

Abstract
Identical studies employing stable isotopes were performed before and after a 3-day trial of oral N-carbamylglutamate (NCG) in 5 subjects with late onset carbamyl phosphate synthetase deficiency. NCG augmented ureagenesis and decreased plasma ammonia in 4 of 5 subjects. There was marked improvement in nitrogen metabolism with long-term NCG administration in one subject.
Keywords: N-acetyl-L-glutamate, acetylglutamate, carbamylglutamate, hyperammonemia, carbamoyl phosphate, urea cycle, stable isotopes
The detoxification of ammonia to urea requires a functional hepatic urea cycle (1), the disruption of which may result in hyperammonemia with ensuing encephalopathy, intellectual disability or death (2). The initial step of the urea cycle is catalyzed by carbamyl phosphate synthetase 1 (CPS1; EC 6.3.4.16) (3, 4). CPS1 is active only in the presence of its essential allosteric activator, N-acetylglutamate (NAG), which in turn is generated by NAG synthase (NAGS; EC 2.3.1.1.). In NAGS deficiency (5, 6), reduced flux through CPS1 results in hyperammonemia which is clinically indistinguishable from CPS1 deficiency (CPSD). In vivo studies have demonstrated that the stable analog of NAG, N-carbamylglutamate (NCG), can restore ureagenesis and normalize blood ammonia in patients with inherited NAGS deficiency (7). We therefore hypothesized that some patients with partial CPSD may respond to NCG supplementation. Current published evidence of the effect of NCG in CPS1 deficiency is limited (8-10). We report herein the results of a 3-day NCG trial in 5 subjects with late onset CPSD and document a favorable long-term outcome in one of the patients who had a positive response to the 3-day trial. We conclude that NCG could be an effective therapeutic option in some patients with CPSD.
Methods
This study was approved by the institutional review boards at Children's National Health System and the Children's Hospital of Philadelphia. Studies were performed as previously described (11, 12) (Figure 1; available at www.jpeds.com). This trial measures ureagenesis by monitoring the conversion of administered [1-13C]acetate to [13C]urea using ion ratio mass spectrometry. Blood ammonia, glutamine and urea were also determined prior to, and at the end of a 3-day treatment with NCG (Carbaglu [Orphan Europe, Paris, France]; 100mg/kg/day or 2.2g/m2/day if ≥25kg of body weight in 4 divided doses).
Figure 1.

Biomarkers, including ammonia, urea, glutamine and [13C]urea were measured before and after a 3-day trial of N-carbamylglutamate (NCG) 100 mg/kg/d or 2.2g/m2/d if ≥ 25kg.
On each study day, after an overnight fast, a baseline sample of heparinized blood was obtained. Then, at time 0, each subject received an enteral dose of [1-13C]sodium acetate, dissolved in water. Blood samples were subsequently obtained at 15, 30, 45, 60, 90, 120, 180, and 240 minutes. In subjects ≤ 5 years (subjects 3, 4 and 5), a shortened 120 min protocol was performed, and only samples at 15, 30, 60, and 120 minutes were collected. (From Heibel et al., Hum Mutat. 2011 Oct;32(10):1153-60)
Longitudinal linear regression analyses were applied to evaluate the time-averaged change in the respective analytes pre- and post-NCG treatment. These analyses allowed the adjustment of variance estimates for correlation between multiple measurements on the same subject. Because [13C]urea was used as the primary endpoint for the impact of NCG, we did not adjust the significance level for multiple comparisons.
Five subjects age 5 to 31 years were studied (Table; available at www.jpeds.com). All presented with hyperammonemia after the first week of life and had a definitive diagnosis of CPSD by liver CPS1 enzyme analysis and/or mutation analysis.
Table 1.
Online Only
| Subject | Age (y) | Sex | CPS1 Mutations | CPS1 Enzyme Activity | Age at Diagnosis | Peak NH3 at Diagnosis (μmol/L) |
|---|---|---|---|---|---|---|
| 1 | 25 | M | N/A | 0.38 umol/mg protein/hr | 17 months | N/A |
| (N: 1.65 ± 0.59) | ||||||
| 2 | 31 | F | c.267C>G (p.Y89X) | N/A | 27 years | 522 |
| c.2845G>A (p.A949T) | ||||||
| 3 | 5 | F | c.3101A>G (p.E1034G) | N/A | 15 months | 184 |
| c.3101A>G (p.E1034G) | ||||||
| 4 | 5 | M | c.2376G>C (p.M792I) | 0.9 μmol/g protein/min | 2 weeks | >500 |
| c.2376G>C (p.M792I) | (N: 5.6 ± 0.3) | |||||
| 5 | 5 | M | c.2548C>T (p.R850C) | N/A | 4 years | 150 |
| c.2882C>T (p.T961I) |
N/A = Not Available
Results
The pre- and post-NCG data were resistant to transformation to achieve normality, thus, the statistic employed is the median. However, the pre- to post-NCG difference, once log transformed (ln), achieved acceptable normality to permit parametric testing.
[13C]Urea
Figure 2 illustrates the [13C]urea trajectory over time for all 5 subjects. In each subject, both prior to, and after NCG treatment, plasma [13C]urea concentration increased rapidly, demonstrating rapid conversion of [13C]acetate to 13CO2 and its subsequent incorporation into urea. After receiving 3-d of NCG, all but subject 4 demonstrated greater levels of [13C]urea at every time point after 15 minutes, with marked differences in subjects 1,3 and 5. Additionally, the combined [13C]urea data revealed a trend towards significance (p=0.086) in the aggregate pre- vs. post-NCG [13C]urea. This became significant (p<0.001) when subject 4 was removed from the analysis.
Figure 2.

Isotopic enrichment in plasma concentrations of [13C]urea in 5 subjects with partial CPS1 deficiency and who were administered 27.5 mg/kg of [1-13C]sodium acetate. Subjects ≤ 5 years (subjects 3, 4 and 5) underwent a shortened study, with samples drawn only at time points 0, 15, 30, 60, and 120 minutes.
Ammonia, Glutamine, and Urea Nitrogen
Figure 3, A illustrates plasma ammonia levels for all subjects before and after NCG treatment. Each bar represents the median ammonia measurement in the pre- and post-NCG blood samples from each subject. Subjects with an elevated baseline ammonia level showed the greatest decline in response to NCG. The overall median illustrates a statistically significant decrease in ammonia from 115 to 82 μmol/L(p=0.02). The median aggregate glutamine (Figure 3, B) decreases slightly from 1009 to 868 μmol/L, but it is not statistically significant (p=0.57). Similarly, no change is observed in plasma urea nitrogen (2.9 vs. 3.2 mmol/L, p=0.99) (Figure 3, C).
Figure 3.
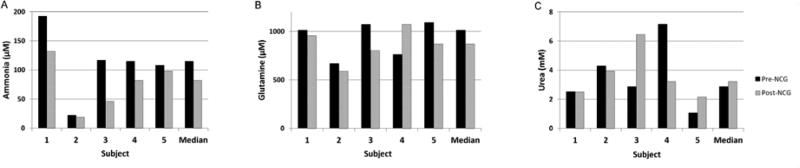
Plasma levels of ammonia (A), glutamine (B), and urea (C) in the 5 subjects before and after a 3-day treatment with NCG.
Prolonged trial of NCG
Following the study, subject 3's ammonia levels returned to her baseline of ~70-80 μmol/L. Because she did not tolerate higher doses of sodium phenylbutyrate and because the results of the 3-day trial were promising, she continued on a longer-term NCG trial at a dose of 100 mg/kg/d. Ammonia levels normalized within 3 days of restarting NCG. Sodium phenylbutyrate was slowly weaned over a 2-month period (Figure 4; available at www.jpeds.com).
Figure 4.

In subject 3, the 3-day trial of NCG showed an encouraging biochemical response that a longer trial was planned. Following baseline (day 0) measurement of ammonia, urea, and amino acids, she was prescribed NCG 100 mg/kg/d.
When a marked decrease in ammonia and glutamine was again observed, the sodium phenylbutyrate (Buphenyl) daily dose was tapered in 500 mg decrements over the ensuing 6 weeks. In a single instance (day 10), due to parental error, the phenylbutyrate dose was unintentionally reduced by 1500 mg/day, and this error was not identified until the subsequent clinical assessment. She remained clinically well throughout.
Discussion
We present evidence of the salutary effect of NCG on ureagenesis and plasma ammonia in some patients with partial CPSD. Results from the aggregate analysis, as well as from select subjects indicate that NCG increased urea production and decreased plasma ammonia, suggesting improved nitrogen disposal and likely reflecting increased CPS1 activity. Subject #3, in whom NCG reduced ammonia levels and increased hepatic ureagenesis, had a marked clinical improvement on NCG. We anticipate the response to NCG in CPSD to be mutation-specific.
In subject 4, [13C]urea decreased following NCG administration. In contrast, in virtually all of our prior studies with NCG, there was either an augmentation in ureagenesis, or no change. Although, no technical cause for this observation was identified, elements such as subclinical illness or dietary changes may explain this seemingly paradoxical response. It is unlikely, but still biochemically possible, that NCG could have paradoxically inhibited mutant CPS1 in this subject.
Until the NCG-responsive CPS1 deficiency genotypes are identified, we recommend that subjects with partial CPS1 deficiency be offered a trial of NCG. As in Subject 3 and other responsive subjects (7, 13), concurrent improvements in blood ammonia, glutamine and urea may be sufficient to predict long-term response to NCG.
We demonstrate the effects of 3-day administration of NCG on ammonia metabolism in patients with partial CPSD. Four of 5 subjects were responsive, and NCG treatment appears to have resulted in long-term clinical improvement due to improved urea cycle function in one subject, but possibly paradoxically decreased urea cycle function in another subject. Future directions of this research include identifying NCG-responsive CPS1 mutations and a clinical trial of NCG in acute hyperammonemia currently in progress.
Acknowledgments
We thank Charles Cottone, Michael Jones, and the Clinical Research Center staff at Children's National Health System for help in performing the study. We also thank Drs Rani Singh, Pasi Nevalainen, Rebecca Mardach, and Henry Lin for their patient referrals to this study.
Supported by the National Institutes of Health (R01HD058567, R01DK47870, R01DK64913, U54RR019453, P01HD26979, NS054900, and RR024134) and the O'Malley Family Foundation. The Urea Cycle Disorders Consortium (U54RR019453) is a part of the Rare Disease Clinical Research Network, supported through a collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, and Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Abbreviations
- CPS1
carbamyl phosphate synthetase 1
- CPSD
carbamyl phosphate synthetase 1 deficiency
- DNA
deoxyribonucleic acid
- NAG
N-acetyl-L-glutamate
- NAGS
N-acetyl-L-glutamate synthase
- NCG
N-carbamyl-L-glutamate
- OTC
ornithine transcarbamylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
References
- 1.Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004. Epub 2008/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310(23):1500–5. doi: 10.1056/NEJM198406073102304. Epub 1984/06/07. [DOI] [PubMed] [Google Scholar]
- 3.Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230(2):1013–21. Epub 1958/02/01. [PubMed] [Google Scholar]
- 4.Haraguchi Y, Uchino T, Takiguchi M, Endo F, Mori M, Matsuda I. Cloning and sequence of a cDNA encoding human carbamyl phosphate synthetase I: molecular analysis of hyperammonemia. Gene. 1991;107(2):335–40. doi: 10.1016/0378-1119(91)90336-a. Epub 1991/11/15. [DOI] [PubMed] [Google Scholar]
- 5.Bachmann C, Krahenbuhl S, Colombo JP, Schubiger G, Jaggi KH, Tonz O. N-acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304(9):543. doi: 10.1056/NEJM198102263040918. Epub 1981/02/26. [DOI] [PubMed] [Google Scholar]
- 6.Ah Mew N, Caldovic L. N-acetylglutamate synthase deficiency: an insight into the genetics, epidemiology, pathophysiology, and treatment. The application of clinical genetics. 2011;4:127–35. doi: 10.2147/TACG.S12702. Epub 2011/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64(2):213–7. doi: 10.1203/PDR.0b013e318179454b. Epub 2008/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guffon N, Schiff M, Cheillan D, Wermuth B, Haberle J, Vianey-Saban C. Neonatal hyperammonemia: the N-carbamoyl-L-glutamic acid test. J Pediatr. 2005;147(2):260–2. doi: 10.1016/j.jpeds.2005.04.059. Epub 2005/08/30. [DOI] [PubMed] [Google Scholar]
- 9.Williams M, Huijmans JGM, van Diggelen OP, van der Louw EJTM, de Klerk JBC, Haeberle J. Carbamoylphosphate Synthase (CPS1) Deficiency: Treatment with Carbglumic Acid (Carbaglu). Journal of Inherited Metabolic Disease. 2010;33(Supplement 1):S118. [Google Scholar]
- 10.Kuchler G, Rabier D, Poggi-Travert F, Meyer-Gast D, Bardet J, Drouin V, et al. Therapeutic use of carbamylglutamate in the case of carbamoyl-phosphate synthetase deficiency. J Inherit Metab Dis. 1996;19(2):220–2. doi: 10.1007/BF01799434. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- 11.Heibel SK, Ah Mew N, Caldovic L, Daikhin Y, Yudkoff M, Tuchman M. N-carbamylglutamate enhancement of ureagenesis leads to discovery of a novel deleterious mutation in a newly defined enhancer of the NAGS gene and to effective therapy. Hum Mutat. 2011;32(10):1153–60. doi: 10.1002/humu.21553. Epub 2011/06/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ah Mew N, Payan I, Daikhin Y, Nissim I, Tuchman M, Yudkoff M. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009;98(4):325–30. doi: 10.1016/j.ymgme.2009.07.010. Epub 2009/08/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 2010;126(1):e208–14. doi: 10.1542/peds.2010-0008. Epub 2010/06/23. [DOI] [PMC free article] [PubMed] [Google Scholar]