

Abstract
The main objective of this review is to provide an appraisal of the current status of the relationship between energy intake and the life span of animals. The concept, that a reduction in food intake, or caloric restriction (CR), retards the aging process, delays the age-associated decline in physiological fitness and extends the life span of organisms of diverse phylogenetic groups, is one of the leading paradigms in gerontology. However, emerging evidence disputes some of the primary tenets of this conception. One disparity is that the CR-related increase in longevity is not universal and may not even be shared among different strains of the same species. A further misgiving is that the control animals, fed ad-libitum (AL), become overweight, prone to early onset of diseases and death, and thus may not be the ideal control animals for studies concerned with comparisons of longevity. Re-examination of body weight and longevity data from a study involving over 60,000 mice and rats, conducted by a National Institute on Aging-sponsored project, suggests that CR-related increase in life span of specific genotypes is directly related to the gain in body weight under the AL feeding regimen. Additionally, CR in mammals and “dietary restriction” in organisms, such as Drosophila, are dissimilar phenomena, albeit they are often presented to be the very same. The latter involves a reduction in yeast rather than caloric intake, which is inconsistent with the notion of a common, conserved mechanism of CR action in different species. Although specific mechanisms by which CR affects longevity are not well understood, existing evidence supports the view that CR increases the life span of those particular genotypes that develop energy imbalance due to AL feeding. In such groups, CR lowers body temperature, rate of metabolism and oxidant production, and retards the age-related pro-oxidizing shift in the redox state.
Keywords: Dietary restriction and aging, energy restriction and life span, oxidative stress and aging, free radicals and aging, mechanisms of aging, redox state and aging, caloric restriction and life span, redox stress hypothesis of aging
Introduction
The notion that caloric restriction (CR), or the curtailment of energy (food) intake without causing under-nutrition, slows the rate of aging, prolongs the duration of youthfulness, postpones the onset of age-associated pathologies and extends the life spans of animals of diverse phylogenies, has been a leading concept in gerontology for several decades [1–5]. It is often contended that because of the ubiquity of the association between CR and increase in life span, the mode of CR action may be an evolutionarily conserved “public” mechanism that modulates the intrinsic rate of aging [5–7]. The conviction that CR has an “anti-aging” effect, or that it is an antidote to the aging process, has indeed gained wide popularity. Nevertheless, there also exists a body of data that is inconsistent with some of the generalizations of the classic CR postulate, especially its universality in the prolongation of life span. It is also presently uncertain how the optimal level of food intake should be established for different genotypes. Additionally, there are qualms about the soundness of ad-libitum (AL) feeding of the control animals, since AL feeding frequently results in overconsumption of energy and consequently an excessive gain of weight. Thus the main objective of this review is to scrutinize the cogency of the CR concept in context of the discordant findings and to discuss whether a revised interpretation of this phenomenon is indeed warranted.
The idea, that the amount of food intake and the resulting rate of growth may affect the potential longevity of animals, seems to have emerged in the early twentieth century, around when Osborne and co-workers [8] reported that among a cohort of female rats, the individuals displaying retarded growth and delayed sexual maturity lived longer than those that grew relatively rapidly. The notion implicit in this observation, was later tested experimentally by McCay et al. [9, 10], using a “stair-step” method of feeding, in which the amount of food provided to the rats was adjusted to hold their body weight at a nearly steady level and upon signs of physical distress enough additional food was provided to obtain a growth rate of 10 g body weight/ 2–3 month. The life span of male but not female rats kept under the restricted feeding regime was found to be longer than those fed AL, which was interpreted to suggest that the “retardation of growth by diets, complete except for calories, affords a means of producing very old animals for studying aging” [9]. The authors also made two additional observations/comments that seem pertinent to the present discourse, namely --- (i) “it is recognized that the group ingesting food ad-libitum, may be subject to injury by the excess above the requirements of the body” [9], and (ii) in the aftermath of a failure of the laboratory heating system -- “animals that were well fed could withstand the drastic drop in temperature of the room while the retarded ones that were being kept at maintenance level had little reserve and part of them perished” [10]. Thus, the potential deleteriousness of excessive energy intake by the AL fed control animals, and the frailty of the CR animals, were both recognized at the advent of the explorations into the effects of CR on life span.
A later study by Ross [11] showed that rats fed a high protein/high carbohydrate AL diet had lower mortality rates at younger ages than those fed a nutrient-poor, low protein/low carbohydrate AL diet; however, in the latter part of life, mortality rates were lower in animals fed nutrient-poor than those given nutrient-rich diets, implying that relatively high energy intake is more optimal for development and reproduction, but less so in protection against age-related disease and shortened longevity. In the same report, Ross discussed another issue of lasting significance and controversy, namely, whether CR increases the maximum life span beyond the biologically established species-specific limit? He reasoned that while the potential life span of AL and CR animals is essentially identical at the time when the animals are placed under either of these two feeding regimens, subsequently the CR population attains relatively longer life span because their nutrition is optimal, whereas the potential longevity of the AL fed animals is cut short due to the deleteriousness of overfeeding. Ross was particularly skeptical of the frequently used phrase -- “extension of life span” to express CR-related increase in longevity, which he suggested is often interpreted to mean an increase in longevity beyond the maximal biological limit or the demographic norm, when in reality longevity of CR animals has not specifically been shown to exceed the biological maximum of the species. To summarize, by the mid-twentieth century it was well recognized that AL feeding may lead to over-eating and shortened life expectancy, and that there exists a certain level of antagonism between the amount of energy intake needed for maximal growth and fitness and the amount that promotes longer survival [12].
A key feature of the classic CR paradigm, that has also heightened its attractiveness, is the belief that longevity prolongation by CR is universal among animals. Indeed, the assumption of universality is considered to be central to the validity of the classic CR paradigm, because the applicability/relevance of CR to human longevity is critically dependent upon the argument that the life-lengthening effects of CR are evolutionarily conserved and any deviation from this principle would breach that rationale [13]. Swindell [14] has discussed several examples in the literature where the effects of CR on longevity do not uphold the assertion of universality. Particularly, studies by Liao et al. [15] on several different strains of mice have demonstrated that under 40% CR, longevity of individual strains is either decreased, increased or remains unaffected, thereby, suggesting that the nature of the CR effect on life span is genotype-specific.
Another misgiving about the classic CR model is that the “control” animals are usually fed AL, which frequently leads to over-eating and excessive gain of body weight (Fig. 1). For instance, following the implementation of CR during adulthood (usually at 3–4 months of age), the body weights of the CR mice/rats remain relatively stable throughout life except towards the terminal phase during which there is a modest decline; in contrast, the peak body weights of AL fed animals often more than double compared to those at 3–4 months of age. [16–19]. A key point to note here is that the amount of energy intake by the AL fed animals often significantly exceeds the amount expended, resulting in a substantial gain in body weight, or positive energy balance, which is often associated with an early onset of disease conditions [2, 20]. Although in some studies the amount of food fed to the controls is reduced by ~ 10% from the AL level, energy imbalance still persists [2, 21, 22]. Consequently, it remains doubtful whether the AL fed over-weight rats or mice are meaningful controls, especially in studies dealing with longevity [23].
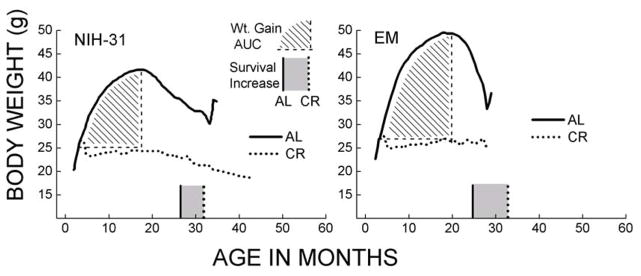
Fig. 1.

Comparison of body weights between ad-libitum fed (AL) and calorically restricted (CR) male C57BL/6 mice from 3–23 months of age. The AL mice were maintained on NIH-31 diet while the CR mice were fed a NIH-31 special diet that compensated for the nutrients. CR (40%) was initiated at ~ 4 months of age and the body weights were recorded at 3-week intervals. The average body weight of CR mice remained relatively stable, whereas it increased by ~ 100% at its peak in the AL fed mice (Adapted from [151]).
In this context, the main objective of this review is to examine the tenability of the classic CR paradigm in the context of the evidence that is apparently discordant with some of its main tenets. Additionally, we discuss the nature of the possible mechanisms that may underlie the CR-related effect on life span. Readers seeking diverse perspectives about the nature of the association between CR and longevity are directed to: [1–3, 6, 14, 24–30].
Variations in the effect of caloric restriction in different genotypes
Although the reports confirming the positive effects of CR on longevity far exceed those that do not, this lop-sided imbalance could be due to a disinclination to document discordant data. Notwithstanding, the literature points to a wide spectrum of CR effect on longevity-- ranging from strongly positive to the negative [14, 15, 31–37]. For instance, meta-analysis of a grand total of 246 studies on mice and rats, spanning from 1934–2012, showed that in half of all experiments on rats, CR-related increase in life span (pooled male and female data) ranged between 13.8– 45.4%, with one quarter showing < 13.8% and one quarter > 45.4% increase [14]. In mice, the corresponding increases were: one half, 4.1–27.0%, one quarter, < 4.1% and one quarter > 27.0%; however, the average longevity-extension by CR per genotype was only 2–3% when the recent study of recombinant inbred strains by Liao et al. [15] was included. In mice, CR related increase in life span was greater in the non-inbred than in the inbred strains. More relevantly, in some genotypes of mice and to a lesser extent of rats, CR elicited a negative or a weak response ([14] and references therein). In their seminal paper, McCay et al. [9] had also noted variability in CR-responsiveness, as CR-related increase in life span was observed only in restricted male but not female rats. The DBA/2 mice [32, 38–40], strains of ILSXISS recombinant inbred mice [15], rhesus monkeys [41] and offspring of wild-caught mice reared in the laboratory [33] have all been reported to be resistant to the longevity extension effect of CR. For instance, a comparison among 41 ILSXISS recombinant inbred strains revealed that a majority of the strains displayed no prolongation of life span under 40% CR. Longevity of male and female mice was extended, respectively, in only 5% and 21% of the strains, whereas, it was shortened by CR in 27% of the strains for males and 26% for the females [15]. Notably, the longest life spans under CR did not significantly surpass the longest under the AL regime, and the average of the mean life spans of the five longest-lived strains under CR did not exceed that of the corresponding strains, fed AL. Thus, in addition to the demonstration that longevity-extension by CR is not universal across genotypes, the latter results also challenge the view that CR, when effective, extends maximum life beyond the norm. Another meta-analysis of 145 studies involving 36 species also indicated a significant heterogeneity in the effect of CR on longevity [37]. One potentially significant inference was that the longevity-extending effect of CR is “only evident when animals have been housed in laboratory conditions for a number of generations”. A similar conclusion was reached previously by Harper et al. [33], who investigated whether 40% CR elicited a similar type of effect on the longevity of the grand-offspring of the wild caught mice as it does in the laboratory adapted mice. Caloric restriction was found to have little effect on the average life span of the wild-derived mice.
A fundamental dichotomy seems to exist between rodents and insects in the nature of the response to CR. In general, a decrease in the amount of caloric intake may shorten rather than prolong the life span in flies [31, 36, 42]; however, a positive effect on survival may be obtained by decreasing the yeast (protein): carbohydrate ratio of the diet [36, 43]. In initial studies, nutrient restriction in Drosophila used to be imposed by the dilution of the food mixture [7, 44], which tended to increase life span in comparison to those fed the more concentrated medium [45–49]. However, the validity of the dilution-based regimen was challenged by Benzer’s group [44], who showed that the increase in life span ascribed to food dilution was abolished if the flies, fed the relatively concentrated medium, were offered ad-libitum access to water, which suggested that longevity extension by food dilution in insects was due to a hydration rather than a caloric restriction effect (also see, a confirmation of this phenomenon by Dick et al. [50] and a rebuttal by Piper et al. [51]). To compensate for the dilution, flies also tend to gulp relatively larger volumes of food. Furthermore, low yeast: carbohydrate ratio rather than the amount of food intake was identified to be the critical factor in the modulation of longevity in Drosophila [44] and other species of fruit flies [52, 53].
Accordingly, in more recent studies, variations in yeast: sucrose ratios are employed to affect “dietary restriction” (DR) in fruit flies. Dipteran flies primarily use carbohydrates as the preferred fuel to generate energy; e.g., the respiratory quotient of D. melanogaster during flight is 1.0, suggesting that carbohydrates constitute the only source of energy during flight [54, 55]. While dietary protein is essential in females for egg production, the male flies require relatively little or no protein during adulthood [52, 56]. However, ingestion of proteins above a certain threshold level, especially by males, has a detrimental/toxic effect on longevity and fitness, suggesting that flies have poor tolerance for over-consumption of proteins [57]. In females, diets with relatively high yeast: carbohydrate ratios tend to increase egg production, whereas those with high carbohydrate:yeast ratios result in longer life span [52, 58]. Analyses of mortality indicate that increase in longevity of yeast restricted flies is associated with a decrease in age-independent mortality, reflected by a lower y-intercept in the Gompertz plot, whereas the slope of the plot, considered to be reflective of the rate of aging, is not affected [46, 59]. Interestingly, it has been reported recently that longevity of AL fed mice also is prolonged by a diet that is low in protein and high in carbohydrate content [42, 60, 61]. Dilution of the food with indigestible cellulose, which affected a 30% decrease in caloric intake, had no salutary effect on life span. The authors’ main contention was that longevity extension of AL fed animals is achieved by alterations in the ratios of the macronutrients rather than the amount of calories.
Unlike in Drosophila and related fruit flies, under caged conditions, food and water for caged adult houseflies are provided separate containers and thus the amount of food consumed can be measured directly by weight [31]. To identify macronutrients that promoted the longest survival, male houseflies were fed seven different diets, namely (i) sucrose alone, (ii) glucose alone, (iii) fructose alone, (iv) trehalose alone, (v) a mixture of sucrose, powdered skim milk and powdered whole egg in 6:6:1 ratio, normally used for the breeder cages, (vi) a mixture of sucrose, powered skim milk, powdered whole egg and powdered whey protein in 6:6:1:6 ratio, and (vii) a mixture of 80% sucrose, 17% powdered skim milk and 3% powdered whole egg--- proportions of the ingredients in this diet were based on the amounts consumed by the flies when each ingredient was offered simultaneously in a separate dish. Results showed that 100% sucrose diet promoted the longest life span, and the presence of any protein in the diet consistently decreased longevity. Furthermore, the foods preferred by the flies (diet vii) were not necessarily those that maximized their longevity. Reduction in sucrose consumption by as little as 10% from the AL fed level decreased the life span of male houseflies [31, 35].
To summarize, studies in the male housefly clearly demonstrated that the longest life span was achieved by the carbohydrate only diet, fed AL. Decrease in energy intake below the AL level or inclusion of protein in the diet reduced longevity. In fruit flies, excessive protein intake, as well as caloric restriction, also decreases longevity [36, 43]. Although the longer survival, resulting from a reduction in the amount of yeast ingestion, has been proffered to constitute a phenomenon akin to CR, a shortcoming of this contention is that the deleterious effects of excessive yeast intake persist even in iso-caloric diets. Thus, DR in flies fundamentally differs from CR in mammals as it involves reduction in yeast (presumably protein) and not energy intake. Therefore, the term DR in reference to yeast restriction is quite misleading, as the designations, DR and CR, have historically been used interchangeably to refer to a reduction in energy intake. Accordingly, we suggest that restrictions of yeast, methionine or any other nutrient should be termed specifically and not grouped with a phenomenon that is fundamentally apart.
Characteristics of CR-responsive versus CR-unresponsive genotypes
Perhaps, because the longevity effect of CR was until recently considered to be a universal phenomenon, little effort was invested in the elucidation of the differences between genotypes whose life spans are increased by CR and those where CR has a negative effect or no effect. Arguably, such comparisons should facilitate the identification of genetic/environmental factors that determine the nature of the effect of CR on life span. One such factor may be that the AL-catered phenotype is a product of adaptations to the laboratory setting which abet excessive food consumption and limit physical activity. Biological effects of such a lifestyle include an increase in body mass, a decrease in the age of fertility and a more prolific litter size [62, 63]. For instance, under the AL regimen, body mass of the CR-responsive mice and rats often doubles during adulthood [16–18] (see also Figs I -3). A comparison between the progeny of recently caught wild mice and a laboratory strain showed that during the first 4 months of life, the latter gained 70% more body mass than the former [64]. Another comparison indicated that, under similar conditions, Sprague-Dawley and Long-Evans rats became 40–60% heavier, reached the first oestrus and the first conception earlier and had larger litter sizes than the captive-reared wild Norway rats [63]. According to Martin et al. [65], failure to recognize that “control rats and mice used in biomedical research are sedentary, obese, glucose intolerant and on a trajectory to premature death, may confound data interpretation and outcomes of human studies”. Over-eating and the resultant energy imbalance are known to enhance oxidative stress, inflammation, diabetes, kidney disease and cancer, among others, leading to an abridgement of potential life span [2, 6, 22, 65, 66]. It may thus be questionable whether the AL fed, over-weight animals are the most suitable controls for studies exploring the relationship between energy intake and longevity. The fact that 20–40% reduction in food intake from the AL levels delays the onset of morbidity [2] would also seem to argue that the animals fed an AL regimen fall short of the standard of a healthy control animal.
It has long been known that unlike most other laboratory strains of mice, the DBA/2 have either a relatively low tolerance for CR or they are unresponsive to it [38–40]. More recently, Forster et al. [32] observed that gradual imposition of 40% CR, starting at 14 weeks of age, increased the average life span of C57BL/6 and B6D2F1 mice by 25% or more, but had no effect in the DBA/2. This result was in contrast to that reported earlier by Bronson and Lipman [67] who found an 18% increase in median life span of male DBA/2 mice maintained under 40% CR. However, the data used in their report and supplied to them by the custodian of the mice, National Center for Toxicological Research (NCTR), was from a study yet to be completed. Upon completion, the CR-associated increase in median longevity (Fig. 2) was found to be only 5% [16]. Besides the atypical response to CR, the DBA/2 mice also exhibit a number of divergent behavioral, neural, immunological and metabolic phenotypes [68–72].
Fig. 2.

Relationship between body weights and longevity in different strains of mice and rats maintained under AL or 40% CR regimens (original data from NCTR/NIA BP program; digitized from figures in [16]). Cumulative weight gain under the AL regimen was calculated as area under the curve (AUC; indicated by the hatched area). AUC is the gain in body weight starting at three months of age until the peak of the body weight is reached. The rationale for quantifying weight gain in this manner is based on the hypothesis that the deleterious effect of energy imbalance should be proportional to both the amount of weight gained and the period of time during which the overweight condition was maintained. The median life spans of the AL and CR groups are indicated by the solid and dotted vertical lines along the abscissa in each panel. The grey area between the two lines indicates the increase in life span attributable to CR. These data demonstrate that different strains of mice and rats significantly vary in the AUC of weight gained under the AL feeding regimen and the increases in life span under CR.
Nevertheless, comparisons between the CR-responsive, C57BL/6 mice, and the CR-unresponsive, DBA/2 mice, have pointed to the role of energy imbalance in determining the nature of the effect of CR on longevity [32, 73–75]. A major difference between the C57BL/6 and DBA/2 mice was that during 4 to 23 months of age, the AL fed C57BL/6 mice maintained a mean body weight that was 20% greater than their weight at 4.5 months of age, whereas the comparable gain in body weight was insignificant in the DBA/2 mice. The DBA/2 mice also had a higher rate of energy expenditure than the C57BL/6 mice, as indicated by resting and in vitro rates of oxygen consumption and the core body temperature [32, 73–75]. Thus, while the amount of energy (food) consumption was similar in the two strains, the C57BL/6 mice, whose longevity is extended by CR, had a relatively lower metabolic rate and gained more weight with age, whereas the DBA/2 mice had a higher metabolic rate, did not exhibit a significant gain in the mean body mass during adult life, and their longevity was not extended by CR. These associations support the hypothesis that CR-induced increase in life span occurs in those genotypes that display weight gain or positive energy balance, when fed AL. Rikke et al. [76] have also noted that fuel efficiency, determined as the ability to maintain growth and body weight, was correlated with longevity of their inbred mice, a finding that is compatible with our analysis.
To test the predictions of this hypothesis, we re-examined the data on the body weights and life spans from a study of over 60,000 mice and rats, belonging to seven commonly studied genotypes, including inbred and F1 hybrid mice, such as C57BL/6 (B6), DBA/2, B6D2F1, and B6C3F1 (B6 x C3H FI), and rats, such as Brown Norway (BN), Fischer 344 (F344), and F344 x BN F1 [16]. These data were gathered during a 10-year study, jointly sponsored by the National Institute on Aging (NIA), Biomarkers of Aging Program (BAP) and the National Center for Toxicological Research (NCTR). The main objective of the project was to identify “biomarkers/indicators of biological age” to evaluate the efficacy of anti-aging interventions [77–80]. The broad conclusion drawn was that “the utility of CR in extending the life span ubiquitously lends further weight to the use of CR as a ‘gold standard’ for evaluating and stimulating new intervention strategies in aging” [16]. One feature of the NIA/NCTR/BAP dataset was that weight gain during adulthood markedly varied among different strains of mice and rats. Further analysis indicated that CR related increases in longevities of different strains were directly related to the level of body weight gained under the AL regimen (Fig. 2). For instance, the genotypes exhibiting relatively low cumulative weight gain under the AL regimen showed a correspondingly small CR-related increase in life span, whereas those with larger weight gains showed much greater CR-related benefit (Fig. 2). While these observations might be interpreted to suggest that CR has a relatively greater effect on longevity in the relatively long-lived genotypes such as the F1 hybrid mice and rats, additional findings of the project indicated that, rather, the weight gain is the critical variable. When C57BL/6 mice were fed the high fat/high protein Emory-Morse 911 (EM) breeding diet, instead of the standard NIH-31 diet, they gained considerably more body weight with age under the AL condition (Fig. 3), and also exhibited the largest CR-related increase in longevity among all of the rodent genotypes used in the NIA/NCTR/BAP program [16]. Such a potent effect of the EM-diet within the same genotype provides compelling support for the hypothesis that weight gain under the AL feeding regime is a critical predictor of longevity extension by CR.
Fig 3.
Effect of diets with dissimilar energy content on body weight and CR- related increase in longevity in C57BL/6 mice. Mice were fed NIH-31 diet (left) or a high fat/protein (EM) diet [16]. The cumulative weight gain in the AL fed groups was calculated as the area under the curve (hatched areas), which represents the increase in body weight from 3 months of age until the peak body weight was reached. The median age of survival for the AL and CR groups is indicated respectively as vertical lines along the x-axis in each panel, with CR associated longevity extension represented by the shaded area. Note that the mice fed EM diet ad-libitum gained relatively greater body weight and also showed a greater increase in longevity under the CR regimen.
The association between energy imbalance and the effect of CR on longevity, summarized in Fig. 4, is also compatible with findings in rhesus monkeys, reported from the Wisconsin National Primate Center (WNPC) [81] and the NIH [41]. Caloric restriction was reported to decrease mortality from age-related [81] as well as aggregate causes at the WNPC [13], whereas, no significant differences in the life spans of control and CR groups were observed at the NIH site. However, a crucial variation in the experimental design at the two sites was that the control monkeys at NIH were fed a diet that did not permit excessive weight gain whereas the AL fed controls in the WNPC study ate more liberally. Consequently, the body weights of the age-matched control groups at WNPC were greater than those in the NIH study, suggesting that a comparatively higher degree of energy imbalance is associated with longevity extension under CR. Comparisons of the body weights of the control groups at these two sites with those available from a national database, indicated that control monkeys at WNPC were approximately 7–10% heavier than the national average, whereas controls in the NIH study were 9–17% below the average [13]. Authors at both sites seem to agree that the relatively higher body weight of the AL fed monkeys at WNPC could account for the apparent disparity [13, 41]. In our view, there may be no conceptual inconsistency in the findings of the two groups. Both confirm that the increase in life span assigned to CR is dependent upon energy imbalance (gain in body weight) in the AL animals, as hypothesized here.
Fig. 4.
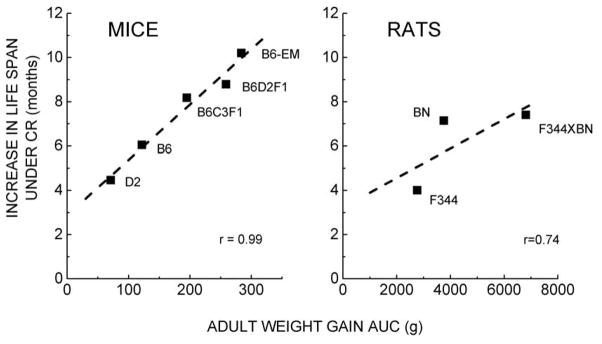
Increase in the maximum life span (10% survival) of different strains of mice (left) and rats (right) that is attributable to 40% CR. Values were calculated from the NIA BAP/NCTR data [16], as described in the legends of Figs. 2 and 3. The data suggest a direct relationship between the weight gain under the AL feeding regimen and the increase in longevity resulting from 40% CR.
While the hypothesis that CR increases longevity by preventing excessive body mass and adiposity has received some attention [82–85], it has historically been downplayed in the rodent biogerontology literature [19, 86–88], partly based on the results of a study by Harrison et al. [87], in which they compared the effect of CR on longevity in genetically obese (ob/ob) and normal C57BL/6 mice. The obese mice were fed AL or placed on a CR regimen that maintained their weight at a level equivalent to the non-obese mice. An additional group of normal mice was fed approximately 33% less food than their AL fed counterparts. Compared to the AL fed normal mice, the food- restricted ob/ob mice had a higher percentage of body fat, yet the life span of the two groups was nearly equal. This outcome was widely interpreted to mean that a reduction in body weight/adiposity should be ruled out as a mechanism for the CR-related increase in longevity. However, this explanation ignores a rather crucial observation in the study, namely that the cumulative weight gain in ob/ob mice under the AL regimen was ~ 4-fold greater than in the normal mice (Fig. 5). Thus, an alternative interpretation of the results of the study by Harrison et al. would be that CR was more effective in lengthening the life span of ob/ob mice than the normal mice because it attenuated their energy imbalance and the consequent deleterious effects of fat accumulation to a relatively much greater extent [66, 82, 89–98]. This explanation accords with the data shown in Figs. 2–4.
Fig. 5.
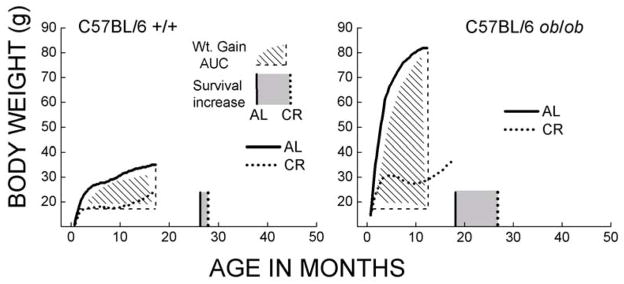
Body weight and longevity extension by caloric restriction in the female C57BL/6 ob/ob congenic and normal mice (re-plotted from [87]). The shaded area represents the cumulative weight gain under AL feeding starting at 1 months of age until the peak body weight was reached. Median age of survival under AL and CR conditions is shown, respectively, as solid and dotted vertical lines along the x-axis. The grey area in between these two lines represents the increase in longevity resulting from CR. Note that increases in body weight under AL feeding as well as the CR-related increase in longevity were much greater in the ob/ob than in the normal mice. This pattern is similar to that shown in Figures 2–4.
Pitfalls of uniform percent-based CR
While the data from the NCTR/BAP project are compatible with the interpretation that positive energy balance associated with AL feeding shortens potential longevity in the laboratory rodents, they also unveil a major weakness in the design of the conventional CR regimen, namely, that the imposition of a fixed percentage-based (usually 30–40%) reduction in the amount of energy intake for all genotypes may be a flawed approach because it ignores variations in metabolic rate, appetite, energy requirements or energy balance. As shown in Fig. 2, imposition of 40% CR in the DBA/2 mice resulted in a prolonged period of weight loss [32, 73, 75], whereas in other mouse and rat genotypes, it only caused a transient negative energy balance, followed by a long period of neutral (F344 rats, C57BL/6 mice) or positive energy balance (B6D2F1, BN, F344 x BN F1) (Figs. 1,2). Consequently, effects of CR on longevity may be obscured in some genotypes when a fixed level of restriction is imposed.
Indeed, complications associated with the percent-based CR regimen have been highlighted by the findings of Liao et al. [15], who showed that the effects of 40% CR were very dissimilar in different mouse genotypes. Some investigators [99] have been dismissive of these results because: (i) The sample size per strain and diet was quite small (fewer than 15), thereby weakening the reliability/accuracy of the estimates of median and maximum life spans. It would therefore be desirable to repeat the life span studies on selected strains using larger sample sizes. (ii) It is quite possible that a fixed level of CR was too severe for certain strains, which would result in under-nutrition [89, 99]. Such criticisms, while valid, also underscore the need to define a satisfactory standard for CR that obviates under-nutrition and can also be employed in studies involving comparisons among genotypes that vary in size, body composition, metabolic rate, energy intake or energy balance. While studies by Liao et al. as well as others suggest the exciting prospect that quantitative genetic approaches may facilitate discovery of loci responsible for the CR-related longevity effect [100, 101], ultimately leading to the identification of the underlying causal mechanisms, any such expectations may have to be tempered until the optimal levels of food intake, that result in the genotype-specific maximal longevity are better defined [25]. Although there is presently no generally accepted criterion for determining the optimal level of CR in rodents, it has long been recognized in the field of animal husbandry, especially poultry science, that the ratio between the quantity of food eaten and the amount of gain in body weight varies among members of a cohort and that AL food intake exacerbates morbidity/mortality [91, 102–105]. Conversely, the loss in body weight following CR also varies among individual mice due to variations in resting metabolic rate and physical activity [106].
Complications arising from the variability in responses to percent-based CR in different genotypes [106] may be avoided by the adoption of a feeding regimen that aims to maintain an optimal body weight during adulthood. Indeed, other authors have also lamented that while food intake of laboratory-confined non-rodent species is routinely restricted as a healthy veterinary practice, the rodents have been exempted perhaps because AL feeding is less laborious [107, 108]. The feeding regimen directed towards the maintenance of a neutral energy balance or a stable body weight, referred to here as “weight-stable caloric restriction” (WSCR), is analogous to an individually tailored weight management regime. In practice, it would involve daily administration of a fixed amount of food, with weekly adjustments for each individual to prevent weight gain above that reached during early adulthood [41, 109–114]. Figure 6 demonstrates the feasibility of WSCR in Sprague-Dawley rats maintained in the co-authors’ (MJF) laboratory for 18 months. The WSCR regimen is widely practiced in neuroscience and behavioral pharmacology laboratories, where food is used as a reward for the performance of specific tasks in a chronic setting (e.g., [115–117]). The WSCR regime would seem to be particularly appropriate in studies on the effects of gene deletion or over-expression, on longevity and health-span, because it would avoid the complications arising from variations in energy balance. Finally, the WSCR regimen should identify the genotypes in which 40% or any other percent-based CR is too much or too little in terms of achieving the longest potential life span.
Fig. 6.

Mean body weights of Sprague Dawley rats fed ad-libitum (AL) or maintained under a modified CR regimen (weight stable caloric restriction; WSCR). The amount of food in the latter group was adjusted weekly to sustain a relatively stable body weight. The target weight was 85% of the weight recorded at 2.5 months. Results demonstrate the feasibility of a CR regimen that aims to maintain a neutral energy balance. The body weight data for the AL fed rats are from Novelli et al. [264].
Mechanisms linking energy imbalance and longevity
Numerous hypotheses have been proposed to explain the biological basis of how CR prolongs the life span of responsive genotypes. The most frequently listed include: the retardation of growth, reduction of body fat, attenuation/postponement of immunologic and hormonal changes, enhanced damage/repair capacity, changes in gene expression, enhanced autophagy and apoptosis, alterations in IGF/insulin/TOR signaling, hormesis, activation of sirtuins, decrease in body temperature/metabolic rate, and attenuation of reactive oxygen species (ROS) generation/oxidative stress, among others (reviewed in [2–5, 26, 28, 30, 118–122]). Nevertheless, the central issue, namely, how CR counteracts the deleterious effects of AL feeding remains controversial. One current school of thought is that the nutrient sensing signaling pathways (mTOR/insulin) mediate the CR effect on longevity by enhancing the activity of genes encoding protective and repair functions, which slows the aging process and lengthens the duration of survival [5, 25, 26, 123]. This hypothesis, if indeed correct, seems to implicitly presuppose that AL feeding results in the generation of deleterious agents that are counteracted by the CR- induced enhancement of protective/repair mechanisms. Hormesis, a concept that the protective responses evoked by low intensity stress/damage, or intermittent moderate stress, lead to a state of physiological invigoration [124, 125], also appears to implicate the causal participation of deleterious factors in the aging process. Indeed, CR-related increase in life span would be difficult to explain without the putative involvement of molecular damage, as the age-related losses in functional capacity are unlikely to be self-initiating [126].
There are several lines of evidence suggesting that the in vivo source of damage in relation to CR, and more broadly the aging process, may be oxygen metabolism and the associated generation of ROS. It is a well-recognized phenomenon that scarcity of food ubiquitously leads to a hypo-metabolic state in the homeotherms, whereby body temperature and metabolic rate are depressed [127–129]. For instance, in C57BL/6 mice, 40% CR lowered the daily mean daytime colonic body temperature by 0.9 to 1.2 °C (Fig. 7A) [73]. Decreases in core body temperature in response to CR have also been observed in several other laboratories [130–134]. Lower core body temperature has been shown to be a significant predictor of survival in both humans and non-human primates [135]. In C57BL/6 mice, a decrease of as little as 0.3–0.5 °C in body temperature has been experimentally demonstrated to increase the life span by 12–20% [136, 137]. The reduction in core temperature in these studies was induced by transgenic over-expression of uncoupling protein 2 in hypocretin neurons, which act as a central thermostat. It is a recognized physiological precept that ~ 95% of the energy generated in the body emanates from oxygen-linked decomposition of organic molecules and that the basal heat production and body temperature reflect the intensity of cellular metabolism [138–140]. A comparison of the resting rate of oxygen consumption between AL fed and CR C57BL/6 mice, following 2 and 19 months of 40% CR, showed that at both of these ages food restriction lowered the rate of oxygen consumption, normalized as per mouse, per unit body weight, lean body mass (Fig. 7B) and organ weight, implying that the effect of CR on metabolic rate is long-lasting and not transitory [73, 75]. Such demonstrations support the view, originally advanced by Sacher [141], that CR-induced decrease in metabolic rate/body temperature may be a factor in the resultant increase in longevity.
Fig. 7.

A. Colonic temperature of 17–18-month-old C57BL/6 mice fed AL or kept under 40% CR starting at 4 months of age (n=12–13). Mice were housed under a 12-h light:dark cycle (indicated by grey/white bars). (Based on Ferguson et al, [73]). B. Rates of resting oxygen consumption of C57BL/6 mice, fed AL or at 40% CR, at 6 and 23 months of age. Mice were placed under increasing level of CR at 14 weeks of age and 40% restriction was implemented at the age of 4 months (n= 6–8 in each group). Rates of oxygen consumption were normalized as per g lean body mass, which was calculated by subtracting 8 times the epididymal white adipose mass from the body mass. CR decreased the rate of oxygen consumption at both ages, suggesting that this effect is chronic rather than transitory [75]. C. Rates of H2O2 release by cardiac mitochondria of C57BL/6 mice fed AL or at 40% CR from 4 months of age. H2O2 generation was measured in isolated mitochondria at 9, 17 and 23 months of age as H2O2 reduction of by horseradish peroxidase [151]. D. Comparison of protein carbonyl content in heart homogenates between AL fed and the CR mice. Carbonyl content was measured spectrophotometrically using dinitrophenylhydrazine at 9, 17 and 23 months of age [151].
There is strong evidence that life span in ectotherms, such as insects, is inversely related to ambient temperature and rate of oxygen consumption [142–145]. In the housefly as well as Drosophila, increase in physical activity and the consequent rate of oxygen consumption decreases life span [146, 147]. In homeotherms, the existence of a relationship between the rate of metabolism and longevity is supported by inter-species comparisons. A study on seven closely related non-primate mammals including, mouse, hamster, rat, guinea pig, rabbit, pig and cow, whose maximal recorded life spans range from 3.5 to 30 years, showed that species life spans were negatively related with the specific metabolic rate, which in turn was directly related to the rate of mitochondrial production of superoxide anion radicals and H2O2 [148]. It may be added parenthetically that such inter-species correlations are usually observed in species that are closely related phylogenetically; the relationship tends not to hold if comparisons are made across distant groups, e.g., between rodents and birds or bats [144, 149, 150]. Besides the rate of respiration, CR also lowers the rates of mitochondrial generation of superoxide anion radical and H2O2 in C57BL/6 mice [151] (Fig. 7C). The significance of such findings is that they point to an association between SMR, mitochondrial oxidant production and longevity. Indeed, the hypothesis that age-related losses in functional capacity of various biological systems are underpinned by the gradual accumulation of macromolecular oxidative damage, was proposed by Harman 58 years ago [152]. The hypothesis was later modified and subsumed by a broader postulate, the “oxidative stress hypothesis”, whose key feature was that there is a residual imbalance between intracellular ROS fluxes and the antioxidant defenses, whereby cells chronically exist under a certain level of oxidative stress, as evidenced by the presence of steady-state amounts of oxidatively-modified macromolecules. The gap between oxidants and antioxidants, or the level of oxidative stress, is postulated to increase during aging, mainly due to the elevation in the rates of mitochondrial superoxide anion radical and H2O2 generation and to a lesser extent due to a decline in antioxidant defenses, resulting in an accelerated accumulation of macromolecular damage (reviewed in [126, 149, 153]. The reduction in the efficiency of antioxidant defenses is surmised on the basis of age-related increases in susceptibility to exogenously-induced oxidant stress [154–156]. Thus, the mechanism by which CR increases longevity was hypothesized to involve attenuation of oxidative stress because CR lowers the rates of mitochondrial production of superoxide anion radical and H2O2 and retards the age-related accrual of macromolecular oxidative damage (Figs. 7C, D; [126]).
Nonetheless, the role of ROS and oxidative stress in the aging process and implicitly in CR- associated modulation of longevity, has recently come under attack because some of its predictions can not apparently be substantiated in certain animal models [29, 157–165]. The most frequently cited evidence is: (i) Inactivation of superoxide dismutases in Caenorhabditis elegans increases susceptibility to oxidants but either has no or a benign effect on life span [166] (ii) Administration of paraquat, a superoxide anion radical generator, increases the longevity of wild type C. elegans [166]; (iii) Over-expression or moderate under-expression of various enzymatic anti-oxidant defenses does not significantly affect the life span of mice [158, 167, 168]; (iv) Mn-SOD heterozygous mice accrue 30–80% higher 8-oxodeoxyguanosine (8-OHdG), a product of DNA oxidation, without showing any effect on longevity [157]. (v) The naked mole rats live considerably longer than the laboratory rats and mice, despite displaying relatively high levels of oxidative stress/damage [165]. As described elsewhere, such phenomena need further investigation, but may also have alternate explanations [149, 150, 169–173]. For instance, the absence of a significant impact of moderate under- or over-expressions of various antioxidant enzymes on longevity does not necessarily disprove the role of oxidative stress in the aging process, as the normally present levels of enzymatic defenses seem to far exceed those needed to sustain a normal life span [167, 174, 175]. In mutant D. melanogaster, compared to the wild type, only 6% of the Cu, Zn-SOD [174] and 15% of the catalase [176] activity were sufficient for the attainment of normal life span. In the same vein, over-expression of anti-oxidant defenses, such as SODs and catalase, systemically or in targeted tissues, also has no significant effect on the life span in D. melanogaster or mice, especially in the relatively long-lived strains [149, 174, 177–181]. Longevity effects of over-expressions, if any, have been found to be sensitive to genetic background and are comparatively smaller if the controls are long-lived [182]. However, results of the studies on the effects of over-expression of antioxidant enzymes should be interpreted cautiously as there may be possible trade-offs between antioxidant defenses and oxidation-induced apoptosis [183, 184].
The assumption that any accumulation of molecular damage should be expected to lead to a functional loss and/or decrease in longevity is contradicted by the evidence that, due to redundancy, relatively high threshold levels of DNA and protein oxidative damage are required for manifest losses in function [150]. For instance, OGG1 null mice exhibited relatively high concentrations of 8-OHdG, a DNA oxidation product, but showed no effect on survival [182, 185], suggesting that oxidative damage below a certain threshold may only be latently causal in functional decline [150]. The naked mole rat indeed presents an intriguing and potentially interesting model for studying an atypical pattern of aging, as it displays relatively high steady-state level of oxidative stress/damage and an aberrant survivorship curve [165]. As pointed out by Liochev [171, 172], elevated oxidant fluxes should not necessarily be expected to result in shortened longevity because they can also induce pro-survival adaptations. For instance, exposure of houseflies to 15 kr irradiation [186], administration of diamide [187] or diethyldiothiocarbamate [188], a SOD inhibitor, were all found to increase the mean life span; however, a critical corollary effect was that the level of physical activity and metabolic rate of the flies were significantly depressed.
Nevertheless, the most strident assertions against the role of ROS and the validity of the oxidative stress hypothesis emanate from studies involving C. elegans, a nematode that normally inhabits the surface layer of the soil, where it is likely to encounter stressful conditions, such as extreme variations in ambient temperature, desiccation, paucity of food and hypoxia/anoxia, among others. This organism has certain unique natural history features that may be responsible for the atypical extensions of longevity in response to manipulations that are normally deleterious in mammals. For instance, C. elegans is highly resistant to a variety of stresses and can survive lengthy periods under adverse conditions by entering a dormant dauer pathway, without future loss of adult life expectancy. Interestingly, it can also be stored in liquid nitrogen indefinitely and revived when desired [189]. Hypomorphic mutations or RNAi induced partial inactivation of a variety of genes, encoding components of the mitochondrial electron transport chain, cell signaling and nutrient sensing, among others, have been found to lead to notable extensions of life span in C. elegans [190–195]. Such mutants are often hypometabolic, diminutive in size, poor competitors and have low fecundity [196–198]. Although their longevity is extended by such factors, many physiological activities are performed at a low ebb. Life history trajectories in which survival is procured at the cost of other biological functions have been metaphorically compared to as “life in the slow lane” [199]. Thus, unlike homeotherms, life span in C. elegans is highly variable, being profoundly dependent on metabolic rate. Although some gerontologists believe that life span is the “gold standard” for quantifying the rate of aging, chronological duration without measures of physiological fitness, may be a potentially misleading indicator of aging, especially in ectotherms [144, 150, 182]. To conclude, while several important questions remain to be answered about the role of oxidative stress in the aging process, pronouncements of the demise of the hypothesis are premature and unwarranted. As explained by various authors [149, 150, 169, 170, 172, 173, 200–203], the rationales presented for the categorical rejection of the free radical/oxidative stress hypothesis [159–161] are often based on data from peculiar model systems, flawed methodology, fallacious assumptions and/or disregard of vast existing literature that may provide alternative explanations for the incongruous findings.
Nonetheless, the argumentation presented above is not meant to assert that the classic free radical/oxidative stress hypothesis is underpinned by unshakable evidence. Rather, the point made is that reasoning presented by some of the critics is itself inadequate. Indeed, there are other more cogent reasons for a modification of the hypothesis [150, 182, 200]. Specifically, the classic free radical/oxidative stress hypothesis consists of two main components: one is that the endogenously-generated oxidants are deleterious, causing macromolecular damage, and the second is that progressive accrual of such damage leads to senescence-associated losses in functional capacity, ultimately resulting in death [126, 153, 204]. However, a large body of evidence suggests that although the amounts of structurally modified macromolecules do tend to increase with age, their steady-state concentrations in cells of the aged animals are often too low in magnitude to convincingly explain the age-related losses in functional capacity [150, 205, 206]. One possible explanation for the presence of relatively low levels of damaged macromolecules in aged animals may be that accrual of modified macromolecules is hindered by their preferential degradation, which may then be followed by nascent biosynthesis [207–210, 210, 211]. On the other hand, molecular damage could indeed play a role in senescent decline of cells if the nascent biosynthesis for the replacement of damaged molecules is insufficient, resulting thereby in reductions in the total amounts of undamaged macromolecules. Another possible explanation of why the presence of relatively low levels of specific adducts may not constitute evidence against the conventional oxidative stress hypothesis, is that the commonly used indicators of damage may be transitory rather than the final products. For instance, 8-OHdG may get converted to point mutations or the carbonylated adducts lead to protein cross-linking. Given that there is presently insufficient information to assess the significance of macromolecular structural damage in the aging process, it does not follow that the death knell of the oxidative stress hypothesis should be invoked. An alternate vision might postulate that the oxidants may cause deleterious alterations via additional mechanisms, as discussed below.
The redox stress hypothesis of aging
The nature of the putative mechanisms by which ROS and oxidative stress play a role in cell physiology has been significantly reshaped during the past 2–3 decades. The classic view that oxidants produced under normal physiological conditions are invariably, potentially deleterious due to their attacks on various macromolecules [212–215], has been supplanted by the notion that some oxidants, particularly H2O2, play a vital physiological role [216–220]. At relatively low concentrations, they regulate the functions of redox-sensitive proteins via reversible oxidation/reduction of cysteinyl thiols, whereas at high concentrations they may be potentially deleterious due to over-oxidation of such proteins, resulting in the impairment of cellular redox potential and cell signaling mechanisms [221–229]. Indeed, multiple redox sensitive proteins have been identified amongst the so-called aging pathways, delineated in model systems (reviewed in [182]). Thus, in the current view, the deleterious effects of oxidative stress, emanating from endogenously- generated oxidants, may occur due to a combination of factors: the disruption of the thiol-based cell signaling networks by non-radical reactions and the free-radical-induced macromolecular damage [150, 173, 182, 216, 218, 222, 230–232]. Accordingly, it has been proposed that the classic oxidative stress hypothesis of aging, which emphasized the primacy of radical-induced damage, should be amended and re-termed as the “redox stress hypothesis” to accommodate the more contemporary understanding of the mechanisms by which ROS may be involved in the aging process [150, 182]. The classic version seems to lack the pliancy to accommodate the physiological role of the oxidants.
Hydrogen peroxide is considered to be the most ubiquitous electrophile involved in redox signaling [216, 217, 220, 226, 231]. A unique aspect of H2O2 is that its production is linked to the mitochondrial electron transport chain and oxygen consumption, and it is thus incessantly produced under basal conditions. Its rate of production can be modulated by factors that affect metabolic rate of the cell. Mitochondrial H2O2 generation occurs primarily due to the auto-oxidation of ubisemiquinones, associated with respiratory complexes I and III, from where it diffuses into other cellular domains by virtue of its ability to permeate through cellular membranes [233–236]. In addition, H2O2 involved in cell signaling is also generated by the activity of NADPH oxidases, located in cellular membranes. The mechanisms of H2O2 involvement in redox signaling have been extensively reviewed [216, 217, 220, 226, 231] and are thus only skeletally described here. The main targets of H2O2 in cell signaling are the specific protein cysteinyl thiolates, which have a relatively low pKa and thus a comparatively higher reactivity with H2O2 than their protonated counterparts. (For a recent comprehensive review of the mechanisms of redox signaling and the role of H2O2 therein, see Forman et al. [220]). Reaction between a cysteinyl protein thiolate and H2O2 results in the formation of a cysteine sulfenic acid (Pr-SOH), which being an unstable molecule may undergo further modifications (Fig. 8). For instance, it may link with GSH to form a protein mixed disulfide (Pr-SSG), a process referred to as glutathionylation, or it may bond with a protein sulfhydryl group (Pr-SH) to create an intra- or inter-protein disulfide bridge (Pr-S-S-Pr). Such oxidative modifications can be reversed by the thioredoxin or peroxiredoxin systems [216, 218, 222, 226, 228, 232, 237]. The oxidation/reduction of specific cysteinyl thiols is believed to act as “on/off” switches controlling protein function. Under conditions of oxidative stress involving increased oxidant (H2O2) production, cysteine sulfenate can also undergo hyper-oxidation by further reactions with oxidants, such as H2O2, to form a sulfinic acid (Pr-SOOH) and a sulfonic acid (Pr-SOOOH), respectively, which are considered to be irreversible [221, 232, 238–240]. A key aspect of cell signaling is the specificity of the interaction between the signal molecule and the target protein, which is particularly relevant to redox signaling via H2O2, due to the latter’s ubiquitous distribution in the cell. The mechanisms that assure such specificity and regulate redox signaling appear to be highly complex and presently not well understood. They include the selective deprotonation of specific cysteinyl residues in the target proteins, location of the target cysteinyl thiolate, the kinetics of the reaction between the electrophile and the target thiolate anion, the termination and reversal of signaling, among others, as recently discussed by Forman et al. [220].
Fig. 8.
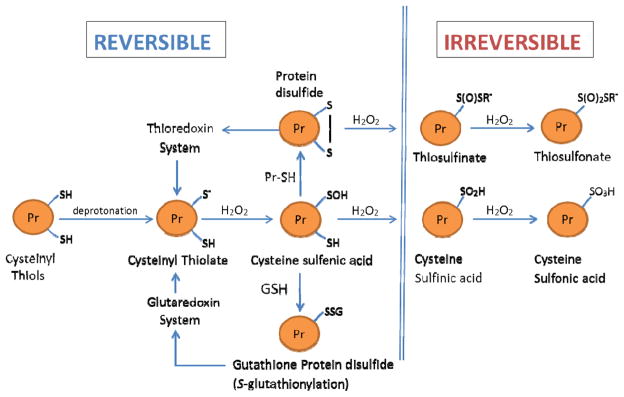
Redox stress: Schema of the modifications of cysteinyl thiols of redox-sensitive proteins in response to elevation in H2O2 fluxes. Under physiological conditions, protein cysteinyl thiols, involved in cell signaling, exist as deprotonated thiolate anions (Pr-S−), which have enhanced reactivity with H2O2. Oxidation of Pr-S− by H2O2 results in the formation of cysteine sulfenic acid (Pr-SOH), which is a relatively unstable molecule. In turn, Pr-SOH may react with GSH to form a mixed protein disulfide (Pr-SSG) or alternatively it may form a disulfide bridge with a Pr-SH to form Pr-S-S-Pr. Such oxidations of Pr-S− are reversed by the glutaredoxin and thioredoxin systems, respectively. At relatively high intra-cellular concentrations of H2O2, Pr-SOH may be further oxidized to cysteine sulfinic acid (Pr-SOOH) and cysteine sulfonic acid (Pr-SOOH), both of which are considered to be irreversible products. Similarly, protein disulfides Pr-S-S-R’ may undergo further irreversible oxidations to thiosulfinate or thiosulfonate, as described by Brandes et al. [221].
The redox stress hypothesis of aging postulates that the rate of mitochondrial H2O2 generation progressively increases in the latter part of life, which initiates a cascade of events that result in the disruption of the redox-based mechanisms for the regulation of protein function [150, 182, 216]. The hypothesis is supported by the following lines of correlative and experimental evidence: (i) During aging, the glutathione redox state, indicated by GSH:GSSG ratios, becomes progressively more pro-oxidizing, or less negative, e.g., ranging from 4.5 mV in the brain to 15 mv, in the heart of mice (Figs. 9A, B; [241–244]. The two main underlying reasons seem to be an increase in GSSG concentration and a decrease in the GSH pool in some tissues. Our studies suggest that the ability for de novo GSH biosynthesis declines during aging in mice due to the loss in the activity of glutamate-cysteine ligase, the rate-limiting enzyme in GSH biosynthesis, probably caused by an age-related increase in homocysteine concentration [245]. (ii) The levels of protein mixed disulfides and GSSG increase (Fig. 9C) and protein sulfhydryl content decreases in aged animals [154] (iii) Transgenic over-expressions of glutamate-cysteine ligase and glucose-6-phosphate dehydrogenase, which enhance GSH and NADPH biosynthesis, respectively, increase the life span of Drosophila without causing negative tradeoff effects, suggesting that augmentation of reductive capacity tends to prolong survival [246, 247]. (iv) Conversely, genetic manipulations that cause a pro-oxidant shift in the redox state, such as the under-expression of glutamate-cysteine ligase, which suppresses GSH biosynthesis, or mitochondrial peroxiredoxins, which eliminate H2O2, decrease life span in Drosophila [248–251].
Fig. 9.
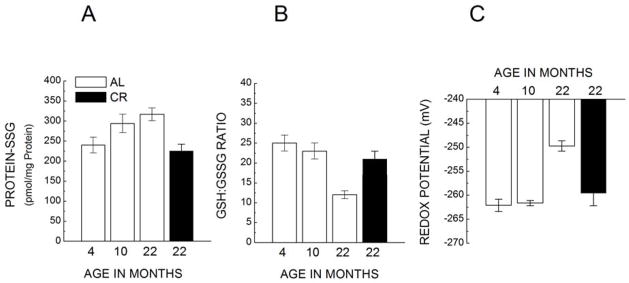
Ratios of glutathione/glutathione disulfide (A); the amounts of protein-glutathione mixed disulfides (B); and glutathione redox potential (C) in the cardiac mitochondria of C57BL/6 mice, fed AL or kept under 40% CR, at 4,10 and 22 months of age. (Adapted from [241]).
There are also several indications that CR attenuates cellular redox stress in those genotypes where it extends longevity. For instance, it lowers the rate of mitochondrial H2O2 production [169, 241, 252–254], increases the GSH:GSSG ratios in mitochondria and tissue homogenates, elevates the protein sulfhydryl content, lowers the levels of GSSG and Pr-SSG and enhances the redox potential [241, 255–257]. In a test of the hypothesis that CR-related increase in longevity is linked to an attenuation of the age-associated pro-oxidant shift in redox state, a comparison was made between the C57BL/6 mice, whose life span is increased by CR, and the DBA/2 mice in which CR has little effect. The amounts of GSSG and Pr-SSG were found to increase and GSH:GSSG ratios to decrease with age in the skeletal muscle and liver of both strains of mice [258]. Caloric restriction, started at 4 months of age, mostly prevented these age-related changes in the C57BL/6 mice, but had little effect in the DBA/2 mice [258]. Caloric restriction induced an increase in the activity of glutamate-cysteine ligase in the liver of C57BL/6 but not in the DBA/2 mice. It is known that the ability for de novo GSH biosynthesis rather than the steady-state concentration of GSH is more critical in defense against an oxidant challenge [259, 260]. Thus, the finding that CR lowers the age-related increases in GSSG and Pr-SSG levels in the mouse genotype whose life span is increased by CR but not in the one where longevity remains unaffected by CR, suggests that one factor in longevity extension by CR may be the enhancement of the ability to synthesize GSH and the consequent attenuation of the age-associated pro-oxidizing shift in the redox state [258].
In addition to its effect on redox state, CR also tends to decrease the amounts of free radical induced structural damage to macromolecules, such as DNA, proteins and lipids [126]. For instance, CR reduces alkane exhalation [261], delays the age-related loss in membrane fluidity [262] and lowers the steady-state amounts of protein carbonyls and 8-hydroxydeoxyguanine [28, 126, 151, 151, 263]. In the skeletal muscle mitochondria of C57BL/6 mice, oxidative damage to proteins, measured as protein carbonyl level and loss of protein sulfhydryl content, increased with age in the AL fed mice, but there was little increase in the CR mice [256]. Crossover studies, involving transfers of mice from AL to CR, or vice versa, at 18–22 months of age, indicated that protein damage, which accrued with age, could not be fully reversed during the time frame of 6 weeks [257]. Such results support the involvement of ROS/molecular damage in CR-related effects on longevity.
Perspective
What began as a rather simple observation that retardation of growth by reduction in food intake increased the length of survival of laboratory rats, compared to their AL-fed counterparts, had a profound long-term effect on the direction of research aimed at understanding the nature of the mechanisms of senescence. Gradually, it became an accepted dogma that CR universally extends the life span of phylogenetically diverse species, possibly including man, by an evolutionarily conserved common mechanism. So strong has been the appeal of the CR paradigm that studies whose results were incongruous with the mainstream view rarely gained traction. Nevertheless, it is now increasingly evident that the longevity-extension effect of CR is not ubiquitous and there exist even intra-species variations in the nature of the response. Although in the original concept, CR involved merely the reduction in the amount of energy intake, regardless of the type of macronutrient, it now seems that at least in flies the total number of calories consumed does not matter, instead it is the reduction in the amount of yeast, presumably protein, intake that results in increased longevity. Such evidence has challenged the classic belief that CR universally extends life span via a common genetically conserved mechanism.
It is often asserted that CR prolongs the maximum life span, which is regarded as a more critical indicator of the rate of aging than the average life span. However, it has not yet been specifically shown that CR increases longevity beyond the maximum species life span. Perhaps, in counterpoint it can also be argued that CR does not increase longevity per se: rather the AL feeding shortens the life span, because it causes a deleterious energy imbalance, which deprives the animals from reaching their potential longevity. The salutary effect of CR on life span mirrors the negative effect of AL feeding. Accordingly, the longevity prolongation effect of CR should be relatively greater if the life spans of the controls become shorter due to energy imbalance. This postulate is supported by the findings of the NIA-NCTR project, where increases in the life spans of the CR-sensitive strains of rats and mice were found to be directly proportional to the level of their corpulence under the AL feeding regimen. The life span extension by CR may thus be ipso facto universal in genotypes that become overweight when fed AL. In this view, CR should have no significant effect on the length of healthy life span in genotypes that maintain an optimal, relatively stable, adult body weight under an AL regimen. Thus, it would seem that the maximum life span of laboratory rodents, recorded under an AL regimen, should not be considered to constitute the norm for the genotype. Instead, life spans determined under conditions of optimal energy intake should be regarded as the norm, against which the efficacy of treatments purporting to affect the aging process can be evaluated. Indeed, this view also accords with the sentiment expressed by the CR pioneer, Clive McCay [9] that “retardation of growth by diets, complete except for calories, affords a means of producing very old animals for studying aging.”
The mechanisms that determine variations in species-specific life spans are presently poorly understood; nevertheless, it can be predicted that genotypes that are better protected from the reactions that initiate and promote age-related physiological decline would tend to survive relatively longer. Virtually all the current hypotheses concerning how CR prolongs longevity invoke an enhancement in putative protective systems, thereby implicitly subscribing to the view that immortality in animals is countermanded by endogenous factors that cause damage or otherwise irreversibly decrease fitness. Thus, ultimately, the various putative mechanisms of aging share the common premise that some type of physiological “damage” is causal to the aging process and by inference, treatments resulting in longer survival decelerate the rate of infliction of such damage. It is postulated that the pathway by which CR affects the longevity of responsive genotypes involves lowering of the body temperature/metabolic rate and ROS production and the enhancement of GSH biosynthesis. In this view, the energy imbalance in particular genotypes, resulting from AL feeding, accelerates the age-related loss in physiological fitness and CR lowers the magnitude of such imbalance. In a nutshell, CR increases life span when it counteracts a significant energy imbalance.
Research Highlights.
The concept that caloric restriction universally prolongs longevity is contested.
Caloric restriction does not prolong life span of all mammalian genotypes.
Ad libitum (AL) feeding shortens potential longevity of over---weight mammals.
Increased longevity under food restriction is related to weight gain under AL.
Caloric restriction is postulated to prolong longevity by decreasing redox stress.
Acknowledgments
The authors are grateful to Drs. Robin J. Mockett, University of South Alabama, William C. Orr of Southern Methodist University, and Nathalie Sumien of University of North Texas Health Science Center for critiquing the manuscript. We are thankful to Marjana Sarker for performing data analysis. Research of the authors, relevant to the topics discussed here, has been supported by grants R01 AG7657 and R01 AG13563 to RSS and P01 AG022550 to MJF from the National Institute on Aging-National Institutes of Health.
Abbreviations
- AL
ad-libitum
- BAP
Biomarkers of Aging Program
- CR
caloric restriction
- EM
Emory-Morse
- GSH
glutathione
- GSSG
glutathione disulfide
- mTOR
mammalian Target of Rapamycin
- NCTR
National Center for Toxicological Research
- NIA
National Institute on Aging
- OGG1
oxoguanine DNA glycosylase
- ROS
reactive oxygen species
- SMR
specific metabolic rate
- SOD
superoxide dismutase
- WNPC
Wisconsin National Primate Center
- WSCR
weight stable caloric restriction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holehan AM, Merry BJ. The experimental manipulation of aging by diet. Biol Rev. 1986;61:329–368. doi: 10.1111/j.1469-185x.1986.tb00658.x. [DOI] [PubMed] [Google Scholar]
- 2.Weindruch R, Walford RL. The retardation of aging and disease by dietary restriction. Springfield, IL: Charles C Thomas Pub. Ltd; 1988. [Google Scholar]
- 3.Masoro EJ. Caloric restriction: A key to understanding and modulating aging. Amsterdam: Elsevier; 2002. [Google Scholar]
- 4.Yu BP. Aging and oxidative stress: Modulation by dietary restriction. Free Radic Biol Med. 1996;21:651–668. doi: 10.1016/0891-5849(96)00162-1. [DOI] [PubMed] [Google Scholar]
- 5.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–22. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 7.Partridge L, Piper MD, Mair W. Dietary restriction in Drosophila. Mech Ageing Dev. 2005;126:938–950. doi: 10.1016/j.mad.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 8.Osborne TB, Mendel LB, Ferry EL. The effect of retardation of growth upon the breeding period and duration of life of rats. Science. 1917;45:294–295. doi: 10.1126/science.45.1160.294. [DOI] [PubMed] [Google Scholar]
- 9.McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of lifespan and upon ultimate body size. J Nutr. 1935;10:63–79. [PubMed] [Google Scholar]
- 10.McCay CM, Maynard LA, Sperling G, Barnes LL. Retarded growth, lifespan, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. J Nutr. 1939;18:1–13. doi: 10.1111/j.1753-4887.1975.tb05227.x. [DOI] [PubMed] [Google Scholar]
- 11.Ross MH. Length of life and nutrition in the rat. J Nutr. 1961;75:197–210. doi: 10.1093/jn/75.2.197. [DOI] [PubMed] [Google Scholar]
- 12.Morrison SD. Nutrition and longevity. Nutr Rev. 1983;41:133–142. doi: 10.1111/j.1753-4887.1983.tb07172.x. [DOI] [PubMed] [Google Scholar]
- 13.Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557. doi: 10.1038/ncomms4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swindell WR. Dietary restriction in rats and mice: a meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res Rev. 2012;11:254–270. doi: 10.1016/j.arr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF. Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell. 2010;9:92–5. doi: 10.1111/j.1474-9726.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turturro A, Witt WW, Lewis S, Hass BS, Lipman RD, Hart RW. Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J Gerontol A Biol Sci Med Sci. 1999;54:B492–501. doi: 10.1093/gerona/54.11.b492. [DOI] [PubMed] [Google Scholar]
- 17.Sheldon WG, Bucci TJ, Blackwell B, Turturro A. Effect of ad libitum feeding and 40% feed restriction on body weight, longevity and neoplasms in B6C3F1, C57BL/6, and B6D2F1 mice. In: Mohr U, Dungworth DL, Capen CC, Carlton WW, Sundberg JP, Ward JM, editors. Pathobiology of the Aging Mouse. Vol. 1. Washington, DC: ILSI Press; 1996. pp. 21–26. [Google Scholar]
- 18.Blackwell BN, Bucci TJ, Hart RW, Turturro A. Longevity, body weight, and neoplasia in ad libitum-fed and diet-restricted C57BL6 mice fed NIH-31 open formula diet. Toxicol Pathol. 1995;23:570–82. doi: 10.1177/019262339502300503. [DOI] [PubMed] [Google Scholar]
- 19.Wang C, Weindruch R, Fernandez JR, Coffey CS, Patel P, Allison DB. Caloric restriction and body weight independently affect longevity in Wistar rats. Int J Obes Relat Metab Disord. 2004;28:357–362. doi: 10.1038/sj.ijo.0802518. [DOI] [PubMed] [Google Scholar]
- 20.Warner HR, Fernandes G, Wang E. A unifying hypothesis to explain the retardation of aging and tumorigenesis by caloric restriction. J Gerontol A Biol Sci Med Sci. 1995;50:B107–9. doi: 10.1093/gerona/50a.3.b107. [DOI] [PubMed] [Google Scholar]
- 21.Weindruch R, Walford RL, Fligiel S, Guthrie D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J Nutr. 1986;116:641–654. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- 22.Weindruch R, Sohal RS. Caloric intake and aging. N Engl J Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayflick L. Dietary restriction: theory fails to satiate. Science. 2010;329:1014–5. doi: 10.1126/science.329.5995.1014. author reply 1015. [DOI] [PubMed] [Google Scholar]
- 24.Cerqueira FM, Kowaltowski AJ. Mitochondrial metabolism in aging: effect of dietary interventions. Ageing Res Rev. 2013;12:22–28. doi: 10.1016/j.arr.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–54. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 26.Masoro EJ. Caloric restriction-induced life extension of rats and mice: a critique of proposed mechanisms. Biochim Biophys Acta. 2009;1790:1040–1048. doi: 10.1016/j.bbagen.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 28.Ramsey JJ, Harper ME, Weindruch R. Restriction of energy intake, energy expenditure, and aging. Free Radic Biol Med. 2000;29:946–68. doi: 10.1016/s0891-5849(00)00417-2. [DOI] [PubMed] [Google Scholar]
- 29.Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med. 2011;32:159–221. doi: 10.1016/j.mam.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Walsh ME, Shi Y, Van Remmen H. The effects of dietary restriction on oxidative stress in rodents. Free Radic Biol Med. 2014;66:88–99. doi: 10.1016/j.freeradbiomed.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper TM, Mockett RJ, Sohal BH, Sohal RS, Orr WC. Effect of caloric restriction on life span of the housefly, Musca domestica. FASEB J. 2004;18:1591–3. doi: 10.1096/fj.03-1464fje. [DOI] [PubMed] [Google Scholar]
- 32.Forster MJ, Morris P, Sohal RS. Genotype and age influence the effect of caloric intake on mortality in mice. FASEB J. 2003;17:690–2. doi: 10.1096/fj.02-0533fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harper JM, Leathers CW, Austad SN. Does caloric restriction extend life in wild mice? Aging Cell. 2006;5:441–9. doi: 10.1111/j.1474-9726.2006.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Bourg E, Minois N. Failure to confirm increased longevity in Drosophila melanogaster submitted to a food restriction procedure. J Gerontol A Biol Sci Med Sci. 1996;51:B280–3. doi: 10.1093/gerona/51a.4.b280. [DOI] [PubMed] [Google Scholar]
- 35.Mockett RJ, Cooper TM, Orr WC, Sohal RS. Effects of caloric restriction are species-specific. Biogerontology. 2006;7:157–60. doi: 10.1007/s10522-006-9004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee KP, Simpson SJ, Clissold FJ, Brooks R, Ballard JW, Taylor PW, Soran N, Raubenheimer D. Lifespan and reproduction in Drosophila: New insights from nutritional geometry. Proc Natl Acad Sci USA. 2008;105:2498–503. doi: 10.1073/pnas.0710787105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakagawa S, Lagisz M, Hector KL, Spencer HG. Comparative and meta-analytic insights into life extension via dietary restriction. Aging Cell. 2012;11:401–409. doi: 10.1111/j.1474-9726.2012.00798.x. [DOI] [PubMed] [Google Scholar]
- 38.Silberberg R, Jarrett SR, Silberberg M. Longevity of female mice kept on various dietary regimens during growth. J Gerontol. 1962;17:239–244. doi: 10.1093/geronj/17.3.239. [DOI] [PubMed] [Google Scholar]
- 39.Cheney KE, Liu RK, Smith GS, Leung RE, Mickey MR, Walford RL. Survival and disease patterns in C57BL/6J mice subjected to undernutrition. Exp Gerontol. 1980;15:237–258. doi: 10.1016/0531-5565(80)90029-7. [DOI] [PubMed] [Google Scholar]
- 40.Fernandes G, Yunis EJ, Good RA. Influence of diet on survival of mice. Proc Natl Acad Sci USA. 1976;73:1279–1283. doi: 10.1073/pnas.73.4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, de Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson SJ, Raubenheimer D. Macronutrient balance and lifespan. Aging 2009. 2014 Mar 16;1(10) doi: 10.18632/aging.100098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson SJ, Raubenheimer D. Caloric restriction and aging revisited: the need for a geometric analysis of the nutritional bases of aging. J Gerontol A Biol Sci Med Sci. 2007;62:707–713. doi: 10.1093/gerona/62.7.707. [DOI] [PubMed] [Google Scholar]
- 44.Ja WW, Carvalho GB, Zid BM, Mak EM, Brummel T, Benzer S. Water- and nutrient-dependent effects of dietary restriction on Drosophila lifespan. Proc Natl Acad Sci USA. 2009;106:18633–18637. doi: 10.1073/pnas.0908016106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bass TM, Grandison RC, Wong R, Martinez P, Partridge L, Piper MD. Optimization of dietary restriction protocols in Drosophila. J Gerontol A Biol Sci Med Sci. 2007;62:1071–1081. doi: 10.1093/gerona/62.10.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mair W, Goymer P, Pletcher SD, Partridge L. Demography of dietary restriction and death in Drosophila. Science. 2003;301:1731–1733. doi: 10.1126/science.1086016. [DOI] [PubMed] [Google Scholar]
- 47.Mair W, Piper MD, Partridge L. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 2005;3:e223. doi: 10.1371/journal.pbio.0030223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Min KJ, Flatt T, Kulaots I, Tatar M. Counting calories in Drosophila diet restriction. Exp Gerontol. 2007;42:247–251. doi: 10.1016/j.exger.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pletcher SD, Macdonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, Partridge L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol. 2002;12:712–723. doi: 10.1016/s0960-9822(02)00808-4. [DOI] [PubMed] [Google Scholar]
- 50.Dick KB, Ross CR, Yampolsky LY. Genetic variation of dietary restriction and the effects of nutrient-free water and amino acid supplements on lifespan and fecundity of Drosophila. Genet Res (Camb) 2011;93:265–273. doi: 10.1017/S001667231100019X. [DOI] [PubMed] [Google Scholar]
- 51.Piper MD, Wong R, Grandison RC, Bass TM, Martinez PM, Partridge L. Water-independent effects of dietary restriction in Drosophila. Proc Natl Acad Sci USA. 2010;107:E54–6. doi: 10.1073/pnas.0914686107. author reply E57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fanson BG, Taylor PW. Additive and interactive effects of nutrient classes on longevity, reproduction, and diet consumption in the Queensland fruit fly (Bactrocera tryoni) J Insect Physiol. 2012;58:327–334. doi: 10.1016/j.jinsphys.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 53.Bruce KD, Hoxha S, Carvalho GB, Yamada R, Wang HD, Karayan P, He S, Brummel T, Kapahi P, Ja WW. High carbohydrate-low protein consumption maximizes Drosophila lifespan. Exp Gerontol. 2013;48:1129–1135. doi: 10.1016/j.exger.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chadwick LE. The respiratory quotient of Drosophila in flight. Biol Bull. 1947;93:229–239. [PubMed] [Google Scholar]
- 55.Chefurka W. Some comparative aspects of the metabolism of carbohydrates in insects. Annu Rev Entomol. 1965;10:345–382. [Google Scholar]
- 56.Cohen AC. Insect diets: Science and technology. Florida: CRC Press; 2004. [Google Scholar]
- 57.Oviedo A, Nestel D, Papadopoulos NT, Ruiz MJ, Prieto SC, Willink E, Vera MT. Management of protein intake in the fruit fly Anastrepha fraterculus. J Insect Physiol. 2011;57:1622–1630. doi: 10.1016/j.jinsphys.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 58.Kaspi R, Yuval B. Post-teneral protein feeding improves sexual competitiveness. Ann Entomol Soc Am. 2000;93:949. [Google Scholar]
- 59.Partridge L, Pletcher SD, Mair W. Dietary restriction, mortality trajectories, risk and damage. Mech Ageing Dev. 2005;126:35–41. doi: 10.1016/j.mad.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 60.Solon-Biet SM, McMahon AC, Ballard JW, Ruohonen K, Wu LE, Cogger VC, Warren A, Huang X, Pichaud N, Melvin RG, Gokarn R, Khalil M, Turner N, Cooney GJ, Sinclair DA, Raubenheimer D, Le Couteur DG, Simpson SJ. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014;19:418–430. doi: 10.1016/j.cmet.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simpson SJ, Raubenheimer D. The nature of nutrition. A unifying framework from animal adaptation to human obesity. Princeton: Princeton University Press; 2012. [Google Scholar]
- 62.Bronson FH. Energy allocation and reproductive development in wild and domestic house mice. Biol Reprod. 1984;31:83–88. doi: 10.1095/biolreprod31.1.83. [DOI] [PubMed] [Google Scholar]
- 63.Clark BR, Price EO. Sexual maturation and fecundity of wild and domestic Norway rats (Rattus norvegicus) J Reprod Fertil. 1981;63:215–220. doi: 10.1530/jrf.0.0630215. [DOI] [PubMed] [Google Scholar]
- 64.Miller RA, Harper JM, Dysko RC, Durkee SJ, Austad SN. Longer life spans and delayed maturation in wild-derived mice. Exp Biol Med (Maywood) 2002;227:500–508. doi: 10.1177/153537020222700715. [DOI] [PubMed] [Google Scholar]
- 65.Martin B, Ji S, Maudsley S, Mattson MP. “Control” laboratory rodents are metabolically morbid: why it matters. Proc Natl Acad Sci USA. 2010;107:6127–6133. doi: 10.1073/pnas.0912955107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turturro A, Duffy P, Hart R, Allaben WT. Rationale for the use of dietary control in toxicity studies--B6C3F1 mouse. Toxicol Pathol. 1996;24:769–775. doi: 10.1177/019262339602400621. [DOI] [PubMed] [Google Scholar]
- 67.Bronson RT, Lipman RD. Reduction in rate of occurrence of age related lesions in dietary restricted laboratory mice. Growth Dev Aging. 1991;55:169–184. [PubMed] [Google Scholar]
- 68.Frankel WN. Genetics of complex neurological disease: challenges and opportunities for modeling epilepsy in mice and rats. Trends Genet. 2009;25:361–367. doi: 10.1016/j.tig.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swindell WR, Johnston A, Gudjonsson JE. Transcriptional profiles of leukocyte populations provide a tool for interpreting gene expression patterns associated with high fat diet in mice. PLoS One. 2010;5:e11861. doi: 10.1371/journal.pone.0011861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsumoto J, Kouguchi H, Oku Y, Yagi K. Primary alveolar echinococcosis: course of larval development and antibody responses in intermediate host rodents with different genetic backgrounds after oral infection with eggs of Echinococcus multilocularis. Parasitol Int. 2010;59:435–444. doi: 10.1016/j.parint.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 71.High KP, Prasad R, Marion CR, Schurig GG, Boyle SM, Sriranganathan N. Outcome and immune responses after Brucella abortus infection in young adult and aged mice. Biogerontology. 2007;8:583–593. doi: 10.1007/s10522-007-9106-6. [DOI] [PubMed] [Google Scholar]
- 72.Vannier E, Borggraefe I, Telford SR, 3rd, Menon S, Brauns T, Spielman A, Gelfand JA, Wortis HH. Age-associated decline in resistance to Babesia microti is genetically determined. J Infect Dis. 2004;189:1721–1728. doi: 10.1086/382965. [DOI] [PubMed] [Google Scholar]
- 73.Ferguson M, Sohal BH, Forster MJ, Sohal RS. Effect of long-term caloric restriction on oxygen consumption and body temperature in two different strains of mice. Mech Ageing Dev. 2007;128:539–45. doi: 10.1016/j.mad.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferguson M, Rebrin I, Forster MJ, Sohal RS. Comparison of metabolic rate and oxidative stress between two different strains of mice with varying response to caloric restriction. Exp Gerontol. 2008;43:757–763. doi: 10.1016/j.exger.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sohal RS, Ferguson M, Sohal BH, Forster MJ. Life span extension in mice by food restriction depends on an energy imbalance. J Nutr. 2009;139:533–9. doi: 10.3945/jn.108.100313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rikke BA, Liao CY, McQueen MB, Nelson JF, Johnson TE. Genetic dissection of dietary restriction in mice supports the metabolic efficiency model of life extension. Exp Gerontol. 2010;45:691–701. doi: 10.1016/j.exger.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Butler RN, Sprott R, Warner H, Bland J, Feuers R, Forster M, Fillit H, Harman SM, Hewitt M, Hyman M, Johnson K, Kligman E, McClearn G, Nelson J, Richardson A, Sonntag W, Weindruch R, Wolf N. Biomarkers of aging: from primitive organisms to humans. J Gerontol A Biol Sci Med Sci. 2004;59:B560–7. doi: 10.1093/gerona/59.6.b560. [DOI] [PubMed] [Google Scholar]
- 78.Reff ME, Schneider EL. Biological markers of aging. Washington, DC: U S Government Printing Office; 1982. [Google Scholar]
- 79.Sprott RL. Biomarkers of aging. J Gerontol A Biol Sci Med Sci. 1999;54:B464–5. doi: 10.1093/gerona/54.11.b464. [DOI] [PubMed] [Google Scholar]
- 80.Sprott RL. Development of animal models of aging at the National Institute of Aging. Neurobiol Aging. 1991;12:635–8. doi: 10.1016/0197-4580(91)90113-x. [DOI] [PubMed] [Google Scholar]
- 81.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barzilai N, Gupta G. Revisiting the role of fat mass in the life extension induced by caloric restriction. J Gerontol A Biol Sci Med Sci. 1999;54:B89–96. doi: 10.1093/gerona/54.3.b89. discussion B97–8. [DOI] [PubMed] [Google Scholar]
- 83.Berg BN, Simms HS. Nutrition and longevity in the rat. II. Longevity and onset of disease with different levels of food intake. J Nutr. 1960;71:255–263. [PubMed] [Google Scholar]
- 84.Berg BN. Nutrition and longevity in the rat. I. Food intake in relation to size, health and fertility. J Nutr. 1960;71:242–254. doi: 10.1093/jn/71.3.242. [DOI] [PubMed] [Google Scholar]
- 85.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 86.Bertrand HA, Lynd FT, Masoro EJ, Yu BP. Changes in adipose mass and cellularity through the adult life of rats fed ad libitum or a life-prolonging restricted diet. J Gerontol. 1980;35:827–835. doi: 10.1093/geronj/35.6.827. [DOI] [PubMed] [Google Scholar]
- 87.Harrison DE, Archer JR, Astle CM. Effects of food restriction on aging: separation of food intake and adiposity. Proc Natl Acad Sci USA. 1984;81:1835–8. doi: 10.1073/pnas.81.6.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Masoro EJ. History of caloric restriction, aging and longevity. In: Everitt AV, Rattan SIS, Le Couteur DG, De Cabo R, editors. Calorie Restriction, aging and longevity. New York: Springer; 2010. 3-14-14. [Google Scholar]
- 89.Ingram DK, Reynolds MA. The relationship of body weight to longevity within laboratory rodent species. In: Woodhead AD, Thompson KH, editors. Evolution of longevity in animals: A comparative approach. New York: Plenum; 1987. pp. 247–282. [DOI] [PubMed] [Google Scholar]
- 90.Keenan KP, Smith PF, Hertzog P, Soper K, Ballam GC, Clark RL. The effects of overfeeding and dietary restriction on Sprague-Dawley rat survival and early pathology biomarkers of aging. Toxicol Pathol. 1994;22:300–315. doi: 10.1177/019262339402200308. [DOI] [PubMed] [Google Scholar]
- 91.Keenan KP, Soper KA, Hertzog PR, Gumprecht LA, Smith PF, Mattson BA, Ballam GC, Clark RL. Diet, overfeeding, and moderate dietary restriction in control Sprague-Dawley rats: II. Effects on age-related proliferative and degenerative lesions. Toxicol Pathol. 1995;23:287–302. doi: 10.1177/019262339502300306. [DOI] [PubMed] [Google Scholar]
- 92.Tobin G, Stevens KA, Russell RJ. Nutrition. In: Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, Smith AL, editors. The Mouse in Biomedical Research. 2. III. Amsterdam: Elsevier; 2007. pp. 321–383. [Google Scholar]
- 93.Haseman JK, Boorman GA, Huff J. Value of historical control data and other issues related to the evaluation of long-term rodent carcinogenicity studies. Toxicol Pathol. 1997;25:524–527. doi: 10.1177/019262339702500518. [DOI] [PubMed] [Google Scholar]
- 94.Haseman JK, Young E, Eustis SL, Hailey JR. Body weight-tumor incidence correlations in long-term rodent carcinogenicity studies. Toxicol Pathol. 1997;25:256–263. doi: 10.1177/019262339702500302. [DOI] [PubMed] [Google Scholar]
- 95.Dirx MJ, Zeegers MP, Dagnelie PC, van den Bogaard T, van den Brandt PA. Energy restriction and the risk of spontaneous mammary tumors in mice: a meta-analysis. Int J Cancer. 2003;106:766–770. doi: 10.1002/ijc.11277. [DOI] [PubMed] [Google Scholar]
- 96.Chavez JA, Summers SA. Lipid oversupply, selective insulin resistance, and lipotoxicity: molecular mechanisms. Biochim Biophys Acta. 2010;1801:252–265. doi: 10.1016/j.bbalip.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 98.Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MK. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801:266–271. doi: 10.1016/j.bbalip.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 99.Mattson MP. Genes and behavior interact to determine mortality in mice when food is scarce and competition fierce. Aging Cell. 2010;9:448–9. doi: 10.1111/j.1474-9726.2010.00561.x. discussion 450–2. [DOI] [PubMed] [Google Scholar]
- 100.Liao CY, Johnson TE, Nelson JF. Genetic variation in responses to dietary restriction--an unbiased tool for hypothesis testing. Exp Gerontol. 2013;48:1025–1029. doi: 10.1016/j.exger.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nadon NL. Exploiting the rodent model for studies on the pharmacology of lifespan extension. Aging Cell. 2006;5:9–15. doi: 10.1111/j.1474-9726.2006.00185.x. [DOI] [PubMed] [Google Scholar]
- 102.Nir I, Nitsan Z, Dror Y, Shapira N. Influence of overfeeding on growth, obesity and intestinal tract in young chicks of light and heavy breeds. Br J Nutr. 1978;39:27–35. doi: 10.1079/bjn19780008. [DOI] [PubMed] [Google Scholar]
- 103.Nir I, Nitsan Z. Metabolic and anatomical adaptations of light-bodied chicks to intermittent feeding. Br Poult Sci. 1979;20:61–71. doi: 10.1080/00071669008417307. [DOI] [PubMed] [Google Scholar]
- 104.Nir I, Nitsan Z, Cherry JA, Dunnington EA, Jones DE, Siegel PB. Growth-associated traits in parental and F1 populations of chickens under different feeding programs. 2. Ad libitum and intermittent feeding. Poult Sci. 1987;66:10–22. doi: 10.3382/ps.0660010. [DOI] [PubMed] [Google Scholar]
- 105.Katanbaf MN, Dunnington EA, Siegel PB. Restricted feeding in early and late-feathering chickens. 3. Organ size and carcass composition. Poult Sci. 1989;68:359–368. doi: 10.3382/ps.0680359. [DOI] [PubMed] [Google Scholar]
- 106.Vaanholt LM, Magee V, Speakman JR. Factors predicting individual variability in diet-induced weight loss in MF1 mice. Obesity (Silver Spring) 2012;20:285–294. doi: 10.1038/oby.2011.279. [DOI] [PubMed] [Google Scholar]
- 107.Gibb RL. Environment. In: Whishaw IQ, Kolb B, editors. The behavior of the laboratory rat: A handbook with tests. New York, NY: Oxford University Press; 2005. pp. 321–331. [Google Scholar]
- 108.Keenan KP, Ballam GC, Haught DG, Laroque P. Nutrition. In: Krinke GJ, editor. The laboratory rat. London: Academic press; 2000. pp. 57–75. [Google Scholar]
- 109.Dirks AJ, Leeuwenburgh C. Caloric restriction in humans: potential pitfalls and health concerns. Mech Ageing Dev. 2006;127:1–7. doi: 10.1016/j.mad.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 110.Moreira EA, Most M, Howard J, Ravussin E. Dietary adherence to long-term controlled feeding in a calorie-restriction study in overweight men and women. Nutr Clin Pract. 2011;26:309–315. doi: 10.1177/0884533611405992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rickman AD, Williamson DA, Martin CK, Gilhooly CH, Stein RI, Bales CW, Roberts S, Das SK. The CALERIE Study: design and methods of an innovative 25% caloric restriction intervention. Contemp Clin Trials. 2011;32:874–881. doi: 10.1016/j.cct.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rochon J, Bales CW, Ravussin E, Redman LM, Holloszy JO, Racette SB, Roberts SB, Das SK, Romashkan S, Galan KM, Hadley EC, Kraus WE CALERIE Study Group. Design and conduct of the CALERIE study: comprehensive assessment of the long-term effects of reducing intake of energy. J Gerontol A Biol Sci Med Sci. 2011;66:97–108. doi: 10.1093/gerona/glq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mattison JA, Lane MA, Roth GS, Ingram DK. Calorie restriction in rhesus monkeys. Exp Gerontol. 2003;38:35–46. doi: 10.1016/s0531-5565(02)00146-8. [DOI] [PubMed] [Google Scholar]
- 114.Mercken EM, Carboneau BA, Krzysik-Walker SM, de Cabo R. Of mice and men: the benefits of caloric restriction, exercise, and mimetics. Ageing Res Rev. 2012;11:390–398. doi: 10.1016/j.arr.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baker TW, Weisman RG, Beninger RJ. Reinforcer devaluation by extinction depends on the food restriction protocol. Behav Processes. 2012;90:124–129. doi: 10.1016/j.beproc.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 116.Clifton PG. Eating. In: Whishaw IQ, Kolb B, editors. The behavior of the laboratory rat: A handbook with tests. New York, NY: Oxford University Press; 2005. pp. 197–206. [Google Scholar]
- 117.Gatch MB, Taylor CM, Forster MJ. Locomotor stimulant and discriminative stimulus effects of ‘bath salt’ cathinones. Behav Pharmacol. 2013;24:437–447. doi: 10.1097/FBP.0b013e328364166d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Merry BJ. Molecular mechanisms linking calorie restriction and longevity. Int J Biochem Cell Biol. 2002;34:1340–1354. doi: 10.1016/s1357-2725(02)00038-9. [DOI] [PubMed] [Google Scholar]
- 119.McDonald RB, Ramsey JJ. Honoring Clive McCay and 75 years of calorie restriction research. J Nutr. 2010;140:1205–1210. doi: 10.3945/jn.110.122804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mobbs CV, Mastaitis JW, Zhang M, Isoda F, Cheng H, Yen K. Secrets of the lac operon. Glucose hysteresis as a mechanism in dietary restriction, aging and disease. Interdiscip Top Gerontol. 2007;35:39–68. doi: 10.1159/000096555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Y, Daniel M, Tollefsbol TO. Epigenetic regulation of caloric restriction in aging. BMC Med. 2011;9 doi: 10.1186/1741-7015-9-98. 98-7015-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Libert S, Guarente L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu Rev Physiol. 2013;75:669–684. doi: 10.1146/annurev-physiol-030212-183800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rattan SI. Biogerontology: the next step. Ann N Y Acad Sci. 2000;908:282–290. doi: 10.1111/j.1749-6632.2000.tb06655.x. [DOI] [PubMed] [Google Scholar]
- 125.Vaiserman AM. Hormesis and epigenetics: is there a link? Ageing Res Rev. 2011;10:413–421. doi: 10.1016/j.arr.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 126.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Withers PC, Cooper CE. Metabolic depression: a historical perspective. Prog Mol Subcell Biol. 2010;49:1–23. doi: 10.1007/978-3-642-02421-4_1. [DOI] [PubMed] [Google Scholar]
- 128.Storey KB. Mammalian hibernation. Transcriptional and translational controls. Adv Exp Med Biol. 2003;543:21–38. [PubMed] [Google Scholar]
- 129.Storey KB, Storey JM. Tribute to PL. Lutz: Putting life on ‘pause’--molecular regulation of hypometabolism. J Exp Biol. 2007;210:1700–1714. doi: 10.1242/jeb.02716. [DOI] [PubMed] [Google Scholar]
- 130.Duffy PH, Feuers RJ, Leakey JA, Nakamura K, Turturro A, Hart RW. Effect of chronic caloric restriction on physiological variables related to energy metabolism in the male Fischer 344 rat. Mech Ageing Dev. 1989;48:117–133. doi: 10.1016/0047-6374(89)90044-4. [DOI] [PubMed] [Google Scholar]
- 131.Lane MA, Baer DJ, Rumpler WV, Weindruch R, Ingram DK, Tilmont EM, Cutler RG, Roth GS. Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc Natl Acad Sci USA. 1996;93:4159–64. doi: 10.1073/pnas.93.9.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carrillo AE, Flouris AD. Caloric restriction and longevity: effects of reduced body temperature. Ageing Res Rev. 2011;10:153–162. doi: 10.1016/j.arr.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 133.Blanc S, Schoeller D, Kemnitz J, Weindruch R, Colman R, Newton W, Wink K, Baum S, Ramsey J. Energy expenditure of rhesus monkeys subjected to 11 years of dietary restriction. J Clin Endocrinol Metab. 2003;88:16–23. doi: 10.1210/jc.2002-020405. [DOI] [PubMed] [Google Scholar]
- 134.DeLany JP, Hansen BC, Bodkin NL, Hannah J, Bray GA. Long-term calorie restriction reduces energy expenditure in aging monkeys. J Gerontol A Biol Sci Med Sci. 1999;54:B5–11. doi: 10.1093/gerona/54.1.b5. discussion B12–3. [DOI] [PubMed] [Google Scholar]
- 135.Roth GS, Lane MA, Ingram DK, Mattison JA, Elahi D, Tobin JD, Muller D, Metter EJ. Biomarkers of caloric restriction may predict longevity in humans. Science. 2002;297:811. doi: 10.1126/science.1071851. [DOI] [PubMed] [Google Scholar]
- 136.Conti B, Sanchez-Alavez M, Winsky-Sommerer R, Morale MC, Lucero J, Brownell S, Fabre V, Huitron-Resendiz S, Henriksen S, Zorrilla EP, de Lecea L, Bartfai T. Transgenic mice with a reduced core body temperature have an increased life span.[see comment] Science. 2006;314:825–8. doi: 10.1126/science.1132191. [DOI] [PubMed] [Google Scholar]
- 137.Conti B. Considerations on temperature, longevity and aging. Cell Mol Life Sci. 2008;65:1626–30. doi: 10.1007/s00018-008-7536-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prosser CL. Oxygen: Respiration and metabolism. In: Prosser CL, editor. Comparative Animal Physiology. Philadelphia: Saunders Comp; 1973. pp. 165–211. [Google Scholar]
- 139.Gillooly JF, Brown JH, West GB, Savage VM, Charnov EL. Effects of size and temperature on metabolic rate. Science. 2001;293:2248–2251. doi: 10.1126/science.1061967. [DOI] [PubMed] [Google Scholar]
- 140.Guyton AC, Hall JE. Textbook of medical physiology. Philadelphia: Elsevier Saunders; 2006. [Google Scholar]
- 141.Sacher GA. Life table modifications and life prolongation. In: Finch E, Hayflick L, editors. Handbook of the Biology of Aging. New York: Van Nostrand; 1977. pp. 582–638. [Google Scholar]
- 142.Loeb J, Northrop JH. What Determines the Duration of Life in Metazoa? Proc Natl Acad Sci USA. 1917;3:382–386. doi: 10.1073/pnas.3.5.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Miquel J, Lundren PR, Bensch KG, Atlan H. Effects of temperature on the life span, vitality and fine structure of Drosophila melanogaster. Mech Ageing Dev. 1976;5:347–370. doi: 10.1016/0047-6374(76)90034-8. [DOI] [PubMed] [Google Scholar]
- 144.Mockett RJ, Sohal R, Orr WC. Roles of oxidative stress in the aging process of drosophila melanogaster. In: Miwa S, Beckman KB, Muller FL, editors. Oxidative Stress in Aging. Totowa, NJ: Humana Press; 2008. pp. 111–128. [Google Scholar]
- 145.McArthur MC, Sohal RS. Relationship between metabolic rate, aging, lipid peroxidation, and fluorescent age pigment in milkweed bug, Oncopeltus fasciatus (Hemiptera) J Gerontol. 1982;37:268–274. doi: 10.1093/geronj/37.3.268. [DOI] [PubMed] [Google Scholar]
- 146.Sohal RS, Buchan PB. Relationship between fluorescent age pigment, physiological age and physical activity in the housefly, Musca domestica. Mech Ageing Dev. 1981;15:243–249. doi: 10.1016/0047-6374(81)90133-0. [DOI] [PubMed] [Google Scholar]
- 147.Magwere T, Pamplona R, Miwa S, Martinez-Diaz P, Portero-Otin M, Brand MD, Partridge L. Flight activity, mortality rates, and lipoxidative damage in Drosophila. J Gerontol A Biol Sci Med Sci. 2006;61:136–145. doi: 10.1093/gerona/61.2.136. [DOI] [PubMed] [Google Scholar]
- 148.Ku HH, Brunk UT, Sohal RS. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radic Biol Med. 1993;15:621–7. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- 149.Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic Biol Med. 2002;33:575–86. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 150.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52:539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–33. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 152.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 153.Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med. 2002;33:37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 154.Agarwal S, Sohal RS. Aging and protein oxidative damage. Mech Ageing Dev. 1994;75:11–19. doi: 10.1016/0047-6374(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 155.Sohal RS, Agarwal S, Sohal BH. Oxidative stress and aging in the Mongolian gerbil (Meriones unguiculatus) Mech Ageing Dev. 1995;81:15–25. doi: 10.1016/0047-6374(94)01578-a. [DOI] [PubMed] [Google Scholar]
- 156.Agarwal S, Sohal RS. Relationship between susceptibility to protein oxidation, aging, and maximum life span potential of different species. Exp Gerontol. 1996;31:365–372. doi: 10.1016/0531-5565(95)02039-x. [DOI] [PubMed] [Google Scholar]
- 157.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 158.Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–14. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gems D, Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009;8:1681–1687. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 160.Van Raamsdonk JM, Hekimi S. Reactive Oxygen Species and Aging in Caenorhabditis elegans: Causal or Casual Relationship? Antioxid Redox Signal. 2010;13:1911–1953. doi: 10.1089/ars.2010.3215. [DOI] [PubMed] [Google Scholar]
- 161.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 162.Gladyshev VN. The free radical theory of aging is dead. Long live the damage theory! Antioxid Redox Signal. 2014;20:727–731. doi: 10.1089/ars.2013.5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Speakman JR, Selman C. The free-radical damage theory: Accumulating evidence against a simple link of oxidative stress to ageing and lifespan. Bioessays. 2011;33:255–259. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- 165.Lewis KN, Andziak B, Yang T, Buffenstein R. The naked mole-rat response to oxidative stress: just deal with it. Antioxid Redox Signal. 2013;19:1388–1399. doi: 10.1089/ars.2012.4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci USA. 2012;109:5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Orr WC, Sohal RS. Does overexpression of Cu, Zn-SOD extend life span in Drosophila melanogaster? Exp Gerontol. 2003;38:227–230. doi: 10.1016/s0531-5565(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 168.Salmon AB, Richardson A, Perez VI. Update on the oxidative stress theory of aging: does oxidative stress play a role in aging or healthy aging? Free Radic Biol Med. 2010;48:642–655. doi: 10.1016/j.freeradbiomed.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Barja G. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid Redox Signal. 2013;19:1420–1445. doi: 10.1089/ars.2012.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Scialo F, Mallikarjun V, Stefanatos R, Sanz A. Regulation of lifespan by the mitochondrial electron transport chain: reactive oxygen species-dependent and reactive oxygen species-independent mechanisms. Antioxid Redox Signal. 2013;19:1953–1969. doi: 10.1089/ars.2012.4900. [DOI] [PubMed] [Google Scholar]
- 171.Liochev SI. Free radical paradoxes. Free Radic Biol Med. 2013;65:232–233. doi: 10.1016/j.freeradbiomed.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 172.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 173.Vina J, Borras C, Abdelaziz KM, Garcia-Valles R, Gomez-Cabrera MC. The free radical theory of aging revisited: the cell signaling disruption theory of aging. Antioxid Redox Signal. 2013;19:779–787. doi: 10.1089/ars.2012.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mockett RJ, Radyuk SN, Benes JJ, Orr WC, Sohal RS. Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc Natl Acad Sci USA. 2003;100:301–306. doi: 10.1073/pnas.0136976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Parker JD, Parker KM, Sohal BH, Sohal RS, Keller L. Decreased expression of Cu-Zn superoxide dismutase 1 in ants with extreme lifespan. Proc Natl Acad Sci USA. 2004;101:3486–3489. doi: 10.1073/pnas.0400222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Orr WC, Arnold LA, Sohal RS. Relationship between catalase activity, life span and some parameters associated with antioxidant defenses in Drosophila melanogaster. Mech Ageing Dev. 1992;63:287–296. doi: 10.1016/0047-6374(92)90006-y. [DOI] [PubMed] [Google Scholar]
- 177.Orr WC, Sohal RS. The effects of catalase gene overexpression on life span and resistance to oxidative stress in transgenic Drosophila melanogaster. Arch Biochem Biophys. 1992;297:35–41. doi: 10.1016/0003-9861(92)90637-c. [DOI] [PubMed] [Google Scholar]
- 178.Orr WC, Sohal RS. The effects of Cu-Zn superoxide dismutase gene overexpression on life span and resistance to oxidative stress in transgenic Drosophila melanogaster. Arch Biochem Biophys. 1993;301:34–40. doi: 10.1006/abbi.1993.1111. [DOI] [PubMed] [Google Scholar]
- 179.Bayne AC, Mockett RJ, Orr WC, Sohal RS. Enhanced catabolism of mitochondrial superoxide/hydrogen peroxide and aging in transgenic Drosophila. Biochem J. 2005;391:277–84. doi: 10.1042/BJ20041872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mockett RJ, Bayne AC, Kwong LK, Orr WC, Sohal RS. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic Biol Med. 2003;34:207–17. doi: 10.1016/s0891-5849(02)01190-5. [DOI] [PubMed] [Google Scholar]
- 181.Orr WC, Mockett RJ, Benes JJ, Sohal RS. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J Biol Chem. 2003;278:26418–22. doi: 10.1074/jbc.M303095200. [DOI] [PubMed] [Google Scholar]
- 182.Orr WC, Radyuk SN, Sohal RS. Involvement of redox state in the aging of Drosophila melanogaster. Antioxid Redox Signal. 2013;19:788–803. doi: 10.1089/ars.2012.5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ran Q, Liang H, Ikeno Y, Qi W, Prolla TA, Roberts LJ, 2nd, Wolf N, Van Remmen H, Richardson A. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62:932–942. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- 184.Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Stuart JA, Bourque BM, de Souza-Pinto NC, Bohr VA. No evidence of mitochondrial respiratory dysfunction in OGG1-null mice deficient in removal of 8-oxodeoxyguanine from mitochondrial DNA. Free Radic Biol Med. 2005;38:737–745. doi: 10.1016/j.freeradbiomed.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 186.Allen RG, Sohal RS. Life-lengthening effects of gamma-radiation on the adult housefly, Musca domestica. Mech Ageing Dev. 1982;20:369–375. doi: 10.1016/0047-6374(82)90104-x. [DOI] [PubMed] [Google Scholar]
- 187.Allen RG, Farmer KJ, Sohal RS. Effect of diamide administration on longevity, oxygen consumption, superoxide dismutase, catalase, inorganic peroxides and glutathione in the adult housefly, Musca domestica. Comp Biochem Physiol. 1984;78C:31–33. doi: 10.1016/0742-8413(84)90042-2. [DOI] [PubMed] [Google Scholar]
- 188.Sohal RS, Farmer KJ, Allen RG, Ragland SS. Effect of diethyldithiocarbamate on life span, metabolic rate, superoxide dismutase, catalase, inorganic peroxides and glutathione in the adult housefly, Musca domestica. Mech Ageing Dev. 1984;24:175–183. doi: 10.1016/0047-6374(84)90069-1. [DOI] [PubMed] [Google Scholar]
- 189.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ. Remarkable longevity and stress resistance of nematode PI3K-null mutants. Aging Cell. 2008;7: 13–22. doi: 10.1111/j.1474-9726.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 191.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 192.Kenyon C. Ponce d’elegans: genetic quest for the fountain of youth. Cell. 1996;84:501–504. doi: 10.1016/s0092-8674(00)81024-7. [DOI] [PubMed] [Google Scholar]
- 193.Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- 194.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 195.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Van Voorhies WA, Ward S. Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate. Proc Natl Acad Sci USA. 1999;96:11399–11403. doi: 10.1073/pnas.96.20.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gershon H, Gershon D. Paradigms in aging research: a critical review and assessment. Mech Ageing Dev. 2000;117:21–28. doi: 10.1016/s0047-6374(00)00141-x. [DOI] [PubMed] [Google Scholar]
- 198.Van Voorhies WA. Is life span extension in single gene long-lived Caenorhabditis elegans mutants due to hypometabolism? Exp Gerontol. 2003;38:615–618. doi: 10.1016/s0531-5565(03)00070-6. [DOI] [PubMed] [Google Scholar]
- 199.Storey KB. Life in the slow lane: molecular mechanisms of estivation. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:733–754. doi: 10.1016/s1095-6433(02)00206-4. [DOI] [PubMed] [Google Scholar]
- 200.Kirkwood TB, Kowald A. The free-radical theory of ageing--older, wiser and still alive: modelling positional effects of the primary targets of ROS reveals new support. Bioessays. 2012;34:692–700. doi: 10.1002/bies.201200014. [DOI] [PubMed] [Google Scholar]
- 201.Trifunovic A, Larsson NG. Mitochondrial dysfunction as a cause of ageing. J Intern Med. 2008;263:167–178. doi: 10.1111/j.1365-2796.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 202.Nemoto S, Finkel T. Ageing and the mystery at Arles. Nature. 2004;429:149–152. doi: 10.1038/429149a. [DOI] [PubMed] [Google Scholar]
- 203.Moosmann B, Behl C. Mitochondrially encoded cysteine predicts animal lifespan. Aging Cell. 2008;7:32–46. doi: 10.1111/j.1474-9726.2007.00349.x. [DOI] [PubMed] [Google Scholar]
- 204.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 205.Li XD, Rebrin I, Forster MJ, Sohal RS. Effects of age and caloric restriction on mitochondrial protein oxidative damage in mice. Mech Ageing Dev. 2012;133:30–36. doi: 10.1016/j.mad.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Walther DM, Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rivett AJ. Preferential degradation of the oxidatively modified form of glutamine synthetase by intracellular mammalian proteases. J Biol Chem. 1985;260:300–305. [PubMed] [Google Scholar]
- 208.Agarwal S, Sohal RS. Aging and proteolysis of oxidized proteins. Arch Biochem Biophys. 1994;309:24–28. doi: 10.1006/abbi.1994.1078. [DOI] [PubMed] [Google Scholar]
- 209.Stadtman ER. Importance of individuality in oxidative stress and aging. Free Radic Biol Med. 2002;33:597–604. doi: 10.1016/s0891-5849(02)00904-8. [DOI] [PubMed] [Google Scholar]
- 210.Jung T, Grune T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life. 2008;60:743–752. doi: 10.1002/iub.114. [DOI] [PubMed] [Google Scholar]
- 211.Ugarte N, Petropoulos I, Friguet B. Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid Redox Signal. 2010;13:539–549. doi: 10.1089/ars.2009.2998. [DOI] [PubMed] [Google Scholar]
- 212.Pryor W. The role of free radical reactions in biological systems. In: Pryor W, editor. Free Radicals in Biology. I. New York: Academic Press; 1976. pp. 1–69. [Google Scholar]
- 213.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 214.Halliwell B, Gutteridge JM. Free radicals, lipid peroxidation, and cell damage. Lancet. 1984;2:1095. doi: 10.1016/s0140-6736(84)91530-7. [DOI] [PubMed] [Google Scholar]
- 215.Davies KJA. Oxidative stress: The paradox of aerobic life. In: Rice-Evans C, Halliwell B, Lunt GG, editors. Free Radical and Oxidative Stress: Environments, Drugs and Food Additives. London: Portland Press; 1995. pp. 1–32. [Google Scholar]
- 216.Jones DP. Radical-free biology of oxidative stress. Am J Phsiol Cell Physiol. 2008;295:C849–68. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 218.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 219.Droge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 220.Forman HJ, Ursini F, Maiorino M. An overview of mechanisms of redox signaling. J Mol Cell Cardiol. 2014 doi: 10.1016/j.yjmcc.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–42. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 224.Rhee SG. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–3. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 225.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–70. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 227.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 228.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Veal E, Day A. Hydrogen peroxide as a signaling molecule. Antioxid Redox Signal. 2011;15:147–151. doi: 10.1089/ars.2011.3968. [DOI] [PubMed] [Google Scholar]
- 230.Ghezzi P, Bonetto V, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid Redox Signal. 2005;7:964–72. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 231.Groeger G, Quiney C, Cotter TG. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal. 2009;11:2655–71. doi: 10.1089/ars.2009.2728. [DOI] [PubMed] [Google Scholar]
- 232.Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16:476–495. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- 233.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 234.Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 235.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sastre J, Pallardo FV, Vina J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med. 2003;35:1–8. doi: 10.1016/s0891-5849(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 237.Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Lillig CH. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal. 2013;19:1539–1605. doi: 10.1089/ars.2012.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci USA. 2004;101:17982–7. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Gallogly MM, Starke DW, Mieyal JJ. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid Redox Signal. 2009;11:1059–1081. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Brigelius-Flohe R, Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rebrin I, Sohal RS. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp Gerontol. 2004;39:1513–9. doi: 10.1016/j.exger.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Rebrin I, Bayne AV, Mockett RJ, Orr WC, Sohal RS. Free aminothiols, glutathione redox state and protein mixed disulphides in aging Drosophila melanogaster. Biochem J. 2004;382:131–6. doi: 10.1042/BJ20040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rebrin I, Sohal RS. Pro-oxidant shift in glutathione redox state during aging. Adv Drug Deliv Rev. 2008;60:1545–52. doi: 10.1016/j.addr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Toroser D, Sohal RS. Age-associated perturbations in glutathione synthesis in mouse liver. Biochem J. 2007;405:583–589. doi: 10.1042/BJ20061868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Orr WC, Radyuk SN, Prabhudesai L, Toroser D, Benes JJ, Luchak JM, Mockett RJ, Rebrin I, Hubbard JG, Sohal RS. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J Biol Chem. 2005;280:37331–8. doi: 10.1074/jbc.M508272200. [DOI] [PubMed] [Google Scholar]
- 247.Legan SK, Rebrin I, Mockett RJ, Radyuk SN, Klichko VI, Sohal RS, Orr WC. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J Biol Chem. 2008;283:32492–9. doi: 10.1074/jbc.M805832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Radyuk SN, Michalak K, Klichko VI, Benes J, Rebrin I, Sohal RS, Orr WC. Peroxiredoxin 5 confers protection against oxidative stress and apoptosis and also promotes longevity in Drosophila. Biochem J. 2009;419:437–45. doi: 10.1042/BJ20082003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Radyuk SN, Rebrin I, Klichko VI, Sohal BH, Michalak K, Benes J, Sohal RS, Orr WC. Mitochondrial peroxiredoxins are critical for the maintenance of redox state and the survival of adult Drosophila. Free Radic Biol Med. 2010;49:1892–902. doi: 10.1016/j.freeradbiomed.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Radyuk SN, Gambini J, Borras C, Serna E, Klichko VI, Vina J, Orr WC. Age-dependent changes in the transcription profile of long-lived Drosophila over-expressing glutamate cysteine ligase. Mech Ageing Dev. 2012;133:401–413. doi: 10.1016/j.mad.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 251.Radyuk SN, Klichko VI, Michalak K, Orr WC. The effect of peroxiredoxin 4 on fly physiology is a complex interplay of antioxidant and signaling functions. FASEB J. 2013;27:1426–1438. doi: 10.1096/fj.12-214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sohal RS, Dubey A. Mitochondrial oxidative damage, hydrogen peroxide release and aging. Free Radic Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 253.Gredilla R, Sanz A, Lopez-Torres M, Barja G. Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 2001;15:1589–1591. doi: 10.1096/fj.00-0764fje. [DOI] [PubMed] [Google Scholar]
- 254.Lambert AJ, Merry BJ. Effect of caloric restriction on mitochondrial reactive oxygen species production and bioenergetics: reversal by insulin. Am J Physiol Regul Integr Comp Physiol. 2004;286:R71–9. doi: 10.1152/ajpregu.00341.2003. [DOI] [PubMed] [Google Scholar]
- 255.Dubey A, Forster MJ, Lal H, Sohal RS. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch Biochem Biophys. 1996;333:189–97. doi: 10.1006/abbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- 256.Lass A, Sohal BH, Weindruch R, Forster MJ, Sohal RS. Caloric restriction prevents age-associated accrual of oxidative damage to mouse skeletal muscle mitochondria. Free Radic Biol Med. 1998;25:1089–97. doi: 10.1016/s0891-5849(98)00144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Forster MJ, Sohal BH, Sohal RS. Reversible effects of long-term caloric restriction on protein oxidative damage. J Gerontol A Biol Sci Med Sci. 2000;55:B522–9. doi: 10.1093/gerona/55.11.b522. [DOI] [PubMed] [Google Scholar]
- 258.Rebrin I, Forster MJ, Sohal RS. Association between life-span extension by caloric restriction and thiol redox state in two different strains of mice. Free Radic Biol Med. 2011;51:225–33. doi: 10.1016/j.freeradbiomed.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Moore WR, Anderson ME, Meister A, Murata K, Kimura A. Increased capacity for glutathione synthesis enhances resistance to radiation in Escherichia coli: a possible model for mammalian cell protection. Proc Natl Acad Sci USA. 1989;86:1461–4. doi: 10.1073/pnas.86.5.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Matsuo M, Gomi F, Kuramoto K, Sagai M. Food restriction suppresses an age-dependent increase in the exhalation rate of pentane from rats: a longitudinal study. J Gerontol. 1993;48:B133–6. doi: 10.1093/geronj/48.4.b133. [DOI] [PubMed] [Google Scholar]
- 262.Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WA. Life and death: metabolic rate, membrane composition, and life span of animals. Physiol Rev. 2007;87:1175–1213. doi: 10.1152/physrev.00047.2006. [DOI] [PubMed] [Google Scholar]
- 263.Sohal RS, Agarwal S, Candas M, Forster MJ, Lal H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech Ageing Dev. 1994;76:215–24. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- 264.Novelli M, Masiello P, Bombara M, Bergamini E. Protein glycation in the aging male Sprague-Dawley rat: effects of antiaging diet restrictions. J Gerontol A Biol Sci Med Sci. 1998;53:B94–101. doi: 10.1093/gerona/53a.2.b94. [DOI] [PubMed] [Google Scholar]