco modified Eagle medium supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin-streptomycin (Cellgro) at 37 °C in a humidified incubator with 5% CO2 atmosphere. Sub-confluent cells grown in 35-, 60-, 100-, or 150-mm dishes were transfected with Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
Antibodies and chemicals
Rabbit polyclonal sera and affinity-purified antibodies specific for JADE1S, JADE1L, and HBO1 were custom generated (Proteintech Group, details are available upon request). FLAG M5 (1:1000; F1804) and α-Tubulin (1:1000; T9026) mouse monoclonal antibodies, as well as rabbit polyclonal antibodies for FLAG (1:1000; F7425), were from Sigma-Aldrich. HA (IF 1:100, WB 1:1000; sc-7392) mouse monoclonal antibodies and HA (IF 1:100, WB 1:1000; sc-805) and cyclin E1 (1:1000; sc-198) rabbit polyclonal antibodies were from Santa Cruz Biotechnology. Rabbit polyclonal antibody for histones; acetyl-H4 (1:1000; AHP418) was from AbD Serotec. Polyclonal rabbit Ki67 (1:1000; IHC-00375) and nucleolin (1:1000; A300–711A) were from Bethyl Laboratories. Rabbit polyclonal topoisomerase IIα antibody (1:1000; 4733) was from Cell Signaling. Mouse monoclonal anti Lamin B2 (1:30; ab8983) was from Abcam. Goat anti-mouse and anti-rabbit IgG-horseradish peroxidase conjugates (1:2000; sc-2005, and sc-2004) were from Santa Cruz Biotechnology. Alexa Fluor dye-labeled secondary antibodies (1:500; A31627, A31623, A31619, A31631) were from Life Technologies. Protein A/G-agarose mix was from Santa Cruz Biotechnology. Anti-FLAG M2-agarose was from Sigma-Aldrich. Protease inhibitor cocktail and phosphatase inhibitor cocktail was from Roche Diagnostics. Nocodazole (M1404) and L-Mimosine (M 0253) was from Sigma-Aldrich, VX-680 (MK0457) was from Selleckchem, and roscovitine (557360) was from EMD Millipore.
Constructs
HA-HBO1 and FLAG-JADE1S were described previously.17 MGC (Mammalian Genome Collection) fully sequenced human PHF17 (JADE1L) cDNA cloned in pCMV-SPORT6 vector (clone 5111727) was obtained from Open Biosystems. PHF17 cDNA was then cloned into pcDNA3.1 vector modified with an N-terminal FLAG tag and was verified by sequencing (MCLAB; http://www.mclab.com).
Analysis of endogenous histones in nuclear fraction
Histone extraction was done as described earlier.17,18 Cultured cells were lysed with cold 10 mM Tris buffer (pH 8), containing 0.6% NP-40, 0.15 M NaCl, 1 mM EDTA, protease inhibitors (Roche Pharmaceuticals), and 5 mM sodium butyrate. After a 5 min incubation on ice, nuclei were pelleted at 1200 × g (accuSpin Micro 17 R; Fisher Scientific) at 4 °C, and re-suspended in 150 μL 0.4 N H2SO4 on ice. After 20 min of incubation, nuclear debris was removed by centrifugation at 13 000 × g for 10 min at 4 °C. The supernatant was treated with 1.5 mL of cold 20% trichloroacetic acid for 10 min at 4 °C. Pellets were separated by centrifugation at 13 000 × g at 4 °C, and rinsed with 0.1% HCl in acetone and, subsequently, with acetone. The final pellet was re-suspended in reducing SDS-sample buffer.
Fractionation of cultured cells
Cells were fractionated into soluble and chromatin-enriched fractions according to the protocol described earlier with some changes.2,32 Cells were lysed for 15 min in situ with 10 mM HEPES (pH 7.8) buffer containing 10 mM KCl, 1.5 mM MgCl2, 0.1% Triton X-100, protease inhibitors, and PMSF using 500 µl per 100 mm cell culture dish. Pellets were separated by low speed centrifugation at 1300 ×g for 5 min. Supernatants were further cleared by centrifuging at 13 000 × g (Centrifuge 5810 R; rotor A-4-81; eppendorf) for 10 min and frozen for further analysis (Supernatants). The pellets obtained after low speed centrifugation were re-suspended in the same buffer except for omitting Triton X-100 and centrifuged at 13 000 × g for 5 min. Supernatants were discarded, and chromatin-enriched pellets were frozen for analysis. To enable comparison of protein abundance between the 2 different fractions when indicated, soluble and chromatin fractions were extracted with equal volumes of appropriate buffers as previously described.32
Synchronization of cultured cells
To arrest cells in G2/M stage of the cell cycle, nocodazole (100 ng/ml) was added to cultures 30 h post-transfection. After 16 h of incubation, arrested cells were separated by mitotic shake off. Nocodazole was removed by 2× centrifugation at 1300 × g (Centrifuge 5810 R; rotor A-4-81; eppendorf) for 4 min. Cells were plated onto fresh culture dishes for cell cycle recovery. To arrest cells in G1/S stage of the cell cycle, L-mimosine (250 μM) was added to cultures for 16 h. Cell cycle was released by adding fresh media or media with nocodazole.
Pharmacological treatments
Roscovitine treatment
HeLa cells grown in six 60-mm dishes at 80% confluence were treated with 50 ng/mL nocodazole or DMSO (vehicle) for 16 h. When indicated, roscovitine (50 µM) was added to the cells 2 h prior to the nocodazole treatment. Cells were harvested and lysed in 50 mM HEPES (pH 7.4), 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.2% Triton X-100, 0.1 mM PMSF, 5 mM sodium butyrate, and protease and phosphatase inhibitor cocktails for 6 min.
Phosphatase and phosphatase inhibitor treatment
HeLa cells grown in 100-mm dishes at 80% confluency were treated with 50 ng/ml nocodazole or DMSO (vehicle) for 16 h. Cells were washed with PBS and lysed in situ in 50 mM HEPES (pH 7.4), 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.2% triton X-100, 0.1 mM PMSF, 5 mM sodium butyrate and protease inhibitor mixture for 6 min. Lysed cells were collected and centrifuged at maximum speed for 5 min. Fifty (50) µL of the lysates were incubated at 30 °C for 7 min in the presence of 2 mM MnCl2, with or without phosphatase, or phosphatase inhibitors (5 mM EDTA, 2 mM Na3VO4, 5 mM NaF).
VX-680 treatment
HeLa cells arrested at G1/S stage of cell cycle with L-mimosine (250 μM) were released into nocodazole (100 ng/ml) and treated with VX-680 (1 μM) for different time periods. Leptomycin B treatment
Leptomycin B (20 ng/ml) was added to the culture media 24 h post-transfection. Cells were processed for immunofluorescence analysis at 2, 6, and 10 h time points after treatment. Leptomycin B (L2913) was from Sigma-Aldrich.
Flow cytometry (FACS)
Cells were washed in PBS, trypsinized, and collected by centrifugation at 1000 rpm for 10 min (accuSpin Micro 17R; Fisher Scientific). Cells resuspended in PBS were fixed in cold 70% ethanol. Cells collected by centrifugation at 1500 rpm for 10 min were stained with 50 µg/ml propidium iodide (Sigma) and treated with 100 µg/mL RNaseA for 20 min in the dark. Equal numbers of cells (10 000–30 000) were subjected to flow cytometry at Flow Cytometry Core Facility (BUSM; http://www.bu.edu/cores) using either BD FACSCalibur or FACScan flow cytometer (BD Biosciences) and was analyzed using either CellQuest (BD Biosciences) or FlowJo (FlowJo LLC).
Immunofluorescent labeling of whole cells (IF)
IF was done as described.18,32 H1299, HeLa, and 293T/17 cells grown on chamber slides or glass coverslips were rinsed with PBS, and then fixed with 4% paraformaldehyde for 20 min at room temperature. After incubation, cells were rinsed with PBS 3 times and then permeabilized with 0.5% Triton X-100 for 15 min at room temperature. Blocking was performed with 3% horse serum and 2% BSA for 1 h at room temperature. Primary antibodies in 2% BSA, 0.05% Tween-PBS, were incubated overnight at 4 °C and secondary antibodies in 2% BSA, 0.05% Tween-PBS were incubated for 1 h at room temperature. Coverslips were mounted using Vectashield (VECTOR) mounting medium with DAPI. Images were analyzed on Olympus IX 70 inverted microscope with DSU spinning disk confocal system (Olympus) at the Cellular Imaging Core in Boston University School of Medicine using 40× and 60× magnification lens. Images were edited in ImageJ 1.45S (NIH) software. All experiments were repeated at least 2 times and in 3 different types of cells specified, yielding essentially the same results.
Immunoprecipitation (IP)
IP was performed as described18 with few modifications. Cultured transfected cells grown in 60-mm dishes were harvested 48 h post-transfection and were lysed in cold buffer containing 50 mM Tris (pH 7.8), 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1 mM PMSF, 10 mM sodium butyrate and protease inhibitor mixture on ice for 15 min. All steps were done at 4 °C. Lysates were cleared by centrifugation at maximum speed (accuSpin Micro 17R; Fisher Scientific) for 10 min. An aliquot was kept for analysis as “input”. Lysates were incubated with primary antibodies overnight at 4 °C with rotation; following incubation with Protein A/G-agarose beads (1:1 mix, 30 µL for each) for 1 h. Beads with immune complexes were pelleted by low centrifugation for 5 min and washed 3 times with above cold buffer. Proteins were analyzed by SDS-PAGE followed by western blotting.
Large-scale sample preparation for mass spectrometry
H1299 cells were split into eight (8) 150-mm plates at 60% confluency. Same day co-transfection with 18 µg of FLAG-JADE1S and HA-HBO1 cDNA was done according to the Lipofectamin 2000 manufacturer’s protocol. Twenty-four hours post-transfection cells were treated with 150 ng/mL nocodazole or with DMSO (control) for 16 h. Cells were harvested in 10 mL of PBS/ 150-mm plate, and centrifuged at 1000 rpm (Centrifuge 5810 R; rotor A-4–81; eppendorf) for 5 min at 4 °C. All following procedures were done at 4 °C unless specified. Cell pellets were lysed in 50 mM Tris (pH 7.8), 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1 mM PMSF, 10 mM Sodium butyrate, 10 mM NaF, protease inhibitor mixture and phosphatase inhibitor cocktail for 10 min (2 mL/150-mm plate), and lysates cleared by centrifugation for 5 min at 4000 × g. The supernatants were pre-cleared by incubation with A/G-agarose beads for 30 min, and beads removed by centrifugation (9000 × g for 30 min). An aliquot of the supernatant was kept for analysis as “input”. The rest of the supernatant was incubated for 4 h with Anti-FLAG M2-agarose (40 µL of resin/150-mm plate). The resin was recovered by centrifugation (9000 × g for 15 min) and washed 3 times with lysis buffer. An aliquot of the supernatant was kept for analysis. The elution was performed using glycine HCl buffer according to the manufacturer’s instructions.
Mass spectrometry analysis and data processing
Mass spectrometry analysis and data processing were performed by MS Bioworks LLC (www.msbioworks.com). Qubit® 2.0 Fluorometer (Invitrogen) was used to quantitate protein concentrations. Twelve µg of each sample was separated on a 4–12% Bis-Tris Novex mini gel (Invitrogen) using the MOPS buffer system and stained with coomassie. The major band corresponding to PHF17 (JADE1S) was excised, washed with 25 mM ammonium bicarbonate followed by acetonitrile, reduced with 10 mM dithiothreitol at 60 °C and alkylated with 50 mM iodoacetamide at room temperature. The sample was then divided into 3 parts and was digested individually with trypsin (Promega), chymotrypsin (Worthington), and elastase (Worthington) at 37 °C for 4 h, quenched with formic acid, and the supernatant was analyzed directly without further processing. Each gel digest was analyzed by nano LC/MS/MS with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher LTQ Orbitrap Velos. Peptides were loaded on a trapping column and eluted over a 75 µm analytical column at 350 nL/min; both columns were packed with Jupiter Proteo resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with MS performed in the Orbitrap at 60 000 FWHM resolution and MS/MS performed in the LTQ. The 15 most abundant ions were selected for MS/MS. Data processing was done using a local copy of Mascot, and the files were parsed into Scaffold software for validation, filtering, and creating a non-redundant list per sample using Swissprot database. Data were filtered using a minimum protein value of 95%, a minimum peptide value of 50% (Prophet scores), and required at least 2 unique peptides per protein. PHF17 was observed with a combined 55% and 51% sequence coverage across the 3 enzymes analysis for the test and control, respectively. Twelve putative phosphorylation sites were detected for the test sample. Four putative phosphorylation sites were detected for the control sample. Six total phosphorylation sites were manually validated by inspection of the product ion data; the remaining spectra did not contain sufficient product ions to allow site assignment. Using the peak area of the unphosphorylated peptide and its phosphorylated analog at each site, the percentage of phosphorylation was calculated, and the fold change between the test and the control sample was determined.
Computational analysis
The phosphorylation sites of JADE1 were obtained from PhosphoSitePlus (http://www.phosphosite.org). Conservation of the phosphorylation sites were studied by aligning amino acid sequences of selected JADE1 orthologs, which were obtained from NCBI (http://www.ncbi.nlm.nih.gov). Alignment was done using ClustalW2 software (http://www.ebi.ac.uk).
Immunohistochemistry (IHC)
Mouse kidney IHC was performed as described.32 Mouse kidneys were fixed in 4% paraformaldehyde, embedded into paraffin blocks, and sectioned onto glass slides (5 micron thickness). Epitope retrieval was achieved by heating tissue sections in 0.1 M sodium citrate buffer (pH 6.0), using a microwave. Immunohistochemistry of kidney sections was performed using Vectastain ABC kit (VECTOR) following the manufacturer’s protocol and as previously described.32 Detection of the antibody was performed with DAB (3, 3′-diaminobenzidine) kit (Vectastain, VECTOR Lab). Nuclei were visualized by staining sections with Hematoxylin. Images of stained tissue sections were analyzed on an Olympus Bx51 Model UDO3 light microscope. Photos of stained kidney images were taken with an Olympus DP71 microscope camera using 20× or 40× magnification lens.
Quantitation of immunohistochemistry staining
Images were analyzed by 2 blinded investigators. Four representative fields (40× lens) per sample were analyzed by counting cells with stained nuclei, cytoplasm, and total nuclei. The number of positive stained nuclei was normalized to the number of total nuclei (ImageJ, NIH). Average of 4 values and standard deviation was calculated in Excel software (Microsoft Office Professional Plus 2010).
Acute kidney injury after ischemia and reperfusion
Ischemia-reperfusion injury (IR) in mice was performed according to established protocols as previously described.32 Briefly, male C57Bl/6 mice underwent sham surgery or bilateral renal artery clamping for 28 min under anesthesia with tribromoethanol. Post-ischemic and sham kidneys were fixed in 4% paraformaldehyde for immunohistochemistry.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Todd Stukenberg from University of Virginia for insightful suggestions. We thank Charlie Aoun for the expert technical help and proofreading the manuscript. This work was supported by National Institutes of Health Grant RO1 DK087910 and CTSA grant UL1-TR000157 (to M. V. P.). This work was supported, in part, by National Institutes of Health Grants R01 GM098367 (to I.D.) and KO8 DK090143 (to A.H.).
References
- 1.Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–92. doi: 10.1016/S0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 2.Iizuka M, Matsui T, Takisawa H, Smith MM. Regulation of replication licensing by acetyltransferase Hbo1. Mol Cell Biol. 2006;26:1098–108. doi: 10.1128/MCB.26.3.1098-1108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iizuka M, Stillman B. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem. 1999;274:23027–34. doi: 10.1074/jbc.274.33.23027. [DOI] [PubMed] [Google Scholar]
- 4.Iizuka M, Takahashi Y, Mizzen CA, Cook RG, Fujita M, Allis CD, Frierson HF, Jr., Fukusato T, Smith MM. Histone acetyltransferase Hbo1: catalytic activity, cellular abundance, and links to primary cancers. Gene. 2009;436:108–14. doi: 10.1016/j.gene.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 6.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–87. doi: 10.1016/S0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 7.Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–99. doi: 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- 8.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–45. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 9.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 11.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 12.Danis E, Brodolin K, Menut S, Maiorano D, Girard-Reydet C, Méchali M. Specification of a DNA replication origin by a transcription complex. Nat Cell Biol. 2004;6:721–30. doi: 10.1038/ncb1149. [DOI] [PubMed] [Google Scholar]
- 13.McConnell KH, Dixon M, Calvi BR. The histone acetyltransferases CBP and Chameau integrate developmental and DNA replication programs in Drosophila ovarian follicle cells. Development. 2012;139:3880–90. doi: 10.1242/dev.083576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 15.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 16.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–59. doi: 10.1128/MMBR.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foy RL, Song IY, Chitalia VC, Cohen HT, Saksouk N, Cayrou C, Vaziri C, Côté J, Panchenko MV. Role of Jade-1 in the histone acetyltransferase (HAT) HBO1 complex. J Biol Chem. 2008;283:28817–26. doi: 10.1074/jbc.M801407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panchenko MV, Zhou MI, Cohen HT. von Hippel-Lindau partner Jade-1 is a transcriptional co-activator associated with histone acetyltransferase activity. J Biol Chem. 2004;279:56032–41. doi: 10.1074/jbc.M410487200. [DOI] [PubMed] [Google Scholar]
- 19.Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Côté V, et al. HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol Cell. 2009;33:257–65. doi: 10.1016/j.molcel.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, Toyoda T, Shinga J, Katsumoto T, Chiba T, et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood. 2011;118:2443–53. doi: 10.1182/blood-2011-01-331892. [DOI] [PubMed] [Google Scholar]
- 21.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–95. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 22.Zhou MI, Wang H, Ross JJ, Kuzmin I, Xu C, Cohen HT. The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J Biol Chem. 2002;277:39887–98. doi: 10.1074/jbc.M205040200. [DOI] [PubMed] [Google Scholar]
- 23.Wan G, Hu X, Liu Y, Han C, Sood AK, Calin GA, Zhang X, Lu X. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. EMBO J. 2013;32:2833–47. doi: 10.1038/emboj.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke TW, Cook JG, Asano M, Nevins JR. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J Biol Chem. 2001;276:15397–408. doi: 10.1074/jbc.M011556200. [DOI] [PubMed] [Google Scholar]
- 25.Georgiakaki M, Chabbert-Buffet N, Dasen B, Meduri G, Wenk S, Rajhi L, Amazit L, Chauchereau A, Burger CW, Blok LJ, et al. Ligand-controlled interaction of histone acetyltransferase binding to ORC-1 (HBO1) with the N-terminal transactivating domain of progesterone receptor induces steroid receptor coactivator 1-dependent coactivation of transcription. Mol Endocrinol. 2006;20:2122–40. doi: 10.1210/me.2005-0149. [DOI] [PubMed] [Google Scholar]
- 26.Johmura Y, Osada S, Nishizuka M, Imagawa M. FAD24 acts in concert with histone acetyltransferase HBO1 to promote adipogenesis by controlling DNA replication. J Biol Chem. 2008;283:2265–74. doi: 10.1074/jbc.M707880200. [DOI] [PubMed] [Google Scholar]
- 27.Sharma M, Zarnegar M, Li X, Lim B, Sun Z. Androgen receptor interacts with a novel MYST protein, HBO1. J Biol Chem. 2000;275:35200–8. doi: 10.1074/jbc.M004838200. [DOI] [PubMed] [Google Scholar]
- 28.Stedman W, Deng Z, Lu F, Lieberman PM. ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J Virol. 2004;78:12566–75. doi: 10.1128/JVI.78.22.12566-12575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WZ, Liu HO, Wu YH, Hong Y, Yang JW, Liu YH, Wu WB, Zhou L, Sun LL, Xu JJ, et al. Estrogen receptor alpha (ERalpha) mediates 17beta-estradiol (E2)-activated expression of HBO1, Journal of experimental & clinical cancer research. CR (East Lansing, Mich) 2010;29:140. doi: 10.1186/1756-9966-29-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avvakumov N, Lalonde ME, Saksouk N, Paquet E, Glass KC, Landry AJ, Doyon Y, Cayrou C, Robitaille GA, Richard DE, et al. Conserved molecular interactions within the HBO1 acetyltransferase complexes regulate cell proliferation. Mol Cell Biol. 2012;32:689–703. doi: 10.1128/MCB.06455-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyon Y, Selleck W, Lane WS, Tan S, Côté J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–96. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Havasi A, Haegele JA, Gall JM, Blackmon S, Ichimura T, Bonegio RG, Panchenko MV. Histone acetyl transferase (HAT) HBO1 and JADE1 in epithelial cell regeneration. Am J Pathol. 2013;182:152–62. doi: 10.1016/j.ajpath.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kueh AJ, Dixon MP, Voss AK, Thomas T. HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol Cell Biol. 2011;31:845–60. doi: 10.1128/MCB.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tzouanacou E, Tweedie S, Wilson V. Identification of Jade1, a gene encoding a PHD zinc finger protein, in a gene trap mutagenesis screen for genes involved in anteroposterior axis development. Mol Cell Biol. 2003;23:8553–2. doi: 10.1128/MCB.23.23.8553-8562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matter K, Bucher K, Hauri HP. Microtubule perturbation retards both the direct and the indirect apical pathway but does not affect sorting of plasma membrane proteins in intestinal epithelial cells (Caco-2) EMBO J. 1990;9:3163–70. doi: 10.1002/j.1460-2075.1990.tb07514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alessi F, Quarta S, Savio M, Riva F, Rossi L, Stivala LA, Scovassi AI, Meijer L, Prosperi E. The cyclin-dependent kinase inhibitors olomoucine and roscovitine arrest human fibroblasts in G1 phase by specific inhibition of CDK2 kinase activity. Exp Cell Res. 1998;245:8–18. doi: 10.1006/excr.1998.4216. [DOI] [PubMed] [Google Scholar]
- 37.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–4. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 38.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 39.Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Jr., Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70:661–87. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantin GT, Yi W, Lu B, Park SK, Xu T, Lee JD, Yates JR., 3rd Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J Proteome Res. 2008;7:1346–51. doi: 10.1021/pr0705441. [DOI] [PubMed] [Google Scholar]
- 42.Chen RQ, Yang QK, Lu BW, Yi W, Cantin G, Chen YL, Fearns C, Yates JR, 3rd, Lee JD. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009;69:2663–8. doi: 10.1158/0008-5472.CAN-08-3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2:ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- 44.Moritz A, Li Y, Guo A, Villén J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4:rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- 46.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 47.Van Hoof D, Muñoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, Krijgsveld J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5:214–26. doi: 10.1016/j.stem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 48.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–21. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doyon Y, Cayrou C, Ullah M, Landry AJ, Côté V, Selleck W, Lane WS, Tan S, Yang XJ, Côté J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 50.Ferrari S, Marin O, Pagano MA, Meggio F, Hess D, El-Shemerly M, Krystyniak A, Pinna LA. Aurora-A site specificity: a study with synthetic peptide substrates. Biochem J. 2005;390:293–302. doi: 10.1042/BJ20050343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moazed D. Mechanisms for the inheritance of chromatin states. Cell. 2011;146:510–8. doi: 10.1016/j.cell.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–74. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 53.Méndez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–12. doi: 10.1128/MCB.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang F, Higgins JM. Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends Cell Biol. 2013;23:175–84. doi: 10.1016/j.tcb.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 55.Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat Cell Biol. 2011;13:1295–304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aggarwal BD, Calvi BR. Chromatin regulates origin activity in Drosophila follicle cells. Nature. 2004;430:372–6. doi: 10.1038/nature02694. [DOI] [PubMed] [Google Scholar]
- 57.Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell. 2010;37:57–66. doi: 10.1016/j.molcel.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karnani N, Taylor CM, Malhotra A, Dutta A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell. 2010;21:393–404. doi: 10.1091/mbc.E09-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin. 2013;6:6. doi: 10.1186/1756-8935-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.John S, Workman JL. Bookmarking genes for activation in condensed mitotic chromosomes. Bioessays. 1998;20:275–9. doi: 10.1002/(SICI)1521-1878(199804)20:4<275::AID-BIES1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 62.Zaidi SK, Young DW, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet. 2010;11:583–9. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–43, 1p, 143. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.