

Abstract
Integrated whole-cell modeling is poised to make a dramatic impact on molecular and systems biology, bioengineering, and medicine — once certain obstacles are overcome. From our group’s experience building a whole-cell model of Mycoplasma genitalium, we identified several significant challenges to building models of more complex cells. Here we review and discuss these challenges in seven areas: (1) experimental interrogation, (2) data curation, (3) model building and integration, (4) accelerated computation, (5) analysis and visualization, (6) model validation, and (7) collaboration and community development. Surmounting these challenges will require the cooperation of an interdisciplinary group of researchers to create increasingly sophisticated whole-cell models and make data, models, and simulations more accessible to the wider community.
1 Introduction
Predictive and comprehensive models of cellular physiology are critical to understanding and engineering biological systems. Such whole-cell models have the potential to guide experiments in molecular biology, enable computer-aided design and simulation in synthetic biology, and inform personalized treatment in medicine. Constructing and validating models with sufficient scope, detail, and predictive power, for a variety of cells, will be a massive undertaking.
Beginning in the late 1970s [1], researchers began modeling cell physiology, primarily using ordinary differential equation (ODE) approaches, creating increasingly detailed models over the next three decades [2, 3, 4]. Later, other groups introduced frameworks that generally require fewer parameters than ODE systems including constraint-based [5, 6] and Boolean methods [7]. Combining these approaches for their respective benefits, our group developed a hybrid methodology: we modeled individual biological processes, each with its own mathematical representation, and merged their outputs to compute the overall state of the cell [8]. Using this approach, we simulated the life cycle of individual Mycoplasma genitalium cells, accounting for every molecule and representing the function of every annotated gene [9].
Several unforeseen obstacles arose during the modeling process, which should inform any future whole-cell modeling efforts. Specifically, modeling larger cells and more complex physiology presents challenges in (1) experimental interrogation, (2) data curation, (3) model building and integration, (4) accelerated computation, (5) analysis and visualization, (6) model validation, and (7) collaboration and community development, shown in Figure 1. No single research group can simultaneously innovate in all these areas. Rather, a broader community will need to coalesce to tackle these problems. We address this article to that community, discussing the challenges and highlighting notable progress in each area.
Figure 1.
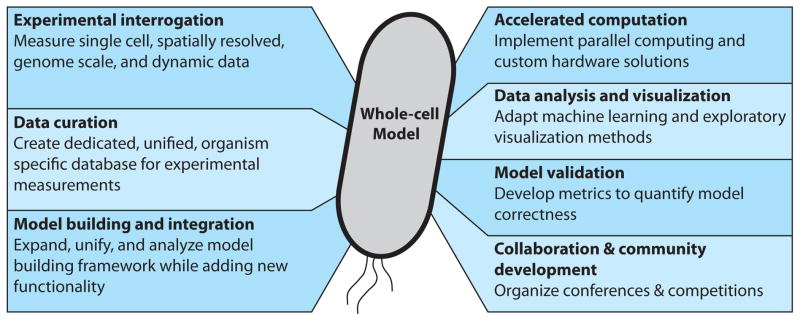
The interdisciplinary challenges faced by future whole-cell modeling efforts. A community of scientists and engineers will need to innovate together to surmount these challenges.
2 Experimental interrogation
Parameterizing and validating the M. genitalium whole-cell model was particularly challenging due to a lack of organism-specific data. Many values were estimated from measurements made in other species. Future efforts will ideally simulate well-characterized organisms, for example Mycoplasma pneumoniae [10, 11, 12, 13], Escherichia coli [14], and Saccharomyces cerevisiae [15, 16]. Because whole-cell models simulate the life-cycle of an individual cell, one would ideally use spatially-resolved, genome-scale, dynamic, single-cell measurements to parameterize and validate the models. However, many published measurements are static ensemble averages representing a population mean at a single time point [17, 18, 19, 20, 21]. This lack of data ultimately presents the modeler with a dilemma: either infer missing data, or create a less detailed model of a particular phenomenon. To create the M. genitalium model, we necessarily inferred some degree of dynamical behavior. Faced with a similar problem, others have found ways to incorporate static spatial data in their efforts to create dynamic 3D cell-scale simulations [22]. Promising work in advancing single-cell measurement techniques and technologies [23, 24, 25, 26] will ultimately drive more detailed and accurate modeling. To make these efforts even more impactful and useful, the experimental community could work to establish standardized conditions and place a higher value on consistent, reproducible measurements.
3 Data curation
No single technology exists which can chronically measure and record the entire state of a single cell. As a result, heterogeneous data sets must be combined and unified for model parameterization and validation. While efforts such as the BioCyc databases have sought to unify genomic and metabolic pathway information [27], separate databases contain functional parameters such as kinetic rates [28, 29] and expression levels [30]. To compile the data required to build the M. genitalium model, which we share via WholeCellKB [31], we had to download and synthesize parameters from these and other databases as well as the primary literature. For larger and more complex organisms, the sheer magnitude of data to collect, and the number of discrepancies to resolve, will present significant hurdles to parameterizing a model.
Since parameterization data increases with organism complexity and known physiology, a part-time manual curation effort will not be tenable. Researchers will need to exploit advances in natural language processing to extract information from the primary literature en masse [32], or outsource part of the effort. Formally interacting with domain experts, as has been done in the flux-balance analysis community [33], will be critical to assembling consensus data sets. Ultimately, a combination of computer-automated and human-augmented approaches will be necessary to gather and assemble the data for larger whole-cell models.
A collection of centralized, organism-specific databases similar to WholeCellKB will be required for subsequent whole-cell modeling efforts. In the best case, researchers would go beyond including raw data for each figure in a paper [34] and would deposit their results to the appropriate database in a machine-readable format. Dedicated curators would update the database schemas to incorporate new types of information as needed. In addition, the databases would alert the community to significant discrepancies between parameters and flag them as critical issues to resolve. By providing these capabilities, the databases would link experimental evidence to whole-cell models.
4 Model building and integration
Comprehensively representing cell physiology in a single computational model requires integrating diverse phenomena over multiple length and time scales, handling the different levels of understanding associated with each phenomenon, and representing the state of the cell in sufficient detail. Our lab’s approach to meeting these requirements relies on the notion of biological modularity [35], allowing us to divide the cell into independent state variables (e.g., representing metabolite counts or the functional state of macromolecules) and cellular processes (e.g., transcription, metabolism) [9]. We create sub-models of each cellular process using a mathematical representation informed by available data and current understanding. We assume that, over a small time step, each sub-model can independently execute and update a subset of the cell state variables. To meaningfully combine sub-models in this fashion, we must (1) establish and link common variables, and (2) ensure that the combined behavior is consistent with physical laws and biological phenotypes.
To avoid duplicating work, it is desirable to incorporate published models of particular biological processes into a whole-cell modeling framework. This often requires that the published models be modified to use the common whole-cell state variables, which may, for example, involve changing the published model’s quantities from concentrations to counts, or linking its variables to the appropriate cell compartment in the whole-cell framework. Establishing mathematical methods for properly converting a spatially-resolved variable, used in a detailed sub-model, to a bulk quantity, or even to a Boolean value, used in a less-detailed sub-model, would ease the data interconversion between sub-models. Numerical analysis of these methods could be performed to examine factors which affect stability and accuracy of the simulations, and to quantify numerical uncertainty in model predictions.
With a collection of sub-models that properly interface with cell state variables, it must further be enforced that their aggregate behavior does not violate physical laws. For example, the aggregate action of multiple sub-models should not result in the consumption of more resources than are present. To avoid this situation, we developed a method to allocate cell state variables to biological processes proportional to each process’s need. In the future, this top-down approach could be replaced with one more grounded in physical laws.
Furthermore, the aggregate behavior of a collection of sub-models should be consistent with biological phenotypes. For instance, the small molecule, RNA, protein, and DNA mass fractions, must approximately double over the exponentially-growing cell’s life cycle. This requirement constrains certain sub-model parameters so that metabolism, for example, produces nucleotides and amino acids in the proportions needed by replication, transcription, and translation. The M. genitalium model performed this adjustment prior to simulation; however, new methods must be developed to update these loosely-coupled parameters during simulation. Importantly, this will enable proper incorporation of regulatory sub-models [36, 37] which modify the nucleotide and amino acid demands as the RNA and protein expression profiles change in response to perturbations.
5 Accelerated computation
Computational simulation is a powerful scientific and engineering tool because it enables rapid and inexpensive exploration of alternative scenarios and hypotheses, as well as design optimization. Such investigations, however, hinge on efficient computation in order to explore a sufficiently large portion of parameter space. The whole-cell simulations of M. genitalium, which each took approximately ten hours to run, do not meet this criteria. We can extrapolate that, without innovation in this area, simulations of more complex organisms will take considerably longer to execute. High-performance parallelized computing technologies, such as the Compute Unified Device Architecture (CUDA) [38] or Message Passing Interface (MPI) [39], or even custom hardware platforms [40], in the spirit of Anton [41] or Neurogrid [42], should be adapted and investigated for their abilities to speed-up the execution of whole-cell simulations.
6 Data analysis and visualization
Raw simulation data, like raw experimental data, typically requires extensive analysis to be adequately understood and communicated. Techniques from machine learning and dynamical systems analysis could be used to explore and interrogate simulated single-cell phenotypes. These analyses could suggest novel hypotheses about the dynamics of single cells that wouldn’t emerge from static, population-averaged data.
To complement analysis technologies, advances are needed in large-data visualization. While our group released WholeCellViz to expose a portion of the M. genitalium data set [43], going forward more sophisticated tools must be developed, particularly for exploration, rather than just communication, of large data sets. This requires the development of not only new visual motifs for biological data, but also improvements in data processing and retrieval to enable interactive interfaces for manipulating entire data sets. Existing tools [44] offer these interactive exploratory interfaces, but generally operate on smaller data sets [45]. Fortunately, these problems are recognized as pressing issues by the visualization community [46]. Preliminary work has begun to explore new visual motifs for biological data [47], [48], [49], and the high-performance computing community is supporting new techniques to improve data retrieval [50].
7 Model validation
Model predictions and experimental validation are linked by an iterative process in which each provides feedback on the other [51]. For the initial validation of the M. genitalium whole-cell model, we simply compared model predictions to as many heterogeneous data sets as possible that were withheld from model reconstruction. We have also used the model to predict the outcome of experiments which are performed subsequently [52]. Nevertheless, the validation process for the M. genitalium model has been guided more by intuition than by a systematic methodology. Ideally, a quantitative metric would exist to specify how much of a model has been validated and would point to data sets needed to improve the coverage of validation. More subtly, methods should be developed which can differentiate novel predictions (e.g., gene essentiality in the M. genitalium model) from outputs arising directly from parameter fitting (e.g., biomass composition in the M. genitalium model). These innovations would support more widespread model adoption by building trust in the predictions.
8 Collaboration and community development
Whole-cell models of more complex microbes and cell types will likely become community endeavors, particularly as the models grow in scope and detail. To facilitate interaction with the broader community, we released the entire code base for the M. genitalium whole-cell model under the MIT license [53], permitting open development and re-use. Going forward, we must engage the broader community in contributing to whole-cell model development. The interface between cell state variables and process sub-models must be explicitly documented in detail to lower the barrier to contribution. Furthermore, a formal plug-in system must be developed to simplify the incorporation of alternate sub-models for a particular process. At the project-management level, metrics to quantify contribution and guidelines for authorship need to be proposed and ratified. At the community level, workshops, conferences, and competitions [54] specifically focusing on whole-cell modeling need to be organized to engage the breadth of contributing researchers.
9 Conclusion
The need to address the aforementioned challenges provides a wealth of opportunities for interdisciplinary contribution by experimentalists, modelers, computer scientists, statisticians, bioinformaticians, and software engineers. We hope a community will form where scientists and engineers from diverse backgrounds can collaborate and innovate together to overcome these obstacles.
Whole-cell modeling can help researchers prioritize experiments by identifying knowledge gaps and by highlighting measurement discrepancies [52]. Additionally, the comprehensive scope of a whole-cell model enables predictions of the pleiotropic effects of perturbation [55], critical to the future of synthetic biology and personalized medicine. Addressing the issues discussed here will enable whole-cell modeling to realize its potential, and in the process make an impact on model-guided science, synthetic biology, and medicine.
Highlights.
Whole-cell models have the potential to impact science, bioengineering, and medicine.
We highlight seven challenges in whole-cell modeling.
We hope an interdisciplinary community will form to address these challenges.
Acknowledgments
We thank Elsa Birch, Ellen Casavant, Shrivats Iyer, and Jonathan Karr for their critical feedback of this manuscript, as well as members of the Covert Lab for enlightening discussions on the topic. This work was supported by an NIH Director’s Pioneer Award (5DP1LM011510-05), an Allen Distinguished Investigator Award, and an award from the Stanford Bio-X Corporate Forum and Agilent to MWC, a Benchmark Stanford Graduate Fellowship and DOE CSGF Fellowship (DE-FG02-97ER25308) to DNM, and an NSF Graduate Research Fellowship to NAR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shuler ML, Leung S, Dick CC. A Mathematical Model for the Growth of a Single Cell. Annals of the New York Academy of Sciences. 1979;326(1):35–52. [Google Scholar]
- 2.Domach MM, Shuler ML. A finite representation model for an asynchronous culture of E. coli. Biotechnology and Bioengineering. 1984 Aug;26(8):877–884. doi: 10.1002/bit.260260810. [DOI] [PubMed] [Google Scholar]
- 3.Tomita M, et al. E-CELL: software environment for whole-cell simulation. Bioinformatics. 1999 Jan;15(1):72–84. doi: 10.1093/bioinformatics/15.1.72. [DOI] [PubMed] [Google Scholar]
- 4*.Shuler Michael L, Foley Patricia, Atlas Jordan. Modeling a minimal cell. Methods in molecular biology (Clifton, NJ) 2012;881:573–610. doi: 10.1007/978-1-61779-827-6_20. Annotations: An ordinary differential equation model of a “minimal” cell with the smallest gene set required to grow and divide. Incorporates diverse aspects of cellular physiology including transcription, translation, metabolism, and replication. [DOI] [PubMed] [Google Scholar]
- 5.Savinell JM, Palsson BO. Network analysis of intermediary metabolism using linear optimization. I. Development of mathematical formalism. Journal of Theoretical Biology. 1992;154(4):421–454. doi: 10.1016/s0022-5193(05)80161-4. [DOI] [PubMed] [Google Scholar]
- 6.Varma A, Palsson BO. Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Applied and environmental Microbiology. 1994;60(10):3724. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson Eric H, et al. A genomic regulatory network for development. Science (New York, NY) 2002 Mar;295(5560):1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 8.Covert Markus W, et al. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24(18):2044–2050. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Karr Jonathan R, et al. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell. 2012 Jul;150(2):389–401. doi: 10.1016/j.cell.2012.05.044. Annotations: The first whole-cell computational model. Simulates the life cycle of the human pathogen Mycoplasma genitalium including all functionally annotated gene products and their interactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Güell Marc, et al. Transcriptome complexity in a genome-reduced bacterium. Science (New York, NY) 2009 Nov;326(5957):1268–1271. doi: 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- 11.Kühner Sebastian, et al. Proteome organization in a genome-reduced bacterium. Science (New York, NY) 2009 Nov;326(5957):1235–1240. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- 12.Yus Eva, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science (New York, NY) 2009 Nov;326(5957):1263–1268. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- 13.Maier Tobias, et al. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Molecular Systems Biology. 2011;7(1) doi: 10.1038/msb.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishii Nobuyoshi, et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science (New York, NY) 2007 Apr;316(5824):593–597. doi: 10.1126/science.1132067. [DOI] [PubMed] [Google Scholar]
- 15.Picotti Paola, et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013 Jan;494(7436):266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller Christian, et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Molecular Systems Biology. 2011 Jan;7(1) doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoheisel Jörg D. Microarray technology: beyond transcript profiling and genotype analysis. Nature reviews Genetics. 2006 Mar;7(3):200–210. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 18.Wang Zhong, Gerstein Mark, Snyder Michael. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews Genetics. 2009 Jan;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahmood Tahrin, Yang Ping-Chang. Western blot: technique, theory, and trouble shooting. North American journal of medical sciences. 2012 Sep;4(9):429–434. doi: 10.4103/1947-2714.100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher Sean R. One-dimensional SDS gel electrophoresis of proteins. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit 10.2A. Chapter 10. Aug, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Furey Terrence S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nature reviews Genetics. 2012 Dec;13(12):840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Roberts Elijah, et al. Noise contributions in an inducible genetic switch: a whole-cell simulation study. PLoS Computational Biology. 2011 Mar;7(3):e1002010. doi: 10.1371/journal.pcbi.1002010. Annotations: Stochastic, single-cell, spatially-resolved kinetic model which simulates the switching of the lac operon in Escherichia coli. Critical steps taken in modeling more complex bacterial cell physiology spatially. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taniguchi Yuichi, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science (New York, NY) 2010 Jul;329(5991):533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee Timothy K, et al. A noisy paracrine signal determines the cellular NF-kappaB response to lipopolysaccharide. Science Signaling. 2009;2(93):ra65. doi: 10.1126/scisignal.2000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Fuchou, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature methods. 2009 May;6(5):377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 26.Ibáñez Alfredo J, et al. Mass spectrometry-based metabolomics of single yeast cells. Proceedings of the National Academy of Sciences. 2013 May;110(22):8790–8794. doi: 10.1073/pnas.1209302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspi Ron, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic acids research. 2010 Jan;38:D473–9. doi: 10.1093/nar/gkp875. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schomburg Ida, et al. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic acids research. 2013 Jan;41:D764–72. doi: 10.1093/nar/gks1049. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wittig U, et al. SABIO-RK–database for biochemical reaction kinetics. Nucleic acids research. 2011 Dec;40.D1:D790–D796. doi: 10.1093/nar/gkr1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett T, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic acids research. 2012 Dec;41.D1:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karr Jonathan R, et al. WholeCellKB: model organism databases for comprehensive whole-cell models. Nucleic acids research. 2013;41.D1:D787–D792. doi: 10.1093/nar/gks1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finkel Jenny, et al. Exploring the boundaries: gene and protein identification in biomedical text. BMC bioinformatics. 2005;6(Suppl 1):S5. doi: 10.1186/1471-2105-6-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thiele Ines, Palsson Bernhard O. Reconstruction annotation jamborees: a community approach to systems biology. Molecular Systems Biology. 2010 Apr;6:361. doi: 10.1038/msb.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Announcement: Reducing our irreproducibility. Nature. 2013 Apr;496(7446):398–398. [Google Scholar]
- 35.Hartwell Leland H, et al. From molecular to modular cell biology. Nature. 1999 Dec;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- 36.Bonneau Richard, et al. The Inferelator: an algorithm for learning parsimonious regulatory net-works from systems-biology data sets de novo. Genome Biology. 2006;7(5):R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrera Javier, Rodrigo Guillermo, Jaramillo Alfonso. Model-based redesign of global transcription regulation. Nucleic acids research. 2009 Apr;37(5):e38. doi: 10.1093/nar/gkp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.CUDA Toolkit Documentation. url: http://docs.nvidia.com/cuda/index.html.
- 39.MPI Documents. url: http://www.mpi-forum.org/docs/
- 40.Gunawardena Jeremy. Silicon dreams of cells into symbols. Nature Biotechnology. 2012 Sep;30(9):838–840. doi: 10.1038/nbt.2358. [DOI] [PubMed] [Google Scholar]
- 41.Dror Ron O, et al. Biomolecular simulation: a computational microscope for molecular biology. Annual Review of Biophysics. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 42.Silver Rae, et al. Neurotech for neuroscience: unifying concepts, organizing principles, and emerging tools. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007 Oct;27(44):11807–11819. doi: 10.1523/JNEUROSCI.3575-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee Ruby, Karr Jonathan R, Covert Markus W. WholeCellViz: data visualization for whole-cell models. BMC bioinformatics. 2013;14(1):253. doi: 10.1186/1471-2105-14-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Business Intelligence and Analytics Software. url: http://www.tableausoftware.com/
- 45.Tableau Technology|Tableau Software. url: http://www.tableausoftware.com/products/technology.
- 46.Wong Pak Chung, et al. The Top 10 Challenges in Extreme-Scale Visual Analytics. IEEE Computer Graphics and Applications. 2012;32(4):63–67. doi: 10.1109/mcg.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer Miriah, et al. MulteeSum: a tool for comparative spatial and temporal gene expression data. IEEE transactions on visualization and computer graphics. 2010 Nov;16(6):908–917. doi: 10.1109/TVCG.2010.137. [DOI] [PubMed] [Google Scholar]
- 48.Meyer M, et al. Pathline: A Tool For Comparative Functional Genomics. Computer Graphics Forum. 2010 Aug;29(3):1043–1052. [Google Scholar]
- 49.Meyer Miriah, Munzner Tamara, Pfister Hanspeter. MizBee: a multiscale synteny browser. IEEE transactions on visualization and computer graphics. 2009 Nov;15(6):897–904. doi: 10.1109/TVCG.2009.167. [DOI] [PubMed] [Google Scholar]
- 50.Ashby Steve, et al. The opportunities and challenges of exascale computing|summary report of the advanced scientific computing advisory committee (ASCAC) subcommittee. US Department of Energy Office of Science. US Department of Energy Office of Science. 2010 [Google Scholar]
- 51.Kitano Hiroaki. Computational systems biology. The American Journal of Gastroenterology. 2002 Nov;420(6912):206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 52.Sanghvi Jayodita C, et al. Accelerated discovery via a whole-cell model. Nature Methods. 2013;10(12):1192–1195. doi: 10.1038/nmeth.2724. Annotations: The Mycoplasma genitalium whole-cell model is demonstrated to accelerate biological discovery by guiding experiments. Turnover rates for three metabolic enzymes are identified as incorrect and subsequently correctly predicted or bounded. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The MIT License (MIT)|Open Source Initiative. url: http://opensource.org/licenses/MIT.
- 54.Whole-cell parameter estimation DREAM challenge - syn1876068. url: https://www.synapse.org/#!Synapse:syn1876068.
- 55*.Purcell Oliver, et al. Towards a whole-cell modeling approach for synthetic biology. Chaos (Woodbury, NY) 2013 Jun;23(2):025112. doi: 10.1063/1.4811182. Annotations: Modifies the existing whole-cell model to allow for genome modification. Engineered a synthetic gene circut for a Goodwin oscillator in silico and examined how codon usage correlates with synthetic gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]