

Abstract
Background
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are complex neurodegenerative disorders that can be either sporadic or familial and can overlap clinically and pathologically. We present the first Central-Eastern European family with ALS-FTD syndrome due to a C9ORF72 repeat expansion.
Methods
We studied a family consisting of 37 family members, and six of these family members were genetically evaluated for C9ORF72 expansions. Family members were evaluated clinically, by history, and by chart review.
Results
Five generations of the family were studied, and six affected family members were identified. All affected members were females and had a different clinical presentation, which was either ALS, FTD, or both. Among genetically evaluated subjects, five carried a C9ORF72 expansion; four of these individuals remain clinically unaffected.
Conclusion
Our report reveals that the hexanucleotide repeat expansion of C9ORF72, which is the most common genetic cause of ALS-FTD complex disorder, is also present in Central-Eastern Europe. Further studies are needed to assess the frequency of this expansion in the Polish population with familial as well as sporadic ALS, FTD, and the ALS-FTD complex disorder.
Keywords: ALS-FTD complex disorder, familial ALS-FTD, C9ORF72 gene mutation, phenotype
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are complex neurodegenerative disorders that usually begin in the fifth or six decade of life. ALS and FTD are known to be fatal and have no effective treatments.
Classically, ALS is recognized as pure motor system degeneration and is not associated with cognitive or behavioral dysfunction [1]. In 1990, Neary et al. first reported motor neuron disease with frontal lobe dementia [2]. It was later confirmed that almost one-half of ALS patients actually have cognitive problems, and about 15% meet the criteria for the behavioral variant of FTD (bvFTD) [3, 4]. Currently, ALS is increasingly recognized as a multisystem disorder. The disease occurs sporadically in the majority of cases, but about 5-10% of patients have a positive family history [4].
FTD is the second most common cause of early-onset dementia in patients younger than 65 years and is characterized by behavioral and personality changes that are accompanied by Parkinsonism and/or language abnormalities [5, 6]. About 50% of FTD patients will also develop symptoms of motor neuron disease during the course of the disease. Unlike ALS, approximately 20-50% of FTD patients have a positive family history [7].
Clinical studies have suggested that ALS and FTD may represent two extremes on a spectrum of the same neurodegenerative disease and that they co-occur in 15% of patients [8]. The link between motor neuron disease and dementia with behavioral and personality changes was first described in two families diagnosed with pallidopontal nigral degeneration and in disinhibition-dementia parkinsonism-amyotrophy complex linked to chromosome 17 [9] and was confirmed by the identification of the tau gene (MAPT) as the pathogenic gene for FTD with parkinsonism linked to chromosome 17 (FTDP-17) [10]. This was followed by the identification of mutations in the progranulin gene (PGRN) [11]. Furthermore, the understanding of the link between ALS and FTD was accelerated by the identification of transactive response DNA-binding protein 43 (TDP-43) inclusions in ALS and FTD patients, which were followed by the identification of TAR DNA binding protein (TARDBP) gene mutations, which encodes TDP-43 [3]. There are also other genetic factors in hereditary ALS and FTD, e.g. superoxide dismutase-1 (SOD1), fused in sarcoma (FUS), optineurin (OPTN), valosin containing protein (VCP), charged multivesicular body protein 2b (CHMP2B), and angiogenin (ANG) [12,13,14]. These genes do not fully explain familial cases, especially those with a heterogeneous clinical presentation. The recent discovery of a hexanucleotide repeat expansion in chromosome 9 open reading frame 72 gene (C9ORF72), as the most common genetic cause of ALS and FTD, confirmed the overlapping clinical and pathological nature of these syndromes, which are known as ALS-FTD complex disorder [15, 16, 17,18]. Here, we report a Polish family with a C9ORF72 expansion and clinical diagnoses that bridge motor neuron disease, Parkinsonism, and the dementia spectrum.
Material and Methods
All members of the family with a history of motor neuron disease, dementia, and Parkinsonism were included in this study. The six family members enrolled in the study provided written informed consent for clinical and molecular genetic studies, which was approved by the Institutional Review Boards of the Silesian Medical University and the Mayo Clinic.
Clinical evaluation was performed on all family members participating in the study. The clinical evaluation consisted of a neurological examination and a neuropsychological test battery. The clinicians who assessed subjects were blinded to the genetic test results.
A genealogical study was performed by reviewing available medical records and by conducting a semi-structured interview that included questions on age at disease onset, age at death, disease duration, and disease symptoms, which included possible cognitive impairment, upper and lower motor neuron dysfunction, and features of Parkinsonism. This was followed by questions on the presence of other possible co-existing non-neurological diseases. Information was provided by all family members participating in the study independently and was then compared to obtain the most accurate genealogical data.
Neuroimaging studies performed in affected subjects included structural examination with 1.5 Tesla (SE, T1, T2, FLAIR) Magnetic Resonance Imaging (MRI) and functional examination with Tc-99m-ECD Single-Photon Emission Computed Tomography (SPECT).
DNA was extracted from frozen peripheral blood samples using standard procedures [15]. Genomic DNA was genotyped with a standard fluorescently labeled polymerase chain reaction (PCR) primer, and the assay was visualized on an automated ABi3730 DNA analyzer (Applied Biosystems). If the genotype was homozygous, then the samples were further analyzed using repeat-primed PCR to determine if the sample was a true homozygote (wildtype), or if a stutter pattern was present, which identifies expansion carriers. These methods have been previously described by DeJesus-Hernandez et al. [15].
The size of the GGGGCC repeat was estimated by Southern blotting, as described elsewhere [19]. In brief, seven to ten micrograms of genomic DNA were digested with a restriction enzyme (XbaI; Promega Corporation, Madison, WI, USA), and electrophoresed for 6 hours at 100 V in a 0.8% agarose gel. The DNA was then transferred to a positively charged nylon membrane (Roche, Penzberg, Germany) and cross-linked by ultraviolet irradiation. After prehybridization in digoxygenin EasyHyb solution (Roche), hybridization with a digoxygeninlabelled probe was performed. Anti-digoxygenin antibody (1:10 000, Roche) was used to detect the probe, which was subsequently visualized with CDP-star substrate (Roche) on autoradiography film. Multiple exposures between 15 minutes and 1 hour were obtained, and the most abundant repeat sizes were estimated using densitometry.
Results
The resulting pedigree consists of 37 individuals spanning 5 generations of one family that originated from Southern Poland. (Fig.1.). Based on the detailed histories, chart reviews, and interviews with family members, we identified six clinically affected subjects with a diverse clinical phenotype (Tab.1.). For the present study, we were able to examine the patients directly and were able to obtain the genetic status of the C9ORF72 expansion in the last living affected subject and five individuals without clinical symptoms from generations IV and V.
Figure 1.
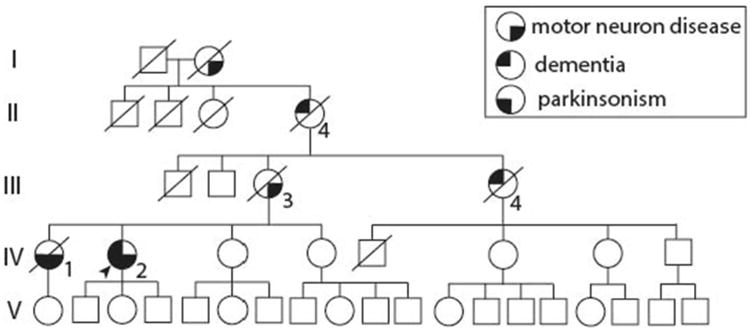
Pedigree. Square: male; circle: female; diagonal line: deceased; arrow: proband. Lower right quarter filled with black, motor neuron disease. Lower left quarter, Parkinsonism. Upper left quarter, dementia.
Table 1. Demographic and clinical data of affected subjects.
| Pedigree# /gender | Year of birth | AAO | AAD | Disease duration (years) | First symptoms | Motor neuron disease symptoms | Parkinsonian symptoms | Cognitive symptoms | Psychiatric symptoms |
|---|---|---|---|---|---|---|---|---|---|
| IV.1 / F | 1951 | 56 | 61 | 5 | bulbar onset ALS | dysarthria, dysphagia, general muscle atrophy, pyramidal sings | bradykinesia, rigidity | UKN | UKN |
| IV.2 / F | 1953 | 54 | AL | 6 | Parkinsonism | dysarthria, dysphagia | bradykinesia, rigidity, postural instability | dementia | pseudobulbar affect |
| III.3 / F | 1931 | 53 | 58 | 5 | bulbar onset ALS | general muscle atrophy | UKN | UKN | UKN |
| III.4 / F | 1932 | 49 | 59 | 10 | dementia | NP | NP | early onset dementia | behavioral symptoms |
| II.4 / F | 1906 | UKN | 59 | UKN | dementia | UKN | UKN | early onset dementia | UKN |
| I.2 / F | approximately 1870 | UKN | [death in 1930]* | UKN | movement disorder | UKN | UKN | UKN | UKN |
AAO age at onset in years, AAD age at death in years, AL alive,
year of death, NP not present, UKN unknown
All affected subjects were females, and the mean age at disease onset was 53 years (±2.5 years). The proband, a 60-year old woman (IV.2), first presented with symptoms of right side predominant Parkinsonism at age 54 years. She was initially diagnosed with Parkinson's disease and demonstrated a typical disease course with a fairly good response to L-dopa. She did not have any cognitive deficits in her neuropsychological evaluation (MMSE 28/30, CDT 10/10). After two years of prospective observation, bulbar signs (flaccid dysarthria and dysphagia), followed by progressive balance difficulties, gait disturbances, mild cognitive impairment, and pseudobulbar affect developed. Other than bulbar signs, no upper or lower motor neuron disease features were identified. By the fifth year of follow-up, which was also when she was genetically tested, her symptoms had progressed rapidly. At that time, the patient had dementia, was unable to walk, and had severe dysarthria and dysphagia, which caused malnutrition. Her parkinsonian syndrome was no longer L-dopa responsive. Head MRI showed bilateral fronto-temporal cortical atrophy (Fig 2). SPECT demonstrated hypoperfusion in both fronto-temporal regions, but this was more prominent on the left side. This patient carried a hexanucleotide repeat expansion of C9ORF72 with an expansion size of ∼900 repeat units [∼7.7 kb] (Fig.3).
Figure 2.
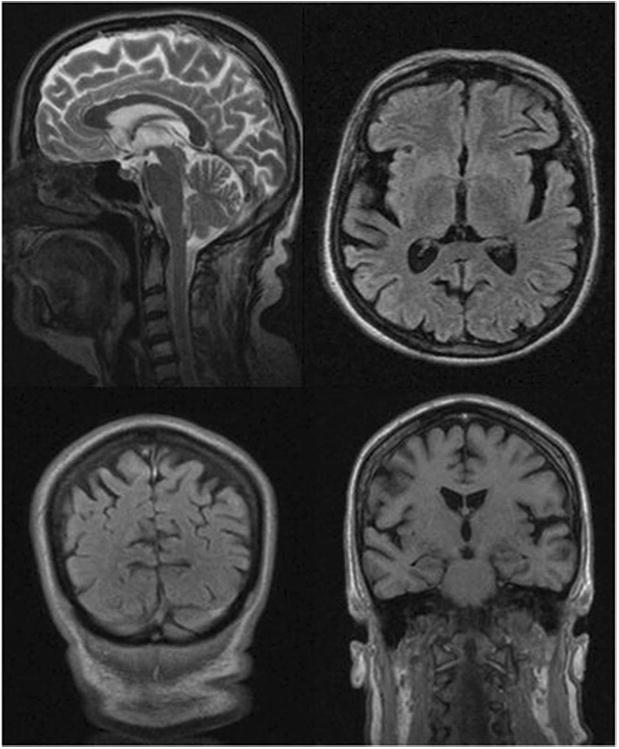
Head MRI of the proband demonstrates cortical atrophy with fronto-temporal predominance.
Figure 3.
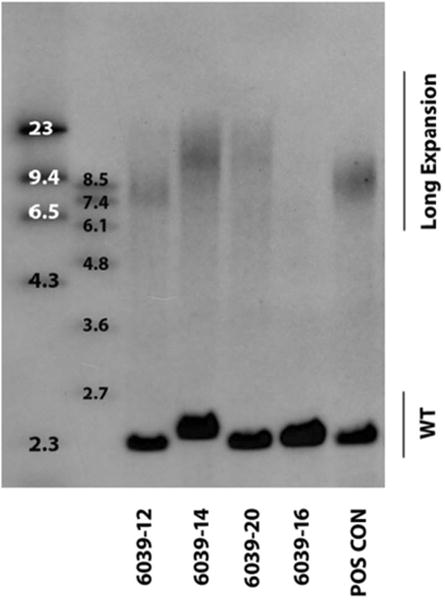
The C9ORF72 expansion sizes assessed in blood of the proband (6039-12: ∼900 repeat units [∼7.7 kb]), two unaffected mutation carriers (6039-14 and 6039-20: ∼1800 repeat units [∼13 kb]) and one non-carrier (6039-16: expansion not detected) displayed on a Southern blot. POS CON, positive control; WT, wild-type C9ORF72 allele.
The proband's sister (IV.1) was diagnosed with ALS at the age of 56 years, and presented with bulbar signs, which included dysarthria, dysphagia, tongue muscle atrophy and fasciculations of the tongue, which were validated by an electromyography study. Additionally, she had cervical spinal cord involvement with shoulder girdle muscle atrophy; the first dorsal interosseous muscle was also atrophied on the right side. No cognitive changes were reported, but she did not have a neuropsychological assessment. After three years, she developed mild parkinsonian signs, which included bradykinesia, rigidity, and impaired balance, which caused a gait disorder. Her upper motor neuron signs progressed and because of severe dysphagia, she developed malnutrition. She was given a percutaneous endoscopic gastrostomy (PEG) to assist with her nutrition. She died two years after PEG implantation from respiratory failure; she was 61-years-old. An autopsy was not performed. Neuroimaging studies revealed general cortical atrophy with no pathology in the cervical spine. Electromyography conducted when she was alive showed that she had chronic neurogenic changes in the muscles of her upper and lower extremities. No abnormalities were seen on her nerve conduction studies. The SOD1 mutation was previously excluded as a genetic cause of ALS in this subject. DNA samples for the present study were not available.
The proband's mother (III.3) first developed symptoms of ALS at the age of 53 years; she presented with dominant bulbar signs followed by general muscle atrophy. She died at the age of 58 years from respiratory failure. The proband's maternal aunt (III.4) developed early onset dementia at the age of 49 years with non-fluent aphasia and behavioral changes. No motor symptoms were reported throughout the disease course. She died at the age of 59 years of unknown cause. The proband's maternal grandmother (II.4) was diagnosed with early-onset dementia of an unknown etiology and died at the age of 59 years. Her mother, the proband's maternal great grandmother (I.2), had a movement disorder with severe gait disturbances and died aged approximately 60 years based on anecdotal information from the family. Information about those subjects was provided by family members. No medical history or charts were available for review. DNA samples from individuals from generations I, II and III were unavailable. No other relatives within the pedigree were known to have dementia, Parkinsonism, or a motor neuron disease.
Other family members who donated blood samples for this study and who agreed to undergo clinical evaluation presented at our institution without any subjective complaints. They did not have any clinically significant medical history. Four of them were carriers of the hexanucleotide repeat expansion in C9ORF72, and the estimated expansion size was approximately 1800 repeat units [∼13 kb] (Fig.3). The participants had a normal neurological examination, normal MMSE (28-30/30) and CDT (10/10) results, but the detailed neuropsychological evaluation revealed several disturbances. Attention deficit disorder was found in one of the mutation carriers. Mild frontal lobe dysfunction with mild language deficits presenting as verbal fluency were found in the remaining three mutation carriers. None of them had clinically significant memory deficits.
The only non-carrier of the hexanucleotide repeat expansion in C9ORF72 that was assessed was normal on the neurological examination and neuropsychological evaluation. The other family members were deceased or refused to undergo genetic testing.
Discussion
This is the first description of a familial ALS-FTD complex disorder associated with a C9ORF72 repeat expansion in the Polish population. We present a multigenerational pedigree with six affected subjects who originated from the Silesia region of Poland. Before genetic test results were available, several members of this family were clinically described as having a “neurodegenerative overlap syndrome” that consisted of Parkinsonism, dementia, and motor neuron disease; “neurodegenerative overlap syndrome” is a term that was first described by Uitti et al. [16]. They were considered to have an alternative manifestation of the same underlying neurodegenerative disease of an unknown etiology.
Indeed, it is now known due to the identification of a series of pathogenic gene mutations that FTD and ALS can be alternative manifestations of the same disease process [3, 18, 20]. The ALS-FTD complex disorder linked to chromosome 9 was first described by Morita et al. in 2006 [21], but in September 2011 a causative expansion in C9ORF72 was reported in two independent publications by DeJesus-Hernandez et al. and Renton et al. [15,17]. This causative mutation is a non-coding hexanucleotide repeat expansion within the promoter region or the first intron of alternatively spliced transcripts of C9ORF72. Healthy individuals generally carry up to 30 GGGGCC repeats; whereas affected individuals can carry hundreds to thousands repeats [15, 17, 19]. Affected individuals may express different phenotypes, including motor neuron disease, bvFTD, Parkinsonism, Alzheimer's-type dementia, and even psychosis [6, 22, 23].
In a recent Southern blot study, Van Blitterswijk et al. revealed that C9ORF72 repeat size is associated with age in the frontal cortex (both age at onset and age at collection) and with survival after disease onset in the cerebellum; however, no association was present in blood (e.g. with age, survival after onset, or disease phenotype) [19]. These findings are well in line with our present study because the C9ORF72 expansion size in the blood did not appear to be associated with the presence or absence of disease symptoms; the most abundant expansion size was clearly smaller in the proband with clinical symptoms (∼900 repeat units) than in unaffected family members carrying the expansion (∼1800 repeat units). These findings, therefore, further substantiate that repeat lengths assessed in blood might not be the most reliable predictors of disease phenotype or severity.
The age at onset and disease duration of C9ORF72 expansion carriers are highly variable. According to the literature, the average age at onset is 55 years, and the disease duration is 4.5 years [22, 23]. It is speculated that there is a tendency toward a younger age at onset and shorter disease duration in C9ORF72 expansion carriers [24, 25, 26]. In our family, each generation had an age at disease onset between 49-56 years without anticipation. Disease duration was 5-10 years depending on initial symptoms. Subjects who were initially diagnosed with ALS tended to die younger, and those with initial symptoms of dementia tended to have longer disease duration. In our family, all affected cases were women. In contrast, male predominance was reported in ALS-FTD caused by C9ORF72 expansions [12, 21, 22, 23].
Most families include patients with mixed phenotypes of ALS, FTD, or both. Initially, the clinical presentation could be “pure,” indicating one phenotype; however, during follow-up most cases have overlapping symptoms [22]. The diverse phenotype includes initial symptoms of Parkinsonism, motor neuron disease with bulbar onset, and dementia, which was also present in affected members of our family throughout all five generations.
L-dopa resistant Parkinsonism was previously described in about 50% of FTD and ALS-FTD patients [25, 26]. Parkinsonian features in cases with ALS-FTD that were caused by a mutation in C9ORF72 usually developed within the first years of the disease course [26]. Parkinsonism was predominantly associated with bvFTD, but was not described in patients with ALS [25, 26, 28]. In contrast, our proband primarily presented with Parkinsonism that initially responded to L-dopa, which was suggestive of Parkinson's disease. She had no typical bvFTD symptoms. However, during the disease course she manifested the signs and symptoms of motor neuron disease with predominant bulbar features. Behavioral changes characterized by pseudobulbar affect were also present. The proband's sister was initially diagnosed as having ALS with a primarily bulbar manifestation. She later developed Parkinsonism, which was unresponsive to L-dopa. No cognitive impairment was reported. In agreement with the previous research, our cases have also shown that Parkinsonism is often present in the clinical spectrum of ALS-FTD linked to chromosome 9. This should be taken into consideration, especially in parkinsonian patients with additional neurological features and a positive family history [29]. All other affected subjects in the family presented either with dementia or motor neuron disease without concomitant symptoms that were suggestive of a complex disorder. Their blood samples were not available for genetic testing, and they were not examined directly. The medical data were based on interviews with family members, and no medical records or charts were available for review.
Structural neuroimaging studies performed in clinically affected subjects have shown cortical atrophy that was predominantly localized in the fronto-temporal regions. Functional Tc-99m-SPECT studies performed in the proband revealed hypoperfusion in both fronto-temporal regions, but this was more profound on the left side. Our findings are consistent with those of large patient cohorts [6, 22, 25].
Other family members who turned out to be carriers of the expansion in C9ORF72 were over 50-years-old and had no subjective complaints when they presented to our institution for genetic testing. They had mild frontal lobe dysfunction without memory deficits; any deficits that they did have neither interfered with their daily activities, nor fulfilled the frontotemporal dementia diagnosis criteria. We know from previous C9ORF72 expansion carrier studies that behavioral changes and features of frontal lobe dysfunction may be subtle and uncharacteristic of fully developed frontotemporal dementia [23, 24, 26]. Genetic test results and neuropsychological evaluation results may identify affected family members; however, these individuals may be functioning normally. We did not receive enough subjective or objective complaints for a clinical diagnosis. It is still unknown, what determines whether someone who carries a C9ORF72 expansion develops ALS, FTD, or both. We plan neurological and neuropsychological follow-up of those subjects, which would allow confirmation of a clinical diagnosis in the early stages of the disease.
Finally, taking into consideration a positive family history, clinical symptoms, and genetic tests results, this family was diagnosed with an ALS-FTD complex disorder linked to chromosome 9. Our report highlights the need for genetic testing of all patients with a positive family history to avoid a misclassification of patients, even if the clinical symptoms are not recognized as part of a spectrum of the same disease. This approach will lead to appropriate genetic counseling for patients and their families.
International studies have already confirmed that a hexanucleotide repeat expansion in C9ORF72 is the most commonly known genetic cause of ALS, FTD, and the ALS-FTD complex disorder in people of European decent [17, 22, 26]. However these expansion carriers were also found to be present in other populations [28, 30, 31], which supports a global presence of the C9ORF72 hexanucleotide repeat expansion.
Our report confirms that this C9ORF72 mutation has a diverse clinical presentation among members of the same family and shows that it is also present in Central-Eastern Europe. Further studies are needed to assess the frequency of this C9ORF72 expansion in the Polish population with familial, as well as sporadic ALS, FTD, and the ALS-FTD complex disorder.
Acknowledgments
We would like to thank all those who have contributed to our research, particularly the patients and all family members who participated in the study. We would like also to thank Audrey J. Strongosky for a pedigree preparation and for logistic and administrative support in collecting material for this study, and Kelly E. Viola, ELS for editorial assistance.
Disclosures/Conflicts: JS is supported by the “Stowarzyszenie na Rzecz Rozwoju Neurologii Wieku Podeszlego.” MvB is supported by the Milton Safenowitz Post-Doctoral Fellowship for ALS research from the ALS Association. ZKW is partially supported by NIH/NINDS P50 NS072187, The Michael J. Fox Foundation for Parkinson's Research, and a gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. RR is supported by NIH/NINDS NS080882 and the ALS Therapy Alliance.
References
- 1.Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases: El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Mann DM, Northen B, Goulding PJ, Macdermott N. Frontal lobe dementia and motor neuron disease. J Neurol Neurosurg Psychiatry. 1990;53(1):23–32. doi: 10.1136/jnnp.53.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris HR, Waite AJ, Williams NM, Neal JW, Blake DJ. Recent advances in genetics of ALS-FTLD complex. Curr Neurol Neurosci Rep. 2012;12:243–250. doi: 10.1007/s11910-012-0268-5. [DOI] [PubMed] [Google Scholar]
- 4.Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O'Brien C, Phukan J, Wynne B, Bokde AL, Bradley DG, Pender N, Al-Chalabi A, Hardiman O. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11(3):232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 6.Kaivorinne AL, Bode MK, Paavola L, Tuominen H, Kallio M, Renton AE, Traynor BJ, Moilanen V, Remes AM. Clinical Characteristics of C9ORF72-linked Frontotemporal Lobar Degeneration. Dement Geriatr Cogn Disord Extra. 2013;3:251–262. doi: 10.1159/000351859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol. 2012 Dec;25(6):689–700. doi: 10.1097/WCO.0b013e32835a3efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ringholz GM, Appel SH, Bradshaw M, Cooke N, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 9.Wszolek ZK, Lynch T, Wilhelmsen KC. Rapidly progressive autosomal dominant parkinsonism and dementia with Pallido-Ponto-Nigral Degeneration (PPND) and Disinhibition-Dementia-Parkinsonism-Amyotrophy Complex (DDPAC) are clinically distinct conditions that are both linked to 17q21-22. Parkinsonism Relat Disord. 1997 Apr;3(2):67–76. doi: 10.1016/s1353-8020(97)00006-0. [DOI] [PubMed] [Google Scholar]
- 10.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol. 1997;41:706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 11.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutation in progranuline cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 12.Achi EY, Rudnicki A. ALS and frontotemporal dysfunction: a review. Neurol Res Int. 2012;2012:806306. doi: 10.1155/2012/806306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan X, Chen X. Clinic, neuropathology and molecular genetics of frontotemporal dementia: a mini-review. Transl Neurodegener. 2013;2:8–17. doi: 10.1186/2047-9158-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Pavani S, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 2013;8:28–43. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uitti RJ, Berry K, Yasuhara O, Eisen A, Feldman H, McGeer PL, Calne DB. Neurodegenerative ‘overlap’ syndrome: clinical and pathological features of Parkinson's disease, Motor Neuron Disease and Alzheimer's disease. Parkinsonism Relat Disord. 1995;1(1):21–34. doi: 10.1016/1353-8020(95)00004-p. [DOI] [PubMed] [Google Scholar]
- 17.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, ITALSGEN Consortium. Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Damme P, Robberecht W. Clinical implications of recent breakthroughs in amyotrophic lateral sclerosis. Curr Opin Neurol. 2013;26:466–472. doi: 10.1097/WCO.0b013e328364c063. [DOI] [PubMed] [Google Scholar]
- 19.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12:978–88. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conte A, Lattante S, Luigetti M, Del Grande A, Romano A, Marcaccio A, Marangi G, Rossini PM, Neri G, Zollino M, Sabatelli M. Classification of familial amyotrophic lateral sclerosis by family history: effects on frequency of genes mutation. J Neurol Neurosurg Psychiatry. 2012;83:1201–1203. doi: 10.1136/jnnp-2012-302897. [DOI] [PubMed] [Google Scholar]
- 21.Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH., Jr A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 22.Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L, Miller J, Lee Y, Troakes C, Scott KM, Jones A, Gray I, Wright J, Hortobágyi T, Al-Sarraj S, Rogelj B, Powell J, Lupton M, Lovestone S, Sapp PC, Weber M, Nestor PJ, Schelhaas HJ, Asbroek AA, Silani V, Gellera C, Taroni F, Ticozzi N, Van den Berg L, Veldink J, Van Damme P, Robberecht W, Shaw PJ, Kirby J, Pall H, Morrison KE, Morris A, de Belleroche J, Vianney de Jong JM, Baas F, Andersen PM, Landers J, Brown RH, Jr, Weale ME, Al-Chalabi A, Shaw CE. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur J Hum Genet. 2013;21:102–108. doi: 10.1038/ejhg.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Jr, Parisi JE, Lucas JA, Petersen RC, Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:768–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boeve BF, Graff-Radford NR. Cognitive and behavioral features of c9FTD/ALS. Alzheimers Res Ther. 2012;4:29. doi: 10.1186/alzrt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedland RP, Shah JJ, Farrer LA, Vardarajan B, Rebolledo-Mendez JD, Mok K, Hardy J. Behavioral variant frontotemporal lobar degeneration with amyotrophic lateral sclerosis with a chromosome 9p21 hexanucleotide repeat. Front Neurol. 2012;3:136. doi: 10.3389/fneur.2012.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savica R, Adeli A, Vemuri P, Knopman DS, Dejesus-Hernandez M, Rademakers R, Fields JA, Whitwell J, Jack CR, Lowe V, Petersen RC, Boeve BF. Characterization of a family with c9FTD/ALS associated with the GGGGCC repeat expansion in C9ORF72. Arch Neurol. 2012;69(9):1164–1169. doi: 10.1001/archneurol.2012.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruts M, Gijselinck I, Van Langenhove T, van der Zee J, Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):450–459. doi: 10.1016/j.tins.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 28.García-Redondo A, Dols-Icardo O, Rojas-García R, Esteban-Pérez J, Cordero-Vázquez P, Muñoz-Blanco JL, Catalina I, González-Muñoz M, Varona L, Sarasola E, Povedano M, Sevilla T, Guerrero A, Pardo J, López de Munain A, Márquez-Infante C, de Rivera FJ, Pastor P, Jericó I, de Arcaya AÁ, Mora JS, Clarimón J, C9ORF72 Spanish Study Group. Gonzalo-Martínez JF, Juárez-Rufián A, Atencia G, Jiménez-Bautista R, Morán Y, Mascías J, Hernández-Barral M, Kapetanovic S, García-Barcina M, Alcalá C, Vela A, Ramírez-Ramos C, Galán L, Pérez-Tur J, Quintáns B, Sobrido MJ, Fernández-Torrón R, Poza JJ, Gorostidi A, Paradas C, Villoslada P, Larrodé P, Capablo JL, Pascual-Calvet J, Goñi M, Morgado Y, Guitart M, Moreno-Laguna S, Rueda A, Martín-Estefanía C, Cemillán C, Blesa R, Lleó A. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mut. 2012;34:79–82. doi: 10.1002/humu.22211. [DOI] [PubMed] [Google Scholar]
- 29.Puschmann A. Monogenic Parkinson's disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19:407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 30.Dobson-Stone C, Hallupp M, Loy CT, Thompson EM, Haan E, Sue CM, Panegyres PK, Razquin C, Seijo-Martínez M, Rene R, Gascon J, Campdelacreu J, Schmoll B, Volk AE, Brooks WS, Schofield PR, Pastor P, Kwok JB. C9ORF72 repeat expansion in Australian and Spanish frontotemporal dementia patients. PLoSONE. 2013;8(2):e56899. doi: 10.1371/journal.pone.0056899. doi:10.1371/ journal.pone.0056899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai CP, Soong BW, Tu PH, Lin KP, Fuh JL, Tsai PC, Lu YC, Lee IH, Lee YC. A hexanucleotide repeat expansion in C9ORF72 causes familial and sporadic ALS in Taiwan. Neurobiol Aging. 2012;33:2232.e11–2232.e18. doi: 10.1016/j.neurobiolaging.2012.05.002. [DOI] [PubMed] [Google Scholar]