

Abstract
T cells devoid of tumor necrosis factor receptor associated factor-3 (Traf3) exhibit decreased proliferation, sensitivity to apoptosis, and an improper response to antigen challenge. We therefore hypothesized that TRAF3 is critical to the growth of malignant T cells. By suppressing TRAF3 protein in different cancerous T cells, we found that anaplastic large cell lymphoma (ALCL) cells require TRAF3 for proliferation. Since reducing TRAF3 results in aberrant activation of the noncanonical nuclear factor-κB (NF-κB) pathway, we prevented noncanonical NF-κB signaling by suppressing RelB together with TRAF3. This revealed that TRAF3 regulates proliferation independent of the noncanonical NF-κB pathway. However, suppression of NF-κB-inducing kinase (NIK) along with TRAF3 showed that high levels of NIK have a partial role in blocking cell cycle progression. Further investigation into the mechanism by which TRAF3 regulates cell division demonstrated that TRAF3 is essential for continued PI3K/AKT and JAK/STAT signaling. In addition, we found that while NIK is dispensable for controlling JAK/STAT activity, NIK is critical to regulating the PI3K/AKT pathway. Analysis of the phosphatase and tensin homolog (PTEN) showed that NIK modulates PI3K/AKT signaling by altering the localization of PTEN. Together our findings implicate TRAF3 as a positive regulator of the PI3K/AKT and JAK/STAT pathways and reveal a novel function for NIK in controlling PI3K/AKT activity. These results provide further insight into the role of TRAF3 and NIK in T cell malignancies and indicate that TRAF3 differentially governs the growth of B and T cell cancers.
Keywords: JAK/STAT, NF-κB, NIK, PI3K/AKT, TRAF3
Introduction
Originally identified in B cells, nuclear factor-κB (NF-κB) is a pleiotropic transcription factor required for orchestrating proper immune and inflammatory responses as well as for the development of secondary lymphoid tissues.1,2 Mammals express 5 NF-κB subunits known as RelA, RelB, cRel, p105/p50, and p100/p52, which form homo- and heterodimers to alter gene expression.3 Prior to activation, NF-κB subunits exist as preformed dimers and are sequestered in the cytoplasm by IκB proteins.2,4,5 Interestingly, the p100 NF-κB subunit also functions as an IκB protein and must undergo partial proteolytic processing to produce p52, the active form of p100.6 NF-κB signaling is controlled by 2 pathways, known as the canonical and noncanonical NF-κB pathways, which are differentially regulated and lead to the activation of distinct NF-κB dimers.7,8 Cellular stresses, the IL-1 receptor, the retinoic acid-inducible gene-1 receptor, toll-like receptors, and most tumor necrosis factor receptors (TNFR) modulate canonical NF-κB signaling by inducing the degradation of IκB proteins to allow nuclear translocation of predominantly p50/RelA and p50/cRel dimers.9-12 Conversely, the noncanonical NF-κB pathway involves the activity of p52/RelB dimers and is regulated by a subset of TNFRs, including LTβR, BAFFR, CD40, CD30, TWEAK, and RANK.8,13-15
The NF-κB-inducing kinase (NIK; encoded by Map3k14) is required for stimulation of the noncanonical NF-κB pathway and is normally continuously targeted for proteasomal degradation by a complex consisting of tumor necrosis factor receptor-associated factor-3 (TRAF3), TRAF2, and the cellular inhibitor of apoptosis proteins 1 and 2.16,17 Initiation of noncanonical NF-κB signaling results in the degradation of TRAF3, which disrupts complex formation and results in the accumulation of NIK, followed by the production of p52.18 Interestingly, deleting Traf3 in murine B and T lymphocytes results in differential effects on cell function, although noncanonical NF-κB signaling is activated in both cell types.19 While Traf3−/− B cells disengage from their dependence on B cell-activating factor (BAFF) for continued viability, Traf3−/− T cells remain dependent on growth factors for survival.20,21 In fact, Traf3−/− T cells become sensitized to cell death and undergo high rates of apoptosis after stimulation of the T cell receptor and co-receptor, CD28.22 In addition, whereas loss of Traf3 has no obvious effect on the division of B cells, Traf3−/− T cells fail to proliferate when challenged with antigen.19
The phenotypes associated with Traf3−/− B and T lymphocytes imply that TRAF3 has opposing functions in regulating the physiology of the 2 cell types and suggests that TRAF3 has differential roles in B and T cell oncogenesis. Because deleting Traf3 in B cells results in uncontrolled growth, it is not surprising that TRAF3 mutations have been identified in several human B cell cancers, including Hodgkin lymphoma, splenic marginal zone lymphoma, and multiple myeloma.23-25 These findings suggest that TRAF3 functions as a tumor suppressor in B cells, which was recently confirmed by the observation that specifically deleting Traf3 in B cells leads to the development of B cell lymphomas in mice.26 In contrast, to our knowledge, TRAF3 mutations have not been identified in human T cell cancers, which would be predicted based on the phenotype of Traf3−/− murine T cells. However, the role of TRAF3 in cancerous T cells remains poorly understood. Therefore, to further characterize the function of TRAF3 in T cell-derived cancers, we suppressed TRAF3 in various human malignant T cell lines. Given that TRAF3 is required for normal T cells to function properly, we hypothesized that reducing TRAF3 protein in cancerous T cells would lead to detrimental effects on growth. Here we report that anaplastic large cell lymphoma (ALCL) cells require TRAF3 to maintain cell growth. Notably, though reducing TRAF3 protein results in activation of the noncanonical NF-κB pathway, our data indicate that TRAF3 controls cell division independently of noncanonical NF-κB activity. We show that ALCL cells depend on PI3K/AKT and JAK/STAT signaling for proliferation, and that TRAF3 is required to sustain cell division by promoting PI3K/AKT and JAK/STAT activity.
Results
TRAF3 is required for the proliferation of ALCL cells
Previous work has demonstrated that while TRAF3 appears to be a tumor suppressor in B cells, deleting Traf3 in T cells has negative effects on normal T cell function.26 Based on these observations, we predicted that TRAF3 is critical to the growth of cancerous T cells. To test this hypothesis, TRAF3 protein was suppressed in malignant T cells derived from ALCL, acute lymphoblastic leukemia (T-ALL), and in a malignant T cell with Hodgkin lymphoma histological characteristics. Cell cycle analysis of treated cells found that reducing TRAF3 protein in ALCL cells (Karpas 299, Michel, SUDHL-1) triggered a dramatic accumulation of cells in the G1 phase of the cell cycle (Fig. 1A). Intriguingly, a proliferation defect was not observed in T cells from T-ALL (Peer, Molt-13) or Hodgkin lymphoma (L540) cancers, though western blot analysis demonstrated effective suppression of TRAF3 protein (Fig. 1B). In an effort to rule out any off-target effects, 2 additional TRAF3 siRNA duplexes were also used to decrease the levels of TRAF3 in Karpas 299 cells and likewise led to G1 cell cycle arrest (Fig. 1C). Together these findings indicate that in ALCL malignant T cells, TRAF3 is essential for G1 to S transition and continued proliferation.

Figure 1. Suppression of TRAF3 triggers cell cycle arrest in ALCL cells. (A) ALCL (Karpas 299, Michel, and SUDHL-1) cells were transfected with either control (c) or TRAF3 (T3) siRNA for 48 h and then stained with PI to examine the cell cycle profile by flow cytometry. (B) T-ALL (Peer and Molt-13) and Hodgkin lymphoma (L540) cells were transfected with control (c) or TRAF3 (T3) siRNA and analyzed by flow cytometry after propidium iodide (PI) staining. (C) Flow cytometry of PI stained Karpas 299 cells transfected with different TRAF3 siRNA duplexes for 48 h. *P < 0.001 compared with siControl.
TRAF3 inhibits noncanonical NF-κB activity in malignant T cells
Ablation of Traf3 has been shown to induce aberrant noncanonical NF-κB signaling.21 However, it is unclear if the degree of induction between cell types differs and whether variations in activity result in unique phenotypes. In view of our result that suppression of TRAF3 did not trigger cell cycle arrest in cells from T-ALL cell lines or a T cell-derived Hodgkin lymphoma cell line (Fig. 1B), we investigated whether this was due to disparities in noncanonical NF-κB activity. Processing of p100 to p52 is induced when the noncanonical NF-κB pathway is stimulated.27 Therefore, the levels of p52 protein were assessed in the different T cell cancer lines after suppressing TRAF3. As shown by immunoblot analysis, reducing TRAF3 protein in the assorted cancerous T cells results in an increase in p52 production (Fig. 2A and C). Quantitative PCR (qPCR) further revealed an increase in expression of noncanonical NF-κB target genes in the different cancer lines with a notably higher level of activity in ALCL cells (Fig. 2B and D). Whereas loss of TRAF3 in normal cells results in induction of the noncanonical NF-κB pathway, for some malignant cells inactivating mutations in TRAF3 have been shown to also lead to stimulation of canonical NF-κB signaling.28,29 Activation of the canonical NF-κB pathway induces proteasomal degradation of IκBα, and, as demonstrated by immunoblot analysis, reducing TRAF3 did not affect the stability of IκBα in any of the cancerous T cells (Fig. 2A and C).30 Taken together, our results indicate that TRAF3 is required to prevent basal noncanonical NF-κB signaling in several T cell cancers, and that suppressing TRAF3 in ALCL cells elicits the greatest increase in activity.
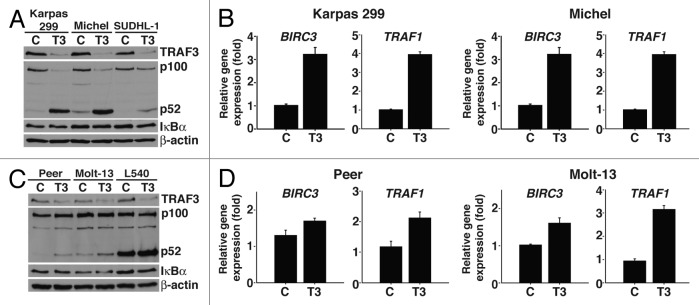
Figure 2. TRAF3 inhibits noncanonical NF-κB activity in malignant T cells. (A) ALCL cells were transfected with control (c) or TRAF3 (T3) siRNA for 48 h and then lysed in RIPA buffer. Lysates were probed with antibodies specific to TRAF3, p100/p52, IκBα, and β-actin. (B) Total RNA was collected from Karpas 299 and Michel cells treated with control (c) or TRAF3 (T3) siRNA for 48 h and subjected to reverse transcription followed by qPCR analysis of BIRC3 and TRAF1 expression. (C) Peer, Molt-13, and L540 cells were transfected with control (c) or TRAF3 (T3) siRNA and lysed in RIPA buffer followed by immunoblotting with antibodies directed to TRAF3, p100/p52, IκBα, and β-actin. (D) Forty-eight hours post-transfection total RNA was isolated from control (c) or TRAF3 (T3) siRNA transfected Peer and Molt-13, which was used to determine the expression levels of BIRC3 and TRAF1 by qPCR.
TRAF3 regulates proliferation independently of NF-κB signaling
To characterize the role of the noncanonical NF-κB pathway in the proliferation defect triggered by suppression of TRAF3, we first determined whether the increase in noncanonical NF-κB activity correlated with the initiation of cell cycle arrest. By conducting a time course experiment in Karpas 299 cells we found that both p52 production and the percentage of cells in G1 began to increase 24 h after TRAF3 siRNA treatment (Fig. 3A and B). In addition, later time points showed further accumulation of p52 as well as higher numbers of cells arresting in G1 (Fig. 3A and B).

Figure 3. Excessive noncanonical NF-κB signaling does not impede cell division. (A) Karpas 299 cells were transfected with control (c) or TRAF3 (T3) siRNA and were lysed in RIPA at the indicated time points. Lysates were probed for TRAF3, p100/p52, and β-actin protein. (B) Karpas 299 cells, treated as in (A), were also stained with PI to evaluate cell cycle profiles. (C) Karpas 299 cells were transfected with control, TRAF3, TRAF3/RelA, TRAF3/RelB, RelA, or RelB siRNA and were lysed 48 h after treatment. Immunoblotting was performed on these lysates using antibodies to TRAF3, RelA, RelB, p100/p52, and β-actin. (D) Total RNA was extracted from Karpas 299 cells 48 h post-transfection with the indicated siRNAs and the expression of BIRC3 and TRAF1 was evaluated by qPCR. (E) Karpas 299 cells were transfected with the indicated siRNA duplexes and after staining with PI cell cycle profiles were analyzed by flow cytometry. *P < 0.001 compared with siControl.
Having found a correlation between activation of the noncanonical NF-κB pathway and induction of cell cycle arrest, we next examined whether preventing noncanonical NF-κB activity would reverse the proliferation defect. RelB is critical for noncanonical NF-κB signaling, and, as shown by qPCR, suppression of RelB together with TRAF3 prevented the increase in expression of noncanonical NF-κB-responsive genes (Fig. 3C and D). However, to our surprise, cell cycle analysis of these cells showed that they still arrested in G1 (Fig. 3E). To ensure that canonical NF-κB activity was not involved in the defect in cell division, RelA was also suppressed with TRAF3. As expected, co-suppression of RelA with TRAF3 did not rescue the arrest phenotype (Fig. 3E). Notably, simultaneous suppression of RelA with TRAF3 did not inhibit the increase in expression of noncanonical NF-κB target genes. Instead, as previously reported, RelA appears to suppress basal noncanonical NF-κB activity, as reducing RelA protein alone led to a modest increase in noncanonical NF-κB signaling (Fig. 3C and D).31 Together, these data demonstrate that TRAF3 controls proliferation independent of its function as a negative regulator of the noncanonical NF-κB pathway.
NIK is important for the division of ALCL cells
TRAF3 has an essential role in targeting NIK for proteasomal degradation. Thus, suppression of TRAF3 leads to an accumulation of NIK protein and activation of the noncanonical NF-κB pathway (Fig. 4A and B).32 Despite the fact that noncanonical NF-κB activity is not involved in the arrest phenotype associated with suppression of TRAF3, NIK has also been implicated in the regulation of other signaling pathways, including the JNK, JAK/STAT, and ERK pathways.33-35 Therefore, co-suppression studies with NIK and TRAF3 were performed to evaluate the role of NIK in controlling cell cycle progression in response to loss of TRAF3. Cell cycle analysis of double suppressed cells showed that reducing NIK protein decreased the number of cells arrested in G1 (Fig. 4C). To confirm that suppression of NIK restored proliferation, the growth of co-suppressed cells was compared with cells with suppressed levels of TRAF3. Assessing growth over several days demonstrated that NIK is critical to inducing cell cycle arrest, as co-suppressed cells grew at a significantly higher rate than those with suppressed TRAF3 alone (Fig. 4D). Along with our observation that noncanonical NF-κB signaling is not involved in the G1 arrest induced by loss of TRAF3, these results indicate that NIK controls the proliferation of ALCL cells independent of the noncanonical NF-κB pathway.

Figure 4. NIK is critical for inducing cell cycle arrest following TRAF3 suppression. (A) Karpas 299 cells were transfected with control, TRAF3, TRAF3/NIK, or NIK siRNA duplexes and were lysed 48 h later. Lysates were analyzed by immunoblotting with antibodies directed to TRAF3, p100/p52, NIK, and β-actin proteins. (B) Total RNA was isolated from Karpas 299 cells 48 h post-transfection with the indicated siRNA duplexes and used to generate cDNA. The expression of Map3K14 (NIK), BIRC3, and TRAF1 was then examined by qPCR. (C) PI staining and flow cytometry was conducted to assess the cell cycle profile of Karpas 299 and Michel cells transfected with the indicated siRNA duplexes. (D) Growth analysis was performed on Karpas 299 and Michel cells transfected with the indicated siRNA duplexes by placing cells at a concentration of 170 000 cells/mL (Karpas 299) or 250 000 cells/mL (Michel) and counting them at 24 h intervals. *P < 0.001 compared with siControl; **P < 0.001 compared with siTRAF3.
NIK alters the localization of PTEN
In order to identify candidate pathway(s) that are regulated by TRAF3 and/or NIK, ALCL cells were treated with chemical kinase inhibitors to ascertain pathways that control cell division. These experiments demonstrated that blocking p38 and ERK activity had no effect on cell growth, while inhibition of JNK, PI3K/AKT, and JAK/STAT signaling resulted in cell cycle arrest (Table 1). Furthermore, preventing PI3K/AKT and JAK/STAT activity led to G1 arrest, similar to reducing TRAF3 (Table 1). Immunoblot analysis was performed to evaluate the activity of the PI3K/AKT and JAK/STAT pathways in cells with suppressed TRAF3 protein. Remarkably, we found that decreasing TRAF3 protein leads to lower levels of AKT and STAT3 phosphorylation (Fig. 5A). Total AKT and STAT3 proteins were also analyzed to evaluate whether the decline in AKT and STAT3 activity resulted from alterations in total protein levels. Interestingly, we observed that while total AKT was not affected by suppressing TRAF3 protein, total STAT3 was dramatically diminished (Fig. 5A). Since concurrent suppression of NIK and TRAF3 partially rescued the proliferation defect that results from reducing TRAF3, we next assessed whether NIK had a role in controlling AKT and/or STAT3 activity. As demonstrated by immunoblot analysis, NIK is required for decreasing AKT phosphorylation but is not involved in regulating the activation of STAT3 (Fig. 5B).
Table 1. The PI3K/AKT and JAK/STAT pathways are vital to the proliferation of ALCL cells.
| Karpas 299 | Michel | SUDHL-1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Kinase Inhibitor | G1 | S | G2/M | G1 | S | G2/M | G1 | S | G2/M |
| DMSO | 33.8 ± 6.3 | 50.6 ± 7.4 | 12.4 ± 1.8 | 28.9 ± 5.9 | 54.1 ± 5.0 | 14.0 ± 3.0 | 26.3 ± 4.0 | 52.5 ± 2.1 | 19.4 ± 3.7 |
| PI3K (LY290042) | 74.1 ± 1.9* | 13.5 ± 1.2 | 7.3 ± 1.3 | 63.1 ± 5.8* | 20.1 ± 2.3 | 11.6 ± 2.8 | 45.0 ± 5.2* | 33.4 ± 2.3 | 17.9 ± 2.3 |
| PI3K (Wortmannin) | 51.2 ± 8.2* | 40.2 ± 8.4 | 5.2 ± 0.43 | 53.4 ± 8.7* | 34.7 ± 6.4 | 7.5 ± 1.7 | 48.6 ± 1.6* | 34.3 ± 0.65 | 14.3 ± 1.0 |
| mTOR (Rapamycin) | 64.5 ± 5.1* | 24.7 ± 4.4 | 6.9 ± 1.0 | 55.7 ± 7.0* | 30.2 ± 3.7 | 9.8 ± 2.6 | 37.4 ± 1.2* | 44.7 ± 1.6 | 15.4 ± 1.9 |
| JAK (WHI-P131) | 67.2 ± 5.0* | 24.9 ± 4.5 | 5.3 ± 0.7 | 60.4 ± 14.1* | 30.6 ± 10.1 | 6.2 ± 2.9 | 46.3 ± 2.9* | 40.4 ± 2.0 | 9.6 ± 1.4 |
| JNK (JNK II) | 52.1 ± 1.7 | 27.3 ± 8.4 | 16.3 ± 7.5 | 21.5 ± 4.4 | 35.1 ± 5.8 | 37.1 ± 6.5 | 3.9 ± 1.8 | 16.7 ± 1.0 | 68.9 ± 2.7 |
| JNK (SP600125) | 51.9 ± 3.5 | 29.5 ± 5.7 | 14.2 ± 5.1 | 24.9 ± 2.2 | 36.1 ± 6.0 | 32.9 ± 5.4 | 4.0 ± 2.0 | 16.4 ± 1.2 | 71.6 ± 2.3 |
| ERK (PD98059) | 35 ± 3.3 | 52.2 ± 4.5 | 9.5 ± 1.1 | 28.4 ± 5.7 | 54.1 ± 5.4 | 13.7 ± 1.0 | 26.7 ± 0.87 | 51.5 ± 1.4 | 18.9 ± 4.3 |
| p38 (SB203580) | 28.3 ± 5.0 | 54.5 ± 8.0 | 12.9 ± 3.2 | 24.0 ± 6.4 | 61.4 ± 6.4 | 10.4 ± 0.9 | 21.7 ± 1.8 | 61.1 ± 1.1 | 14.0 ± 1.2 |
Karpas 299, Michel, and SUDHL-1 cells were treated with the indicated kinase inhibitor and 24 h later were stained with propidium iodide to visualize cell cycle profiles. *P < 0.001.

Figure 5. TRAF3 controls PI3K/AKT signaling in a NIK dependent mechanism. (A) Immunoblot analysis of lysate obtained from Karpas 299 and Michel cells transfected with control (c) or TRAF3 (T3) siRNA for 24 h. Protein specific antibodies to TRAF3, AKT-p (S473), total AKT, STAT3-p (Y705), and total STAT3 were used for immunoblotting. (B) Karpas 299 and Michel cells were transfected with control, TRAF3, TRAF3/NIK, or NIK siRNA duplexes for 24 h. Cells were then lysed in RIPA lysate and probed with antibodies against TRAF3, p100/p52, AKT-p (S473), STAT3-p (Y705), and β-actin. (C) RIPA lysate was collected from Karpas 299 and Michel cells 48 h after transfection with control (c) or TRAF3 (T3) siRNA duplexes and probed with antibodies directed to TRAF3, PTEN, and β-actin. Total RNA was also isolated from these cells and the expression of PTEN was evaluated by qPCR. (D) Digitonin was used to fractionate Karpas 299 cells 36 h after transfection with control or TRAF3 siRNA. Fractions were then immunoblotted using PTEN, TRAF3, CD30, and α-tubulin antibodies. (E) The indicated siRNA duplexes were used to transfect Karpas 299 cells and cells were lysed 48 h after treatment. Antibodies against PTEN, TRAF3, p100/p52, NIK, and β-actin were used to probe lysates. These cells were also examined for PTEN expression by performing qPCR. (F) Karpas 299 and Michel cells were transfected with the indicated siRNA duplexes and 36 h (Karpas 299) or 48 h (Michel) later cells were fractionated with digitonin to isolate cytosolic and heavy membrane proteins. These fractions were probed with PTEN, TRAF3, CD30, and α-tubulin antibodies.
Having found that NIK is dispensable for modulating STAT3 signaling, we focused on understanding how TRAF3 and NIK regulate the PI3K/AKT pathway. Stimulation of PI3K/AKT signaling induces PI3K to phosphorylate phosphatidylinositol (4,5)-bisphosphate (PtdIns[4,5]P2) to phosphatidylinositol (3,4,5)-triphosphate (PtdIns[3,4,5]P3), which serves as a docking phospholipid for AKT phosphorylation and activation.36,37 Considering that suppressing TRAF3 leads to a decrease in AKT phosphorylation, we assessed whether TRAF3 regulates the phosphatase and tensin homolog (PTEN), a phosphatase that inhibits PI3K/AKT activity.38,39 Markedly, both immunoblot and qPCR analysis demonstrated that reducing TRAF3 leads to an increase in PTEN protein and expression (Fig. 5C). PTEN translocates to the plasma membrane, where it blocks PI3K/AKT signaling by dephosphorylating PtdIns(3,4,5)P3 back to PtdIns(4,5)P2.39,40 To examine whether TRAF3 alters the localization of PTEN, we isolated cytosolic and heavy membrane proteins by fractionating cells with digitonin detergent.41 Visualization of PTEN protein revealed that decreasing TRAF3 leads to increased levels of PTEN in the heavy membrane fraction (Fig. 5D). Using CD30 as a marker verified that plasma membrane proteins are found in the heavy membrane fraction, which suggests that suppression of TRAF3 triggers the translocation of PTEN to the plasma membrane. Given that NIK controls AKT phosphorylation, we next investigated whether NIK facilitates the expression and/or translocation of PTEN. Interestingly, both immunoblot analysis and qPCR indicated that NIK is not required for the increase in PTEN protein (Fig. 5E). However, fractionation of cells with digitonin revealed that NIK is critical to the translocation of PTEN (Fig. 5F). These data demonstrate that TRAF3 controls the proliferation of ALCL cells, in part, by preventing the translocation of PTEN to the plasma membrane through inhibition of NIK.
TRAF3 requires PTEN to modulate AKT activity and cell division
In light of our observation that NIK regulates the localization of PTEN, we investigated whether PTEN functions to block AKT signaling and proliferation in response to reducing TRAF3 protein. To this end, PTEN and TRAF3 were co-suppressed, and, as shown by immunoblot analysis, this prevented the decrease in AKT phosphorylation that is associated with suppression of TRAF3 (Fig. 6A and C). Suppressing PTEN and TRAF3 together also led to a significantly lower percentage of cells arrested in G1, indicating that PTEN is critical to blocking proliferation (Fig. 6B and D). Importantly, suppression of PTEN rescued the arrest induced by reduction of TRAF3, similar to that observed for NIK suppression (Fig. 4C). Together these results indicate that one mechanism through which TRAF3 drives the growth of ALCL cells is by inhibiting NIK from targeting PTEN to the plasma membrane and thereby promoting PI3K/AKT signaling.

Figure 6. TRAF3 requires PTEN to modulate AKT activity and cell division. (A) Immunoblot analysis of lysate from Karpas 299 cells, 24 h after transfection with control, TRAF3, TRAF3/PTEN, or PTEN siRNAs. Lysates were probed with antibodies directed to TRAF3, AKT-p (S473), PTEN, and β-actin. (B) Flow cytometry was performed on PI-stained Karpas 299 cells transfected with the indicated siRNA duplexes to analyze the cell cycle profile. (C) Immunoblot analysis of lysates from Michel cells transfected with the indicated siRNAs. Antibodies against TRAF3, AKT-p (S473), PTEN, and β-actin were used to probe the lysates. (D) Michel cells transfected with the indicated siRNA duplexes were stained with PI and analyzed by flow cytometry to ascertain cell cycle profiles. (E) Model depicting the mechanism by which TRAF3 regulates ALCL proliferation. *P < 0.001 compared with siControl; **P < 0.001 compared with siTRAF3.
Discussion
Ablation of Traf3 in normal murine T cells has negative effects on function. Therefore, we predicted that reducing TRAF3 protein in malignant T cells would adversely affect cell growth. By suppressing TRAF3 in a panel of T cell cancer lines, we observed that ALCL cells require TRAF3 for continued proliferation (Fig. 1A). Albeit somewhat surprising that the other cancerous T cells did not exhibit a block in cell division, it is not completely unexpected, since the function of TRAF3 is context-specific.17,22 Consistent with Traf3−/− T cells, reducing TRAF3 protein in the different malignant T cell lines leads to aberrant noncanonical NF-κB signaling (Fig. 2B and D). Interestingly, we observed that, unlike TNFR activation of the noncanonical NF-κB pathway, lowering TRAF3 protein leads to an increase in NIK transcription (Fig. 4B). We speculate that this is a consequence of signal duration, where TNFRs transiently activate the noncanonical NF-κB pathway, and suppressing TRAF3 induces chronic stimulation. Nonetheless, we find that suppression of TRAF3 in ALCL cells elicits the greatest increase in noncanonical NF-κB signaling. Based on this result, we surmised that excessive noncanonical NF-κB activity triggers the arrest of ALCL cells in response to suppression of TRAF3. However, preventing noncanonical NF-κB signaling in cells with reduced TRAF3 protein failed to rescue the cell cycle arrest, indicating that TRAF3 controls proliferation independent of the noncanonical NF-κB pathway. Other NF-κB-independent roles have been identified for TRAF3, including functioning as a scaffolding protein during T cell receptor signaling and as an E3 ubiquitin ligase for regulating NFAT activity.22,42 Thus, together with the previous observations, our findings expand the role of TRAF3 in integrating a multitude of signals to modulate specific cellular activities.
As an inhibitor of noncanonical NF-κB signaling, TRAF3 prevents pathway activation by continuously targeting NIK for degradation. Consequently, we detect increased levels of NIK protein after suppression of TRAF3 (Fig. 4A). Despite the fact that noncanonical NF-κB activity is not involved in the arrest induced by suppression of TRAF3, NIK has been shown to block the proliferation of regulatory T (Treg) cells.43 Interestingly, ALCL cells are known to exhibit a Treg-like phenotype that results from expression of nucleophosmin–anaplastic lymphoma kinase, an oncogenic fusion protein found in the majority of ALCL cancers.44-46 We therefore examined the role of NIK in inhibiting cell division after suppression of TRAF3, and determined that NIK is critical to halting cell cycle progression. Notably, lowering NIK levels together with TRAF3 did not completely rescue the proliferation defect, implying that TRAF3 also controls cell division in a NIK-independent manner.
In an effort to obtain candidate pathway(s) that are governed by TRAF3, we inhibited different kinase activated pathways in ALCL cells and compared the effects on proliferation to those induced by suppressing TRAF3. Of the various pathways examined we found that blocking PI3K/AKT and JAK/STAT activity led to a G1 arrest, while inhibition of JNK signaling resulted in a G2/M arrest. Importantly, the JNK pathway has previously been reported to be required for G2/M to G1 transition in ALCL cells and serves to demonstrate the specificity of PI3K/AKT and JAK/STAT activity in controlling G1 to S transition.47 Given that suppressing TRAF3 also triggers a G1 arrest, we assessed the role of TRAF3 in modulating PI3K/AKT and JAK/STAT signaling, and found that TRAF3 positively regulates both pathways. Intriguingly, the maintenance of normal T cells also depends on the PI3K/AKT and JAK/STAT pathways, which proposes that reliance on certain growth signals by specific cells continues after transformation.48-50 Together with our finding that TRAF3 promotes PI3K/AKT and JAK/STAT signaling, this hypothesis may explain the apparent lack of TRAF3 mutations in human T cell cancers. Though additional investigation is required to determine whether TRAF3 controls PI3K/AKT and JAK/STAT signaling in normal T cells, our results may also offer insight into the physiological role of TRAF3.
Knowing that NIK is important to triggering cell cycle arrest in response to suppression of TRAF3, we investigated whether NIK controls PI3K/AKT and/or JAK/STAT signaling. Lowering TRAF3 and NIK levels together showed that, although NIK is dispensable for modulation of the JAK/STAT pathway, NIK is required to inhibit PI3K/AKT signaling (Fig. 5B). PTEN is an inhibitor of PI3K/AKT activity, and further investigation demonstrated that NIK blocks PI3K/AKT signaling by targeting PTEN to the plasma membrane. While the manner by which NIK controls PTEN localization remains unclear, it is known that phosphorylation has a critical role in regulating the translocation of PTEN. NIK is a kinase, and it may be that the accumulation of NIK that occurs after reducing TRAF3 protein alters PTEN localization through a mechanism that involves phosphorylation. In any case, our results offer further support to the notion that in some circumstances NIK prevents proliferation.43
Interestingly, TRAF3 does not require NIK to inhibit the JAK/STAT pathway, which is presumed to account for the percentage of cells that remain arrested after co-suppression of TRAF3 and NIK. Our current data demonstrate that TRAF3 controls JAK/STAT activity by regulating the levels of total STAT3 protein (Fig. 5A). However, it remains unknown whether TRAF3 modulates STAT3 transcription, or whether TRAF3 alters the degradation of STAT3 protein. TRAF3 contains E3 ubiquitin ligase activity, and while our results are inconsistent with TRAF3 controlling the stability of STAT3 protein directly, TRAF3 may control the half-life of a negative regulator of STAT3. Research is ongoing to characterize the manner by which TRAF3 regulates STAT3 protein, which is important for a more complete comprehension of the mechanisms that TRAF3 utilizes to drive the growth of malignant T cells. This future work notwithstanding, our results demonstrate that TRAF3 is crucial to maintaining PI3K/AKT and JAK/STAT activity and, thus, essential for the proliferation of ALCL cells (Fig. 6E). In addition, our data implicate NIK as a novel regulator of PTEN localization and PI3K/AKT signaling. Notably, these unexpected functions for TRAF3 and NIK are separate from their known roles in controlling the noncanonical NF-κB pathway, and suggest that TRAF3 may be a target for anticancer therapies to certain types of T cell malignancies.
Material and Methods
Cell culture and Reagents
Karpas 299, Michel, SUDHL-1, Peer, Molt-13, and L540 cell lines were cultured in RPMI-1640 medium (Corning) containing 10% fetal bovine serum (Atlas Biologicals, Inc) and 2 mM Glutamax (Life Technologies) at 37 °C and 5% CO2.
Antibodies and immunoblotting
Lysates from cells were prepared by incubating cells on ice in radioimmune precipitation assay (RIPA) buffer (PBS containing 1% Nonidet P-40, 0.5% [w/v] deoxycholic acid, 0.1% SDS, 1 mM PMSF, and 1 mM DTT) supplemented with complete mini protease inhibitor tablets (Roche Diagnostics). Protein samples were resolved on denaturing polyacrylamide gels, transferred to nitrocellulose (Whatman), and blocked with 5% powdered milk (w/v) in TBS containing 0.1% Tween 20. The membranes were incubated with the specified antibodies, washed, and incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare). Peroxidase activity was detected by the enhanced chemiluminescence western blot analysis system (GE Healthcare). Antibodies used: TRAF3, RelB, RelA, and α-tubulin (Santa Cruz Biotechnologies); PTEN, NIK, AKT, AKT-p, STAT3, and STAT3-p (Cell Signaling Technology); CD30 (BD PharMingen); p100/p52 (Millipore); IκBα (Upstate Biotechnology); β-actin (Sigma-Aldrich).
RNA interference
Cells were electroporated as previously described, with either 2 μM or 5 μM of target siRNA duplexes.13 Double RNAi experiments were performed by transfecting both targets simultaneously, except PTEN and TRAF3 co-suppression in Karpas 299 cells. For this double RNAi, RNA duplexes for PTEN were transfected first, and then 24 h later, cells were re-transfected with TRAF3 RNA duplexes. In all double RNAi experiments, the single RNAi samples were normalized with control siRNA. Twenty hours post-transfection cells were transferred to RPMI-1640 medium containing 10% fetal bovine serum and 2 mM Glutamax at 0.5 × 106/mL for various times. Control, TRAF3, RelB, RelA, and PTEN siRNA duplexes were obtained from Sigma-Aldrich. The NIK siRNA duplex was purchased from Life Technologies. Target sequences: control (5′-CATGCCTTGC TTTACGCAT-3′), TRAF3 (5′-GCCACATGCA GCCACTGCA-3′), TRAF3.1 (5′-AGAGTCAGGT TCCGATGAT-3′), TRAF3.2 (5′-GTCATCATGC GTGGAGAAT-3′), RelB (5′-GACTGCACCG ACGGCATCT-3′), RelA (5′-GCCCTATCCC TTTACGTC-3′), NIK (5′-GCCAGTCCGA GAGTCTTGAT CAGAT-3′), and PTEN (5′-AUGCACAUAU CAUUACACC-3′).
Cell cycle analysis
After 48 h of siRNA treatment the indicated cells were harvested and washed with PBS. Next, 1 × 106 cells were fixed in 1 mL of 50% ethanol in PBS for 1 h at −20 °C. The 50% ethanol was then removed, and cells were placed in PBS containing 50 μg/mL propidium iodide (PI) and 100 μg/mL RNase A. Flow cytometry was then performed using the Beckman Coulter Cytomics FC500, and data analysis was performed using FlowJo (Tree Star Inc). Cell cycle analysis of cells treated with chemical kinase inhibitors was performed as described above, except cells were treated with the following inhibitors for 24 h: 5 nM Wortmannin, 25 μM LY294002, 50 μM SB203580, 20 μM PD98059, 25 μM SP600125, 25 μM JNK inhibitor II, 50 nM Rapamycin, or 20 μM WHI-P131.
Cell growth analysis
Karpas 299 or Michel cells were electroporated with control, TRAF3, TRAF3/NIK, and NIK siRNA duplexes as described above. Twenty-four hours after treatment, cells were placed at 170 000 cells/mL (Karpas 299) or 250 000 cells/mL (Michel) per well in 6-well plates. Cells were then counted at 24-h intervals for a total of 72 h using a Z2 Beckman Coulter Particle Count and Size analyzer.
Digitonin fractionation
At 36 h (Karpas 299) or 48 h (Michel) after electroporation, 7 × 106 cells were harvested by centrifugation at 300 × g for 5 min. Cells were then washed twice with PBS. After the final wash, cells were resuspended in 200 μL of Digitonin lysis buffer (200 μg/mL digitonin, 250 mM Sucrose, 1 mM EDTA, 1.5 mM MgCl2, 10 mM KCl, and 20 mM HEPES). Cells were then incubated on ice for 5 min and centrifuged at 10 000 × g for 10 min at 4 °C. The supernatant was saved as cytosolic proteins, and the pellet was lysed in 120 μL RIPA buffer as described above. After centrifugation at 14 000 × g for 20 min at 4 °C, the supernatant was kept as heavy membrane proteins.
Quantitative PCR
Cells were harvested and washed with PBS followed by total RNA isolation using the PureLink RNA mini kit (Life Technologies) according to the manufacturer's instructions. One hundred nanograms of total RNA was subjected to a reverse transcription reaction using random hexamer primers and TaqMan MultiScribe Reverse Transcriptase (Applied Biosystems). The resulting cDNA was analyzed with the indicated TaqMan probe using the Bio-Rad CFX96 real-time system C100 thermal cycler. Each target assay was normalized to glyceraldehyde-3-phosphate dehydrogenase levels and performed in triplicate.
Statistical analysis
All experiments were performed at least 3 times in triplicate, and the data are graphed as mean ± s.e.m. Quantitative PCR experiments are shown as one representative experiment. Comparisons between 2 groups were performed using Student t test. For non-normal data, or data with significantly different variances, the non-parametric Mann–Whitney Ranks Sum test was used. Comparisons between 3 or more groups were analyzed by one-way ANOVA followed by Tukey post-hoc analysis. Non-parametric groups of 3 or more were analyzed with Kruskal–Wallis ANOVA on Ranks followed by Tukey post-hoc analysis. All statistical analyses were performed using SigmaPlot 11 (Systat Software, Inc). Results were considered significant when P value < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs Shawn Bratton and Edward Mills for reagents and critical reading of the manuscript and members of the Wright laboratory for helpful discussion.
Grant Support
This work was supported in part by Cancer Prevention Research Institute of Texas Grant RP101501 (to I.M.), National Institute of Health NIEHS Training Grant T32 ES07247 (to K.A.G.) and Research Starter Grant from the American Cancer Society 121549-RSG-11-180-01-TBE (to C.W.W.).
Glossary
Abbreviations:
- NF-κB
nuclear factor-κB
- TNFR
tumor necrosis factor receptor
- NIK
NF-κB-inducing kinase
- TRAF3
tumor necrosis factor receptor associated factor-3
- BAFF
B cell activating factor
- ALCL
anaplastic large cell lymphoma
- PtdIns[4,5]P2
phosphatidylinositol (4,5)-bisphosphate
- PtdIns(3,4,5)P3
phosphatidylinositol (3,4,5)-triphosphate
- PTEN
phosphatase and tensin homolog
- RIPA
radioimmune precipitation assay buffer
- PI
propidium iodide
- T-ALL
T cell acute lymphoblastic leukemia
- qPCR
quantitative PCR
References
- 1.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–8. doi: 10.1016/0092-8674(86)90807-X. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karin M, Cao Y, Greten FR, Li ZWNF. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin ASJ., Jr. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- 5.Ganchi PA, Sun SC, Greene WC, Ballard DW. I kappa B/MAD-3 masks the nuclear localization signal of NF-kappa B p65 and requires the transactivation domain to inhibit NF-kappa B p65 DNA binding. Mol Biol Cell. 1992;3:1339–52. doi: 10.1091/mbc.3.12.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–81. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 8.Sun SC. The noncanonical NF-κB pathway. Immunol Rev. 2012;246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung K, Betts JC, Xu L, Nabel GJ. The cytoplasmic domain of the interleukin-1 receptor is required for nuclear factor-kappa B signal transduction. J Biol Chem. 1994;269:1579–82. [PubMed] [Google Scholar]
- 10.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–67. doi: 10.1016/0092-8674(93)90134-C. [DOI] [PubMed] [Google Scholar]
- 12.Ak P, Levine AJ. p53 and NF-κB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24:3643–52. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 13.Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF-kappaB pathways in anaplastic large cell lymphoma cells. J Biol Chem. 2007;282:10252–62. doi: 10.1074/jbc.M608817200. [DOI] [PubMed] [Google Scholar]
- 14.Morrison MD, Reiley W, Zhang M, Sun SC. An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappaB signaling pathway. J Biol Chem. 2005;280:10018–24. doi: 10.1074/jbc.M413634200. [DOI] [PubMed] [Google Scholar]
- 15.Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor. J Biol Chem. 2010;285:12971–8. doi: 10.1074/jbc.M109.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhlik M, Good L, Xiao G, Harhaj EW, Zandi E, Karin M, Sun SC. NF-kappaB-inducing kinase and IkappaB kinase participate in human T-cell leukemia virus I Tax-mediated NF-kappaB activation. J Biol Chem. 1998;273:21132–6. doi: 10.1074/jbc.273.33.21132. [DOI] [PubMed] [Google Scholar]
- 17.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–70. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–50. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–15. doi: 10.1016/S1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 20.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–67. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA. TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol. 2011;186:143–55. doi: 10.4049/jimmunol.1000290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto C, Giefing M, Massow A, Vater I, Gesk S, Schlesner M, Richter J, Klapper W, Hansmann ML, Siebert R, et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br J Haematol. 2012;157:702–8. doi: 10.1111/j.1365-2141.2012.09113.x. [DOI] [PubMed] [Google Scholar]
- 25.Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, Spina V, Bruscaggin A, Monti S, Cerri M, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118:4930–4. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- 26.Moore CR, Liu Y, Shao C, Covey LR, Morse HC, 3rd, Xie P. Specific deletion of TRAF3 in B lymphocytes leads to B-lymphoma development in mice. Leukemia. 2012;26:1122–7. doi: 10.1038/leu.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercurio F, Didonato J, Rosette C, Karin M. Molecular cloning and characterization of a novel Rel/NF-kappa B family member displaying structural and functional homology to NF-kappa B p50/p105. DNA Cell Biol. 1992;11:523–37. doi: 10.1089/dna.1992.11.523. [DOI] [PubMed] [Google Scholar]
- 28.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010;115:3541–52. doi: 10.1182/blood-2009-09-243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiDonato JA, Mercurio F, Karin M. Phosphorylation of I kappa B alpha precedes but is not sufficient for its dissociation from NF-kappa B. Mol Cell Biol. 1995;15:1302–11. doi: 10.1128/mcb.15.3.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem. 2003;278:19852–60. doi: 10.1074/jbc.M301945200. [DOI] [PubMed] [Google Scholar]
- 32.Xiao G, Harhaj EW, Sun SCNF. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–9. doi: 10.1016/S1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 33.Akiba H, Nakano H, Nishinaka S, Shindo M, Kobata T, Atsuta M, Morimoto C, Ware CF, Malinin NL, Wallach D, et al. CD27, a member of the tumor necrosis factor receptor superfamily, activates NF-kappaB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-kappaB-inducing kinase. J Biol Chem. 1998;273:13353–8. doi: 10.1074/jbc.273.21.13353. [DOI] [PubMed] [Google Scholar]
- 34.Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277:7920–8. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadiminty N, Chun JY, Hu Y, Dutt S, Lin X, Gao AC. LIGHT, a member of the TNF superfamily, activates Stat3 mediated by NIK pathway. Biochem Biophys Res Commun. 2007;359:379–84. doi: 10.1016/j.bbrc.2007.05.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–8. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 38.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 39.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 40.Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 41.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–34. doi: 10.1016/S0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Huang Y, Li L, Wei Q. TRAF3 negatively regulates calcineurin-NFAT pathway by targeting calcineurin B subunit for degradation. IUBMB Life. 2012;64:748–56. doi: 10.1002/iub.1060. [DOI] [PubMed] [Google Scholar]
- 43.Lu LF, Gondek DC, Scott ZA, Noelle RJNF. NF kappa B-inducing kinase deficiency results in the development of a subset of regulatory T cells, which shows a hyperproliferative activity upon glucocorticoid-induced TNF receptor family-related gene stimulation. J Immunol. 2005;175:1651–7. doi: 10.4049/jimmunol.175.3.1651. [DOI] [PubMed] [Google Scholar]
- 44.Casares N, Rudilla F, Arribillaga L, Llopiz D, Riezu-Boj JI, Lozano T, López-Sagaseta J, Guembe L, Sarobe P, Prieto J, et al. A peptide inhibitor of FOXP3 impairs regulatory T cell activity and improves vaccine efficacy in mice. J Immunol. 2010;185:5150–9. doi: 10.4049/jimmunol.1001114. [DOI] [PubMed] [Google Scholar]
- 45.Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci U S A. 2006;103:9964–9. doi: 10.1073/pnas.0603507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolke C, Tadje J, Bukowska A, Täger M, Bank U, Ittenson A, Ansorge S, Lendeckel U. Assigning the phenotype of a natural regulatory T-cell to the human T-cell line, KARPAS-299. Int J Mol Med. 2006;17:275–8. [PubMed] [Google Scholar]
- 47.Leventaki V, Drakos E, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Claret FX, Rassidakis GZ. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood. 2007;110:1621–30. doi: 10.1182/blood-2006-11-059451. [DOI] [PubMed] [Google Scholar]
- 48.Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. 2010;130:418–26. doi: 10.1111/j.1365-2567.2009.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel ES, Chang LJ. Synergistic effects of interleukin-7 and pre-T cell receptor signaling in human T cell development. J Biol Chem. 2012;287:33826–35. doi: 10.1074/jbc.M112.380113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson SE, Shah N, Bajer AA, LeBien TW. IL-7 activates the phosphatidylinositol 3-kinase/AKT pathway in normal human thymocytes but not normal human B cell precursors. J Immunol. 2008;180:8109–17. doi: 10.4049/jimmunol.180.12.8109. [DOI] [PubMed] [Google Scholar]