

Abstract
Non-alcoholic fatty liver disease NAFLD is closely associated with the dysregulation of lipid homeostasis. Diet-induced hepatic steatosis, which can initiate NAFLD progression, has been shown to be dramatically reduced in mice lacking the electroneutral Na+/H+ exchanger NHE1 (Slc9a1). In this study, we investigated if NHE1 deficiency had effects in liver that could contribute to the apparent protection against aberrant lipid accumulation. RT-PCR and immunoblot analyses of wild-type and NHE1-null livers revealed an expression profile that strongly suggested attenuation of both de novo lipogenesis and hepatic stellate cell activation, which is implicated in liver fibrosis. This included upregulation of the farnesoid X receptor FXR, peroxisome proliferator-activated receptor PPARγ, its co-activator PGC1α, and sestrin 2, an antioxidant protein involved in hepatic metabolic homeostasis. Furthermore, expression levels of the pro-lipogenic liver X receptor LXRα, and acety CoA carboxylases 1 and 2 were downregulated. These changes were associated with evidence of reduced cellular stress, which persisted even upon exposure to a high-fat diet, and the better preservation of insulin signaling, as evidenced by protein kinase B/Akt phosphorylation (Ser473). These results indicate that NHE1 deficiency may protect against NAFLD pathogenesis, which is significant given the availability of highly specific NHE1 inhibitors.
Keywords: NHE1, steatohepatitis, liver fibrosis, insulin resistance, metabolic syndrome, obesity
1. INTRODUCTION
Abnormal lipid accumulation in the liver (hepatic steatosis) is considered the first hit in the development of non-alcoholic fatty liver disease (NAFLD) [1], which is an intimate participant in the metabolic syndrome [2]. NAFLD can progress to non-alcoholic steatohepatitis (NASH) with the development of fibrosis and inflammation and ultimately lead to cirrhosis and cancer [3, 4]. Consistent with the increasing prevalence of obesity and diabetes, NAFLD is now the most common liver disease in the western world [3], which highlights an urgent need to identify novel therapeutic targets that can limit its initiation and progression.
Recent analysis of a genetically-modified mouse line lacking expression of the electroneutral Na+/H+ exchanger NHE1 (gene symbol Slc9a1) has revealed that long-term NHE1 ablation attenuates high-fat diet (HFD)-induced lipid accumulation in liver [5]. This effect was associated with lower fasting plasma glucose levels and a blunting of diet-induced body weight gain. It was therefore unclear if the attenuation of hepatic steatosis reflected changes within NHE1 deficient livers. Multiple factors including elevated lipid availability, de novo lipogenesis, and reduced fatty acid oxidation (FAO) can contribute to hepatic steatosis. In addition, progression from hepatic steatosis to NASH is associated with increased cellular stress, the activation of hepatic stellate cells (HSCs), and the development of fibrosis [6–11].
In this study we assessed the effects of NHE1 deficiency on regulators of hepatic lipid handling, cellular stress and on insulin sensitivity. Our results reveal that loss of NHE1 has wide-ranging effects in liver that are directly relevant to the development and progression of NAFLD.
2. MATERIALS AND METHODS
2.1. Animals
Development and husbandry of the global NHE1-null knockout mouse line has been previously described [5, 12]. Maintenance of mice on high-fat diet (60% kcal fat content; D12492, Research Diets, Inc.) for 8 weeks, and treatment with insulin (13.5 IU/kg bodyweight i.p.) has been previously described [5]. All procedures conformed to guidelines published by the National Institutes of Health (Guide for the Care and Use of Laboratory Animals; Publication No. 86-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati.
2.2. Immunoblot and Real-time PCR analyses
Tissue-harvesting and processing of samples for immunoblot and real-time PCR (RT-PCR) analyses was carried out exactly as previously described [5]. All primary and secondary antibodies were the same as previously used [5]. Primer sequences for RT-PCR analysis were obtained from PrimerBank (CCIB, Harvard Medical School) [13] and validated using the NCBI Primer-BLAST program. The following primer pairs were used: FXR (PrimerBank I.D. no. 6677831a1); LXRα (I.D. no. 7305321a1); PGC1α (I.D. no. 6679433a1); PPARγ (I.D. no. 187960104c1); ACC1 (I.D. no. 14211284a1); ACC2 (I.D. no. 18606146a1); SESN2 (I.D. no. 21450289a1); SESN3 (I.D. no. 12856711a1); and, GAPDH (I.D. no. 6679937a3).
2.3. Statistics
Values are presented as means ± standard error (SE). Two-sided Student's t-test were used, and P < 0.05 was considered significant.
3. RESULTS
3.1. Expression of metabolic regulators in NHE1-null livers
Plasma levels of non-esterified fatty acids and triglycerides were comparable between WT and KO mice [5], indicating that altered lipid availability was unlikely to be a major factor in the reduction of hepatic steatosis in KO livers. RT-PCR analysis revealed that mRNA levels of the farnesoid X receptor (FXR, Nr1h4), which has been shown to attenuate hepatic lipogenesis [14], were elevated in KO livers (141 ± 14% of WT; Fig. 1A). In contrast, expression of the liver X receptor alpha (LXRα, Nr1h3), which promotes lipogenesis [15], was downregulated in KO livers (80 ± 5 % of WT; Fig. 1B). Levels of the acetyl CoA-carboxylases (ACC1, Acaca and ACC2, Acacb), which catalyze the first step of de novo lipogenesis [16], were also downregulated in KO livers (ACC1: 73 ± 9 % of WT, Fig. 1C; ACC2 52 ± 7 % of WT; Fig. 1D). In contrast, expression of the peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC1α, Ppargc1a; 216 ± 31% of WT) and peroxisome proliferator-activated receptor gamma (PPARγ, Pparg; 227 ± 38% of WT) was upregulated in KO livers (Figs. 1E, 1F).
Fig. 1.

Expression of metabolic regulators in NHE1-null livers. RT-PCR analysis was carried out as previously described [5] on cDNA samples generated from wild-type (WT) and NHE1-null (KO) livers. Results show that, when normalized to GAPDH, mRNA levels for farnesoid X receptor, FXR (A); peroxisome proliferator-activated receptor gamma coactivator 1, PGC1α (E); and, peroxisome proliferator-activated receptor gamma, PPARγ (F) were elevated while expression of liver X receptor alpha, LXRα (B); acetyl CoA carboxylase 1, ACC1 (C); and, acetyl CoA carboxylase 2, ACC2 (D) was reduced in KO livers. n = at least 8 for each genotype. Values are mean ± SEM. *p < 0.05, #p = 0.05, KO vs WT.
3.2. Effect of NHE1 ablation on regulators and markers of cellular stress
Hepatic lipid homeostasis is closely associated with the regulation of cellular oxidative and endoplasmic reticulum (ER) stress [7, 8, 17]. Sestrins (SESNs) are a family of antioxidant molecules implicated in the maintenance of energy metabolism [18]. Specifically, SESN2 (Sesn2) and SESN3 (Sesn3) have been shown to regulate hepatic lipid accumulation and insulin resistance [19]. mRNA levels for SESN2 were upregulated in KO livers (149 ± 17% of WT; Fig. 2A), while there was no change in SESN3 levels (Fig. 2B). Oxidative stress can also be attenuated by increased scavenging of reactive oxygen species (ROS). Immunoblot analysis however revealed that expression of the cytosolic ROS scavenging enzyme superoxide dismutase 1 (SOD1) was reduced in KO livers (79 ± 4% of WT), whereas there was no change in levels of the mitochondrial SOD2 (Fig. 2C). Protein levels of the small heat shock protein alpha B-crystallin (CRYAB), which are upregulated during HSC activation and liver fibrosis [20, 21], were also reduced in KO livers (51 ± 12% of WT; Fig. 2D).
Fig. 2.

Effect of NHE1 ablation on regulators and markers of cellular stress. RT-PCR analysis was carried out on wild-type (WT) and NHE1-null (KO) samples as described in Fig. 1. Results show that mRNA levels encoding sestrin 2 (SESN2) were elevated (A) while sestrin 3 (SESN3) was unaltered (B) in KO livers. Immunoblot analysis of total protein homogenates revealed that, when normalized to actin, levels of superoxide dismutase 1 (SOD1) (C) and α-Bcrystallin (CRYAB) (D) were reduced in KO livers when compared to WT controls. Expression of mitochondrial SOD2, cytochrome C oxidase subunit IV (COX4) and heat shock protein HSP60 was unaltered. n = at least 8 for each genotype for RT-PCR analysis; at least 4 of each genotype for immunoblot analysis. Values are mean ± SEM. *p < 0.05, KO vs WT.
3.3. Effect of high-fat diet on expression of hepatic NHE1
Studies have revealed a role for increased NHE activity in liver injury and fibrosis [22, 23]. Development of fibrosis is a key event in NAFLD pathogenesis [9, 10] raising the possibility that HFD-induced liver disease may also involve increased NHE expression/activity. We therefore analyzed livers from WT mice maintained on either a normal chow diet or HFD (60% Kcal from fat) for 8 weeks. Immunoblot analysis revealed that NHE1 expression was increased (191 ± 17% of chow-fed controls; Fig. 3) in livers from HFD-fed mice, which had previously been shown to be steatotic [5].
Fig. 3.
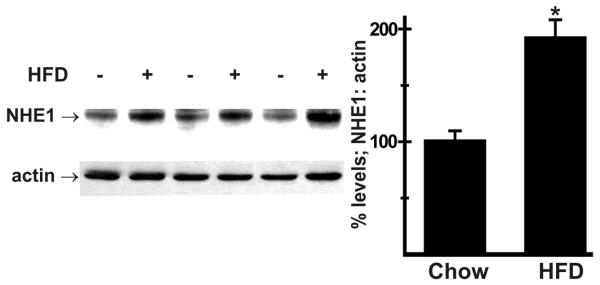
Effect of high-fat diet on NHE1 expression in liver. Wild-type (WT) mice were maintained on a normal chow diet or a high-fat diet (HFD; 60% of kcals in fat) for 8 weeks as previously described [5]. Immunoblot analysis of liver homogenates showed that, when normalized to actin, expression of NHE1 was increased in livers from HFD-fed mice. n = 4 for each diet treatment. Values are mean ± SEM. **p < 0.005.
3.4. Insulin signaling is better preserved in NHE1-null livers
WT and KO mice were maintained on a HFD for 8 weeks, and then treated with insulin (13.5 IU/kg bodyweight) and livers harvested as described above. Phosphorylation levels of Akt (protein kinase B) on Ser473 were determined by immunoblot analysis of liver homogenates in order to assess hepatic insulin sensitivity. Results showed that, when normalized to total Akt expression, phosphorylation of Akt was higher in KO livers (156 ± 13% of WT levels; Fig. 4A). This increase in insulin signaling was associated with a reduction in expression of SOD1 (77 ± 6% of WT) and CRYAB (44 ± 14% of WT) in KO livers (Fig. 4B).
Fig. 4.

Hepatic insulin signaling is better preserved in NHE1-null mice. Wild-type (WT) and NHE1-null (KO) mice were maintained on a high-fat diet (60% of kcals in fat) for 8 weeks and then treated with insulin as described above. Immunoblot analysis of total protein homogenates of livers showed that (A) when normalized to total Akt levels, phosphorylation of Akt on Ser473 (p-Akt) was higher in KO livers when compared to similarly treated WT controls. Results also showed that (B) expression of SOD1 and CRYAB, when normalized to actin, was reduced in KO livers even after exposure to a high-fat diet. n = at least 3 for each genotype. Values are mean ± SEM. *p < 0.05, KO vs WT.
4. DISCUSSION
Activation of FXR attenuates development of hepatic steatosis [24, 25], and treatment with FXR agonists has also been shown to attenuate NASH-related fibrosis [26, 27]. More recently, FXR signaling was identified as the molecular mechanism underlying the beneficial effects of vertical sleeve gastrectomy [28]. In contrast, activation of LXR has been found to promote hepatic steatosis, which has complicated the use of LXR agonists in antiatherogenic and antidiabetic therapies [29–31]. Griffet et al. (2013) have demonstrated that treatment with a liver-specific LXR inverse agonist can reduce the development of hepatic steatosis [32]. Therefore, both the increase in FXR expression and the downregulation of LXRα levels in KO livers were consistent with the observed attenuation of HFD-induced steatosis in KO livers [5].
These findings also raise the possibility that de novo lipogenesis, which is implicated in steatosis [6], is suppressed in KO livers. This is supported by the finding that mRNA levels for ACC1 and AAC2, both of which are key regulators of lipogenesis were reduced in KO livers. Downregulation of ACC1 and ACC2 expression has been shown to reverse hepatic steatosis and insulin resistance [33]. The malonyl-CoA generated by ACC2 also plays a major role in limiting FAO by inhibiting carnitine palmitoyltransferase I [34], which mediates the mitochondrial uptake of long chain fatty acids. Although liver predominantly expresses ACC1, ACC2 deficiency has been shown to reduce triglyceride levels and sharply increase FAO in livers [35, 36]. Therefore, the reduction in ACC2 expression has the potential to facilitate increased fatty acid utilization in KO livers and thereby limit lipid accumulation. It is noteworthy that ACC2 deficiency raises FAO and lipolysis in adipocytes as well, contributing to a leaner phenotype in ACC2 null mice [37]. A similar effect in NHE1 deficient adipocytes could, at least in part, account for the blunting of HFD-induced body weight gain in KO mice [5]. Analyses of adipose-tissue specific NHE1 knockout mice will be necessary to fully elucidate such effects.
Although PGC1α has a critical role in promoting energy metabolism and mitochondrial biogenesis [38, 39], mitochondrial numbers were unlikely to be increased in KO livers, as indicated by the unaltered COX4 protein and Tfam mRNA (data not shown) levels. Loss of PGC1α has been shown to reduce FAO, impair gluconeogenesis and cause hepatic steatosis upon fasting [40, 41]. Conversely, hepatic glucose production was found to be elevated in a global PGC1α overexpression model [42]. This finding, in the context of increased muscle glucose utilization, was interpreted as evidence that PGC1α activity helps balance hepatic glucose production with peripheral glucose disposal. Similar regulatory mechanisms involving PGC1α overexpression could be elicited in KO livers given the evidence that energy expenditure may be increased in NHE1-deficient skeletal muscle (Prasad et al., unpublished results). Consistent with this possibility, mRNA levels of phosphoenolpyruvate carboxylase (Pck1), which catalyzes the rate-limiting step in gluconeogenesis, trended higher (147 ± 16% of WT; p = 0.052) in KO livers.
The effects of PPARγ activation in liver remain to be fully understood and are likely to be complex and cell-type dependent. PPARγ is expressed at relatively low levels in liver and is upregulated in fatty livers [43]. Although thiazolidinediones (TZDs), which activate PPARγ, have been shown to reduce hepatic steatosis [44], this effect is believed to be mediated via the insulin-sensitizing effects of TZDs on adipocytes, which causes a preferential partitioning of lipids from liver and muscle to adipose tissue. In fact, hepatocyte-specific deletion of PPARγ has been shown to decrease diet-induced lipid accumulation [45], indicating a pro-steatotic role for PPARγ in liver parenchymal cells. Elevated Pparγ expression did not however lead to an exacerbation of diet-induced steatosis in KO livers; on the contrary, HFD-induced hepatic lipid accumulation was sharply reduced in KO mice [5]. Consistent with a protective role for Pparγ in liver, there is evidence that it helps maintain HSC quiescence [46]. Activation of HSCs, which constitute 5–8% of total liver cells [47], is strongly implicated in the development of liver fibrosis [10, 11], which is integral to the development of steatohepatitis [48]. PPARγ has been shown to reverse the activation and proliferation of HSCs [46], with rosiglitazone, a PPARγ agonist, preventing the development of diet-induced steatohepatitis [49]. Upregulation of PPARγ therefore has the potential to protect against liver fibrosis. Any such protection afforded in KO livers would mimic findings in heart where long-term treatment with the NHE1-inhibitor cariporide has been shown to limit age-related fibrosis [50].
Also implicated in the attenuation of age-related diseases are the SESNs [18]. SESN2 is induced in obesity [19], and may be particularly relevant to hepatic metabolic homeostasis. Loss of SESN2 exacerbates diet-induced hepatic steatosis and insulin resistance [19]. The increased expression of SESN2 was therefore consistent with the reduction of HFD-induced steatosis in KO livers [5]. SESNs are also known to alleviate oxidative stress and a reduction in baseline oxidative stress could account for the downregulation of SOD1 and CRYAB protein levels in KO livers. A similar reduction in levels of SOD2, HSP60 and HSP25 was found to be associated with reduced oxidative stress in KO hearts [5].
Upregulation of CRYAB is associated with HSC activation and liver fibrosis [20, 21] and there is early evidence that CRYAB may promote cell survival in hepatocellular carcinomas (HCCs) [51]. In fact, CRYAB expression has been shown to serve as a negative prognostic marker of survival in human HCC patients [52]. The reduction in CRYAB levels in KO livers, seen under both normal and HFD-fed conditions, raises the possibility that NHE1 deficiency may antagonize diet-induced hepatocarcinogenesis as well. Indeed, increased NHE activity has been associated with hepatocyte proliferation, regeneration and growth [53]. NHE1 expression and activity are increased in HCC, with NHE1 inhibition limiting HCC tumor growth [54, 55]. Inhibition of NHE activity has also been reported to attenuate liver fibrosis [56], indicating a role for augmented Na+/H+ exchange in HSC activation and proliferation [57]. These findings highlight a potential role for NHE activity in various stages of NAFLD progression, and indicate that the nearly 2-fold increase in NHE1 expression seen in livers of HFD-fed WT mice may serve a pathological role.
The metabolic regulatory mechanisms impacted by NHE1 are not fully understood as yet; studies in heart have revealed that NHE1 expression impacts substrate flexibility and the coupling of glycolysis to glucose oxidation [5, 58]. In addition, evidence that NHE1 may also be localized to the mitochondrial inner membrane [59, 60] has raised the possibility of a more direct role in regulating energy expenditure. It is also possible that in liver, NHE1 may serve distinct roles in hepatocytes and HSCs, all of which remains to be elucidated. Nevertheless, the better preservation of insulin signaling in livers from HFD-fed KO mice strongly supports the idea that NHE1 deficiency can protect the liver from the deleterious effects of HFD. Taken together, these findings highlight the possibility that NHE1 is an effective therapeutic target against NAFLD.
HIGHLIGHTS.
FXR, PGC1α and PPARγ levels are upregulated in NHE1 deficient livers.
NHE1 deficiency downregulates expression of pro-lipogenic genes in liver.
Chronic exposure to high-fat diet upregulates hepatic NHE1 expression.
Loss of NHE1 better preserves hepatic insulin signaling in high-fat diet-fed mice.
ACKNOWLEDGEMENTS
This work was funded by grants from the Cincinnati Diabetes and Obesity Center (V.P.) and American Heart Association (11BGIA7720005; V.P.), and National Institutes of Health (DK050594; G.E.S.). Technical assistance by Sowmya S. Balusu (supported by the University of Cincinnati Honors Biomedical Research and Mentoring Program) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES None
REFERENCES
- [1].Day CP, James OF. Steatohepatitis: a tale of two “hits”. Gastroenterol. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- [2].Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- [3].Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J. Hepatol. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- [5].Prasad V, Lorenz JN, Miller ML, et al. Loss of NHE1 activity leads to reduced oxidative stress in heart and mitigates high-fat diet-induced myocardial stress. J. Mol. Cell Cardiol. 2013;65:33–42. doi: 10.1016/j.yjmcc.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008;34:643–648. doi: 10.1016/S1262-3636(08)74599-3. [DOI] [PubMed] [Google Scholar]
- [7].Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta. 2011;412:1297–1305. doi: 10.1016/j.cca.2011.04.013. [DOI] [PubMed] [Google Scholar]
- [8].Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- [9].Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH) Int. J. Mol. Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rombouts K, Marra F. Molecular mechanisms of hepatic fibrosis in non-alcoholic steatohepatitis. Dig. Dis. 2010;28:229–235. doi: 10.1159/000282094. [DOI] [PubMed] [Google Scholar]
- [11].Washington K, Wright K, Shyr Y, et al. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum. Pathol. 2000;31:822–828. doi: 10.1053/hupa.2000.8440. [DOI] [PubMed] [Google Scholar]
- [12].Bell SM, Schreiner CM, Schultheis PJ, et al. Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am. J. Physiol. 1999;276:C788–795. doi: 10.1152/ajpcell.1999.276.4.C788. [DOI] [PubMed] [Google Scholar]
- [13].Wang X, Spandidos A, Wang H, et al. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucl. Acids Res. 2012;40:D1144–1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Trauner M, Claudel T, Fickert P, et al. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig. Dis. 2010;28:220–224. doi: 10.1159/000282091. [DOI] [PubMed] [Google Scholar]
- [15].Schultz JR, Tu H, Luk A, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Numa S, Nakanishi S, Hashimoto T, et al. Role of acetyl coenzyme A carboxylase in the control of fatty acid synthesis. Vitam. Horm. 1970;28:213–243. doi: 10.1016/s0083-6729(08)60895-x. [DOI] [PubMed] [Google Scholar]
- [17].Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front. Genet. 2014;5:112. doi: 10.3389/fgene.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee JH, Budanov AV, Karin M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013;18:792–801. doi: 10.1016/j.cmet.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JH, Budanov AV, Talukdar S, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012;16:311–321. doi: 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cassiman D, Roskams T, van Pelt J, et al. Alpha B-crystallin expression in human and rat hepatic stellate cells. J. Hepatol. 2001;35:200–207. doi: 10.1016/s0168-8278(01)00122-2. [DOI] [PubMed] [Google Scholar]
- [21].Lang A, Schrum LW, Schoonhoven R, et al. Expression of small heat shock protein alphaB-crystallin is induced after hepatic stellate cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G1333–1342. doi: 10.1152/ajpgi.2000.279.6.G1333. [DOI] [PubMed] [Google Scholar]
- [22].Wang D, Dou K, Song Z, et al. The Na+/H+ exchange inhibitor: a new therapeutic approach for hepatic ischemia injury in rats. Transplant Proc. 2003;35:3134–3135. doi: 10.1016/j.transproceed.2003.10.021. [DOI] [PubMed] [Google Scholar]
- [23].Svegliati-Baroni G, Di Sario A, Casini A, et al. The Na+/H+ exchanger modulates the fibrogenic effect of oxidative stress in rat hepatic stellate cells. J. Hepatol. 1999;30:868–875. doi: 10.1016/s0168-8278(99)80141-x. [DOI] [PubMed] [Google Scholar]
- [24].Cipriani S, Mencarelli A, Palladino G, et al. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid. Res. 2010;51:771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma Y, Huang Y, Yan L, et al. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013;30:1447–1457. doi: 10.1007/s11095-013-0986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang S, Wang J, Liu Q, et al. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 2009;51:380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- [27].Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–582. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- [28].Ryan KK, Tremaroli V, Clemmensen C, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Joseph SB, McKilligin E, Pei L, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. U S A. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baranowski M, Zabielski P, Błachnio-Zabielska AU, et al. Insulin-Sensitizing Effect of LXR Agonist T0901317 in High-Fat Fed Rats is Associated with Restored Muscle GLUT4 Expression and Insulin-Stimulated AS160 Phosphorylation. Cell Physiol. Biochem. 2014;33:1047–1057. doi: 10.1159/000358675. [DOI] [PubMed] [Google Scholar]
- [31].Ducheix S, Montagner A, Theodorou V, et al. The liver X receptor: a master regulator of the gut-liver axis and a target for non alcoholic fatty liver disease. Biochem. Pharmacol. 2013;86:96–105. doi: 10.1016/j.bcp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- [32].Griffett K, Solt LA, El-Gendy Bel-D., et al. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol. 2013;8:559–567. doi: 10.1021/cb300541g. [DOI] [PubMed] [Google Scholar]
- [33].Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- [35].Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, et al. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- [36].Abu-Elheiga L, Oh W, Kordari P, et al. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. U S A. 2003;100:10207–10212. doi: 10.1073/pnas.1733877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oh W, Abu-Elheiga L, Kordari P, et al. Glucose and fat metabolism in adipose tissue of acetyl-CoA carboxylase 2 knockout mice. Proc. Natl. Acad. Sci. U S A. 2005;102:1384–1389. doi: 10.1073/pnas.0409451102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv. Physiol. Educ. 2006;30:145–151. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- [39].Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta. 2011;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Burgess SC, Leone TC, Wende AR, et al. Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha)-deficient mice. J. Biol. Chem. 2006;281:19000–19008. doi: 10.1074/jbc.M600050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Estall JL, Kahn M, Cooper MP, et al. Sensitivity of lipid metabolism and insulin signaling to genetic alterations in hepatic peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression. Diabetes. 2009;58:1499–1508. doi: 10.2337/db08-1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liang H, Balas B, Tantiwong P, et al. Whole body overexpression of PGC-1alpha has opposite effects on hepatic and muscle insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2009;296:E945–954. doi: 10.1152/ajpendo.90292.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Invest. 1997;99:2416–2422. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ratziu V, Charlotte F, Bernhardt C, et al. LIDO Study Group Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–453. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- [45].Morán-Salvador E, López-Parra M, García-Alonso V, et al. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- [46].Hazra S, Xiong S, Wang J, et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J. Biol. Chem. 2004;279:11392–11401. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- [47].Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- [48].Bataller R, Brenner DA. Liver fibrosis. J. Clin. Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nan YM, Fu N, Wu WJ, et al. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand. J. Gastroenterol. 2009;44:358–365. doi: 10.1080/00365520802530861. [DOI] [PubMed] [Google Scholar]
- [50].Linz WJ, Busch AE. NHE-1 inhibition: from protection during acute ischaemia/reperfusion to prevention/reversal of myocardial remodeling. Naunyn Schmiedebergs Arch. Pharmacol. 2003;368:239–246. doi: 10.1007/s00210-003-0808-2. [DOI] [PubMed] [Google Scholar]
- [51].Huang XY, Ke AW, Shi GM, et al. αB-crystallin complexes with 14-3-3ζ to induce epithelial-mesenchymal transition and resistance to sorafenib in hepatocellular carcinoma. Hepatology. 2013;57:2235–2247. doi: 10.1002/hep.26255. [DOI] [PubMed] [Google Scholar]
- [52].Tang Q, Liu YF, Zhu XJ, et al. Expression and prognostic significance of the alpha B-crystallin gene in human hepatocellular carcinoma. Hum. Pathol. 2009;40:300–305. doi: 10.1016/j.humpath.2008.09.002. [DOI] [PubMed] [Google Scholar]
- [53].Kiela PR, Ghishan FK. Na+/H+ exchange in mammalian digestive tract. In: Johnson LR, editor. Physiology of the gastrointestinal tract. fifth ed. Vol. 2. Elsevier Inc.; London, UK: 2012. pp. 1781–1818. [Google Scholar]
- [54].Strazzabosco M, Poci C, Spirlì C, et al. Intracellular pH regulation in Hep G2 cells: effects of epidermal growth factor, transforming growth factor-alpha, and insulinlike growth factor-II on Na+/H+ exchange activity. Hepatology. 1995;22:588–597. [PubMed] [Google Scholar]
- [55].Yang X, Wang D, Dong W, et al. Expression and modulation of Na(+) /H(+) exchanger 1 gene in hepatocellular carcinoma: A potential therapeutic target. J. Gastroenterol. Hepatol. 2011;26:364–370. doi: 10.1111/j.1440-1746.2010.06382.x. [DOI] [PubMed] [Google Scholar]
- [56].Benedetti A, Di Sario A, Casini A, et al. Inhibition of the NA(+)/H(+) exchanger reduces rat hepatic stellate cell activity and liver fibrosis: an in vitro and in vivo study. Gastroenterology. 2001;120:545–556. doi: 10.1053/gast.2001.21203. [DOI] [PubMed] [Google Scholar]
- [57].Di Sario A, Bendia E, Taffetani S, et al. Selective Na+/H+ exchange inhibition by cariporide reduces liver fibrosis in the rat. Hepatology. 2003;37:256–266. doi: 10.1053/jhep.2003.50028. [DOI] [PubMed] [Google Scholar]
- [58].Mraiche F, Wagg CS, Lopaschuk GD, et al. Elevated levels of activated NHE1 protect the myocardium and improve metabolism following ischemia/reperfusion injury. J. Mol. Cell Cardiol. 2011;50:157–164. doi: 10.1016/j.yjmcc.2010.10.016. [DOI] [PubMed] [Google Scholar]
- [59].Javadov S, Rajapurohitam V, Kilić A, et al. Expression of mitochondrial fusion–fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res. Cardiol. 2011;106:99–109. doi: 10.1007/s00395-010-0122-3. [DOI] [PubMed] [Google Scholar]
- [60].Alvarez BV, Villa-Abrille MC. Mitochondrial NHE1: a newly identified target to prevent heart disease. Front. Physiol. 2013;28:152. doi: 10.3389/fphys.2013.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]